

Abstract
Histones mediate dynamic packaging of nuclear DNA in chromatin, a process that is precisely controlled to guarantee efficient compaction of the genome and proper chromosomal segregation during cell division and to accomplish DNA replication, transcription, and repair. Due to the important structural and regulatory roles played by histones, it is not surprising that histone functional dysregulation or aberrant levels of histones can have severe consequences for multiple cellular processes and ultimately might affect development or contribute to cell transformation. Recently, germline frameshift mutations involving the C-terminal tail of HIST1H1E, which is a widely expressed member of the linker histone family and facilitates higher-order chromatin folding, have been causally linked to an as-yet poorly defined syndrome that includes intellectual disability. We report that these mutations result in stable proteins that reside in the nucleus, bind to chromatin, disrupt proper compaction of DNA, and are associated with a specific methylation pattern. Cells expressing these mutant proteins have a dramatically reduced proliferation rate and competence, hardly enter into the S phase, and undergo accelerated senescence. Remarkably, clinical assessment of a relatively large cohort of subjects sharing these mutations revealed a premature aging phenotype as a previously unrecognized feature of the disorder. Our findings identify a direct link between aberrant chromatin remodeling, cellular senescence, and accelerated aging.
Keywords: linker histone, HIST1H1E, linker histone H1.4, cellular senescence, replicative senescence, accelerated aging, chromatin compaction, chromatin remodeling, chromatin dynamics, methylation profiling
Introduction
In eukaryotic cells, nuclear DNA is organized in a complex and dynamic structure called chromatin, which allows efficient packaging of the genome and proper chromosomal segregation during mitosis; chromatin also controls the genome’s accessibility for essential processes such as replication, transcription, and repair.1 The basic unit of chromatin is the nucleosome, in which a stretch of about 147 bp of DNA wraps around an octamer of the core histones (i.e., H2A, H2B, H3, and H4), constituting the nucleosomal core particle (NCP).2 A number of H1 linker histone isoforms bind to short DNA segments at the entry and exit sites on the surface of the NCP, stabilizing the nucleosomal structure and contributing to higher-order chromatin folding.3, 4 Although H1 histones have traditionally been associated with chromatin compaction and a regulatory function favoring transcriptional repression,5 a more complex and plastic role of these proteins in the control of accessibility to DNA has gradually been appreciated.6, 7, 8 In mammals, H1 histones are encoded by 11 genes. Among these, seven are expressed in somatic cells, and the remaining four are transcriptionally active in the germline.3, 4 Of note, even among the seven somatic subtypes, the levels of these proteins appear to be regulated during the cell cycle and development, and in different tissues and cell types, suggesting specific functions for individual isoforms.9 On the other hand, redundancy in their function has been reported.10, 11 In higher eukaryotes, H1 histone isoforms share a tripartite structure consisting of a central highly conserved globular domain and two less-conserved, unstructured N-terminal and C-terminal tails. The globular domain has high sequence homology among H1 subtypes and mediates binding to the nucleosome. The two tails are moderately conserved among orthologs but differ among isoforms, suggesting functional specificity. Notably, both tails encompass a number of residues that are subjected to reversible modifications with regulatory function. Among these, the extent of phosphorylation in serine/threonine residues at the C-terminal tail has been proposed to regulate chromatin dynamics in interphase as well as chromosome condensation during mitosis.4, 12
Aberrant histone function, whether due to mutations in genes coding proteins that participate in histone-modifying complexes or mutations directly affecting histone-coding genes, has been established as contributing to oncogenesis and causing multisystem syndromes that affect growth and cognitive function.13, 14, 15, 16, 17, 18 Recently, germline frameshift mutations affecting HIST1H1E (MIM: 142220), which codes a member of the somatic, replication-dependent linker histone subfamily, have been causally linked to an as-yet poorly defined syndrome that includes intellectual disability (ID) (MIM: 617537).19 Mutations were mapped at the C-terminal tail of HIST1H1E and were predicted to have an equivalent functional impact by generating the same change in the reading frame. The five affected individuals belonged to a cohort of subjects with overgrowth, and were reported to have a similar facial appearance but variable height, head circumference, and degree of ID.19 Notably, the growth pattern of these individuals appeared to be complex and characterized by a height that was above average during infancy but that became progressively closer to average over time until it could be characterized as average or short in adulthood. This peculiar pattern of growth has been highlighted by a subsequent report, which also confirmed the association of this class of HIST1H1E mutations with ID and specific facial features.20
Here, we report that this homogeneous class of disease-causing frameshift mutations affecting the C-terminal tail of HIST1H1E results in stable proteins that reside in the nucleus and bind to chromatin but disrupt proper compaction of DNA and are associated with a specific methylation profile. We also provide data indicating that cells expressing these mutant proteins have a dramatically reduced proliferation rate and competence, hardly enter into the S phase, and undergo accelerated senescence. Remarkably, clinical assessment of 13 newly identified individuals who are heterozygous for this class of mutations and of those previously reported allowed us to identify premature aging as a previously unrecognized feature of the disorder. Collectively, these data highlight a strict link between aberrant chromatin remodeling, cellular senescence, and accelerated aging.
Material and Methods
Subjects
This study was approved by the Committee for Medical Ethics, University of Antwerp, Antwerp, and Ethical Committee, Ospedale Pediatrico Bambino Gesù, Rome. Clinical data and DNA specimens from the subjects included in this study were collected according to procedures conforming to the ethical standards of the declaration of Helsinki protocols and approved by the review boards of all involved institutions, and signed informed consent was obtained from the participating subjects and/or families. Explicit permission was obtained to publish the photographs of the subjects as shown in Figure 1.
Figure 1.

Facial Appearance of Subjects with HIST1H1E Frameshift Mutations and Protein Structure
(A) In affected individuals, facial appearance is characterized by a high anterior hairline, prominent forehead, bitemporal narrowing, sparse temporal hair, hypertelorism, hooded eyelids, short palpebral fissures, a high and broad nasal bridge, and a full nasal tip; small, widely spaced teeth; and low-set ears. A facial appearance compatible with a more advanced age (e.g., hypotrichosis and ptosis) is evident in S1 and S3. (S1, 49 years; S3, 30 years at last evaluation; S4, 14 months; S5, 12 years at last evaluation; S6, 3 years; S7, 12 years; S9, 2 years at last evaluation; S11, 6 years at last evaluation; and S12, 4 years)
(B) Schematic diagram representing the HIST1H1E structure, which is composed of a globular domain flanked by N- and C-terminal tails. The position of the disease-causing frameshift mutations is shown above the cartoon. (Novel mutations are highlighted in red.) The number of independent cases identified in the present study is in brackets. The domain boundaries and cyclin-dependent kinase phosphorylation sites (black triangles) are reported below the cartoon. All mutations are expected to result in a shorter protein with an identical divergent C-terminal tail. (The new stop codon is shown below the cartoon, 194.∗)
Exome Sequencing
WES was performed with DNA samples obtained from leukocytes. In most cases, a trio-based strategy was used. Exome capture was carried out with the Nimblegen SeqCap EZ Exome v3 (Roche) (subjects 1 and 5), Clinical Research Exome (Agilent) (subjects 2 and 3), SeqCap EZ VCRome v2.1 (Roche) (subjects 6 and 7), SureSelect Human All Exon v4 (Agilent) (subjects 8, 9 and 13), SureSelect Human All Exon v5 (Agilent) (subjects 4 and 12), and SureSelect XT Human All Exon v6 (Agilent) (subjects 10 and 11) target enrichment kits, and sequencing was performed on HiSeq 2000/2500/4000 and NextSeq550 platforms (Illumina) through the use of paired-end reads. WES data processing, sequence alignment to GRCh37, and variant filtering and prioritization by allele frequency, predicted functional impact, and inheritance models were performed as previously described.21, 22, 23, 24, 25, 26, 27 The de novo origin of the HIST1H1E frameshifts was confirmed by Sanger sequencing in all cases.
RNA Stability
Total RNA was isolated from circulating leukocytes of subjects 1 and 2 (S1 and S2, hereafter) through the use of the RNeasyMiniKit (QIAGEN). Reverse transcription was performed with the SuperScriptIII first strand kit (Invitrogen) according to the manufacturer’s instructions. Primer sequences used for RT-PCR are available upon request.
Constructs
The c.441dupC (p.Lys148Glnfs∗48), c.464dupC (p.Lys157Glufs∗39), and c.441_442insCC (p.Lys148Profs∗82) were introduced by site-directed mutagenesis in a HIST1H1E cDNA (RefSeq: NM_005321.2, NP_005312.1) tagged with Xpress at the N terminus and cloned in pcDNA6/His version C (Invitrogen).
Cell Cultures and Transfections
Skin fibroblasts isolated from skin biopsies (subjects S1 and S2 and healthy donors), and COS-1 and HeLa lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum (GIBCO) and 1% penicillin-streptomycin, at 37°C with 5% CO2.
SCGE Assay
Samples were processed according to the alkaline single-cell gel electrophoresis (SCGE) assay protocol, as previously described.28 In brief, cells were suspended in 0.7% low-melting agarose. Slides were prepared in duplicates with control cells and fibroblasts derived from affected individuals placed on opposite sides of the same slide, immersed in cold lysis solution, and kept at 4°C overnight. After lysis, slides were transferred in alkaline buffer for 20 min. Electrophoresis was carried out for 20 min at 20 V and 300 mA (0.8 V/cm) at 4°C. We explored the basal level of nucleoid relaxation by applying longer electrophoresis run times (40 and 60 min). Slides were neutralized in 0.4 M Tris (pH 7.5) for 5 min, treated with absolute ethanol, and stored at room temperature. Slides were then stained with GelRed (Biotium) and scored at a fluorescence microscope (Leica). To evaluate induced DNA damage and DNA repair capability, we irradiated fibroblasts with 1 or 2 Gy γ-rays from a 137Cs source at a dose rate of 0.8 Gy/min. During treatment, cells were maintained at 0°C so that DNA repair would be prevented. The kinetics of DNA repair were assessed by SCGE assay, as described above. The residual DNA damage was measured after 15 and 30 min of incubation at 37°C. For each experimental point, at least 75 cells were analyzed.
Histone Modifications and Nucleolus Morphology Assessment
For immunofluorescence, fibroblasts from S1 and control subjects were seeded at a density of 20 × 103 in 24-well cluster plates onto 12 mm cover glasses. After 24 h of culture in complete medium, cells were fixed with 3% paraformaldehyde. Following permeabilization with 0.5% Triton X-100 (10 min at room temperature), fibroblasts were stained with anti-dimethyl-Histone H3 (lys4) rabbit polyclonal antibody, anti-trimethyl-histone H3 (lys9) rabbit polyclonal antibody, anti-trimethyl-histone H3 (lys27) rabbit polyclonal antibody (Millipore), and anti-heterochromatin protein-1β mouse monoclonal antibody (Chemicon) followed by the appropriate secondary antibody (Invitrogen) and DAPI. To study nucleolus morphology, we stained fibroblasts with C23 (MS-3) mouse monoclonal antibody (Santa Cruz) followed by the appropriate secondary antibodies (Invitrogen) and DAPI. Observations were performed on a Leica TCS SP2 AOBS apparatus. Cells stained only with the fluorochrome-conjugated secondary antibodies were used for setting up acquisition parameters. Signals from different fluorescent probes were taken in sequential scanning mode, several fields (>200) were analyzed for each labeling condition, and representative results are shown.
Proliferation and Cell-Cycle Assays
Cells were seeded at 200,000 cells per well in a 6-well plate and incubated a 37°C. Cell numbers (mean of three replicates ± SD) were counted by trypan blue exclusion after 4 and 7 days. The percentages of cells in different cell-cycle phases were determined by dual flow-cytometry analysis of BrdU-positive cells stained with the fluorescent DNA probe propidium iodide (PI). In brief, cells were incubated for 1 h with BrdU (Sigma Aldrich) at a final concentration of 30 μM. Then BrdU was removed, and cells were rinsed with PBS, harvested, and permeabilized in ice-cold 100% EtOH. Cells were incubated with HCl 3N to denature DNA and then with 0.1 M sodium tetraborate to stop this reaction. Fibroblasts were incubated with the anti-BrdU antibody (Invitrogen) followed by goat anti-mouse Alexa Fluor 488 secondary antibody. Finally, cells were resuspended in a buffer containing 10 μg/mL RNase A and 20 μg/mL PI and then immediately analyzed through the use of FACSCalibur (BD Biosciences).
SA-β-gal Activity and P53 Evaluation
SA-β-gal activity was assessed as reported.29 In brief, cells were fixed with 3.6% formaldehyde in PBS for 4 min at room temperature. Fixed cells were washed and then incubated overnight with freshly prepared staining solution at 37°C in the absence of CO2. After being washed, the coverslips were mounted with the antifade Dako fluorescence mounting medium (Agilent Technologies). P53 endogenous levels were evaluated on fibroblast lysates collected at different passages using an anti-p53 mouse monoclonal antibody (Invitrogen). Membranes were probed with an anti-GAPDH mouse monoclonal antibody (Santa Cruz) so that protein content would be normalized.
Analysis of Nuclear Morphology
After 24 h of culture in complete medium, fibroblasts were treated with 2 mM thymidine (Sigma) for 24 h, washed with 1× PBS, recovered with complete medium for 3 h, and then treated with 100 ng/mL nocodazole (Sigma) for 12 h. Afterward, fresh drug-free medium was added, and recovery was allowed for 120 min during which cells were fixed every 15 min in PHEMO buffer for 10 min at room temperature, as previously described.30 Finally, cells were stained with lamin A/C mouse monoclonal antibody (Santa Cruz) followed by the appropriate secondary antibody (Invitrogen) and DAPI. Observations were performed on a Leica TCS SP2 AOBS apparatus as described above.
Evaluation of rRNA Content
Total RNA was extracted from the same amount of control fibroblasts and cells derived from affected individuals (400,000 fibroblasts) at different cellular passages. Three μL of total RNA was loaded for size separation on 1% agarose gel and stained with ethidium bromide.
Evaluation of Protein Stability
COS-1 cells were seeded in 6-well plates the day before transfection. Cells were transfected at 70% confluency with Fugene 6 transfection reagent (Promega) with wild-type or mutant Xpress-tagged HIST1H1E expression constructs. Forty-eight h after transfection, cells were treated with cycloheximide (20 μg/mL) for 8 and 16 h or left untreated before lysis. Immunoblotting was performed so that Xpress-tagged HIST1H1E levels could be assessed. Probing membranes with an anti-GAPDH antibody (Santa Cruz) allowed normalization of protein content.
CSK Assay
Twenty thousand fibroblasts and 30 × 103 HeLa cells were seeded on a glass coverslip and maintained in culture complete medium for 24 h. HeLa cells were transfected with 100 ng of vectors expressing Xpress-tagged HIST1H1E. Forty-eight h after transfection, HeLa cells were either fixed with 3% paraformaldehyde or treated with CSK buffer before being fixed.31 After permeabilization with 0.5% Triton X-100 (for 10 min at room temperature), HeLa cells were stained with monoclonal antibody to Xpress (Invitrogen) and then with the appropriate secondary antibody (Invitrogen) and DAPI for DNA. Analyses were performed in three independent experiments on a Leica TCS SP2 AOBS apparatus.
Analysis of Cell Morphology
Fibroblasts from subject S1 and a control individual were seeded in a 100 mm dish cell culture. Optical microscope images at different passages were taken with Floid Cell Imaging Station (Life Technologies).
γH2AX Immunofluorescence Staining
Cells were seeded on a glass in a Petri dish for 24 h. The slides were fixed with 4% paraformaldehyde (Sigma Aldrich), permeabilized in 0.2% Triton X-100, and blocked in PBS/BSA 1% for 30 min at room temperature. Slides were incubated with a mouse monoclonal anti-phospho-histone H2AX antibody (Millipore) overnight at 4°C, washed in PBS/BSA 1%, and then exposed to the secondary Alexa 488-labeled donkey anti-mouse antibody (Invitrogen, Life Technologies) for 1 h at 37°C. After washes in PBS/BSA, 1% DNA were counterstained with DAPI (Sigma Aldrich) in Vectashield (Vector Laboratories). Cells were analyzed with fluorescence microscopy using an Axio Imager Z2 microscope (Zeiss). The frequency of foci per cell was scored in 150 nuclei in two independent experiments.
Telomere Dysfunction-Induced Foci (TIF) ImmunoFISH Staining
Cells were seeded on glass slides that were processed as reported for the γH2AX immunofluorescence staining up to the secondary antibody (Alexa 488-labeled donkey anti-mouse, Invitrogen). Immediately after, slides were washed in PBS/Triton X-100 0.05%, fixed in 4% formaldehyde for 2 min and dehydrated through graded alcohols (70%, 80%, and 100%). Slides and probes (Cy3 linked telomeric PNA probe, DAKO) were co-denatured at 80°C for 3 min and hybridized for 2 h at room temperature in a humidified chamber. After hybridization, slides were washed twice for 15 min in 50% formamide, 10 mM Tris, pH 7.2, and 0.1% BSA followed by three 5-min washes in 0.1 M Tris, pH 7.5; 0.15 M NaCl; and 0.08% Tween 20. Slides were then dehydrated with an ethanol series and air dried. Finally, slides were counterstained with DAPI (Sigma Aldrich) in Vectashield (Vector Laboratories). Co-localization between γH2AX foci and telomere were analyzed using an Axio Imager Z2 microscope (Zeiss). The frequency of co-localization dots per cell was scored in 150 nuclei in two independent experiments.
Quantitative-Fluorescence In Situ Hybridization Analysis (Q-FISH)
Chromosome spreads were obtained as reported above. Q-FISH staining was performed as previously described,32 with minor modifications. In brief, slides and probes (Cy3 linked telomeric, PANAGENE, and chromosome 2 centromeric PNA probes, DAKO) were co-denatured at 80°C for 3 min and hybridized for 2 h at room temperature in a humidified chamber. Slides were counterstained with DAPI (Sigma Aldrich) in Vectashield (Vector Laboratories). Images were captured at a 63X magnification with an Axio Imager Z2 (Zeiss). ISIS software (MetaSystems) was used to assess telomere size (defined as ratio between total telomeres’ fluorescence [T] and fluorescence of the centromeres of the two chromosomes 2 [C]). Data were expressed as T/C %.33 Experiments were repeated two times and at least 10–15 metaphases were scored each time.
Detection of Aneuploidy
Chromosome spreads were obtained as described before. Metaphases were captured at 63× magnification using an Axio Imager Z2 microscope (Zeiss). We considered euploidy to be the metaphases with 46 chromosomes and aneuploidy to be all of the metaphases that had a different number of chromosomes. The frequency of aneuploidy was scored in 40 metaphases. Two independent experiments were performed.
Chromosome Condensation Assay
After 30 min incubation with 30 μM Calyculin-A (Wako), a 28 min incubation at 37°C with hypotonic solution (75 mM KCl), and fixation in freshly prepared Carnoy solution (3:1 v/v methanol/acetic acid), chromosome spreads were obtained. Slides were stained with DAPI (Sigma Aldrich) in Vectashield (Vector Laboratories), and an Axio Imager Z2 microscope (Zeiss) was used. To analyze the frequency of chromosomal condensation, we counted the number of metaphases in 150 nuclei per sample in two independent experiments.
Methylome Profiling
We assessed the DNA methylation levels of one adult and five pediatric affected individuals through the use of the Infinium Human Methylation EPIC BeadChip assay (Illumina), which allows comprehensive genome-wide coverage, and genomic DNA extracted from leukocytes. Methylation levels were measured as beta values, percentages of methylation at each CpG site, and ranged from 0 (i.e., no methylation) to 1 (full methylation). Raw data were processed with the bioconductor package ChAMP34 and the default parameters were used for filtering low-quality signals and normalizing data. Methylation profiles were compared with those obtained from a series of 35 healthy adult male controls of European descent; these included 31 individuals selected from a European cohort study (average age ± SD: 56.1 ± 7.4 years).35 Unsupervised multidimensional scaling (MDS) analysis was used, and the 1,000 most variable probes among samples were taken into account. Statistical analyses were conducted with R software (v. 3.5.0). We performed gene and pathway enrichment analyses of the differentially methylated probes by using the Gene SeT AnaLysis online tool36 on the associated genes. Functional annotation was carried out by means of GO terms including biological processes, molecular functions, and cellular components classes. Pathway analysis was performed using KEGG. Statistical significance was assumed at 0.05 following multiple testing adjustment (FDR).
Results
Spectrum of HIST1H1E Mutations and Associated Clinical Features
Using whole-exome sequencing (WES), we identified a novel de novo HIST1H1E frameshift variant, c.441dupC (p.Lys148Glnfs∗48), in an adult individual with hypotrichosis, cutis laxa, and ID (S1) (Figure 1A). Mutation analysis performed on primary skin fibroblasts and hair bulb epithelial cells from the affected individual supported our hypothesis of a germline origin of the frameshift. The variant had not been reported in ExAC/gnomAD, and this variant affected a relatively large and highly conserved portion of the encoded protein. In the subject, WES data analysis excluded the presence of other functionally relevant variants compatible with known Mendelian conditions on the basis of the expected inheritance model and clinical presentation. Through networking and a GeneMatcher search,37 we identified 12 additional individuals with similar de novo frameshift HIST1H1E mutations (Figure 1B, Table 1). All changes were short out-of-frame indels resulting in almost identical shorter proteins that contained a shared divergent C-terminal tail (Table S1); these results were consistent with previous reports.19, 20, 38, 39 None of the frameshifts had been reported in gnomAD, all were predicted to dramatically impact protein function, and two of them had already been reported in ClinVar as disease-causing mutations (Table 1).
Table 1.
Frameshift HIST1H1E Mutations Identified in This Study
| Nucleotide Changea | gnomAD | ClinVar | Amino Acid Change | Domain | CADDb | Number of Cases | Origin |
|---|---|---|---|---|---|---|---|
| c.408dupG | - | - | p.Lys137Glufs∗59 | C-terminal tail | 34 | 1 | de novo |
| c.414dupC | - | - | p.Lys139Glnfs∗57 | C-terminal tail | 35 | 2 | de novo (1) |
| c.425_431delinsAGGGGGTT | - | - | p.Thr142Lysfs∗54 | C-terminal tail | 31 | 1 | de novo |
| c.425delinsAG | - | - | p.Thr142Lysfs∗54 | C-terminal tail | 29 | 1 | undetermined |
| c.430dupG | - | reported | p.Ala144Glyfs∗52 | C-terminal tail | 26.8 | 2 | undetermined |
| c.441dupC | - | reported | p.Lys148Glnfs∗48 | C-terminal tail | 34 | 4 | de novo |
| c.447dupG | - | - | p.Ser150Glufs∗46 | C-terminal tail | 34 | 1 | de novo |
| c.464dupC | - | - | p.Lys157Glufs∗39 | C-terminal tail | 35 | 1 | de novo |
Nucleotide numbering reflects cDNA numbering with 1 corresponding to the A of the ATG translation initiation codon in the HIST1H1E reference sequence (RefSeq: NM_005321.2, NP_005312.1).
ClinVar submission IDs: c.408dupG, SCV000925231; c.414dupC, SCV000925232; c.425_431delinsAGGGGGTT, SCV000925233; c.425delinsAG, SCV000925234; c.430dupG, SCV000925235; c.441dupC, SCV000925236; c.447dupG, SCV000925237; and c.464dupC, SCV000925238.
CADD v1.4.
We analyzed the clinical records of the 13 identified subjects (S1 to S13) and collected the information available for the seven previously published individuals (S14 to S20). All subjects had variable developmental delay (DD)/ID and a distinctive facies characterized by bitemporal narrowing (in 100% of individuals), a prominent forehead (93%), and a high anterior hairline (88%) as main features, which were reminiscent of Pallister-Killian syndrome (MIM: 601803) (Figure 1A). In four children, Sotos syndrome (MIM: 117550) had been suggested, and Weaver syndrome (MIM: 277590), Simpson-Golabi-Behmel syndrome (MIM: 312870), and Pallister-Killian syndrome had been suspected in single cases (Table S2). Half of the individuals had scaphocephaly and sparse frontotemporal hair. The craniofacial appearance was characterized by hypertelorism (91%), downslanted palpebral fissures (67%), a broad nasal tip (83%), and low-set and posteriorly rotated ears (57%). Forty-three percent of subjects showed small, widely spaced teeth. Skin hyperpigmentation was present in one-third of individuals. Neonatal problems included hypotonia, feeding difficulties, failure to thrive, jaundice, congenital hypothyroidism, and micrognathia. Three individuals showed single palmar creases possibly related to prenatal hypotonia. Finger abnormalities included short fourth metacarpals, camptodactyly, fifth finger clinodactyly, brachydactyly, and broad and low-set thumbs. Toe abnormalities included long halluces, broad toes, and fifth toes overlapping fourth toes. Two individuals had pectus excavatum. Growth parameters at birth were unremarkable. At last evaluation, only one subject (S10) had macrosomy (>3.65 SD), and 12 individuals (63%) had a large head circumference (>+2.0 SD), including two children with relative macrocephaly at the age of 4 years. DD was present in all individuals. They were mildly to moderately intellectually disabled. Fourteen individuals had speech delay, and gross motor delay was a common feature. In young individuals, hypotonia and a stiff, clumsy, and uncoordinated gait were relatively common findings. The 49-year-old individual (S1) had gait ataxia. Behavioral features included ADHD (two individuals), autistic features (five individuals), and psychotic episodes (one individual). One child had focal seizures in early childhood, and another child was treated for recurrent status epilepticus. Brain MRI revealed aspecific abnormalities, including mild inferior vermian hypoplasia, delayed myelination, partial agenesis of the corpus callosum, and mild to moderately enlarged third and lateral ventricles. Five individuals presented with mild hearing loss. Visual problems included hypermetropia, myopia, astigmatism, and strabismus. Sixty percent of the individuals had feeding or eating difficulties, ranging from satiety problems at younger ages to problems with swallowing fluids or learning to eat solid food. One child was fed by G-tube at the age of 2.5 years. She had gastro-esophageal reflux disease and rumination disorder. Other remarkable features in this cohort included pancytopenia accompanying systemic lupus erythematosus in the oldest individual. A summary of the major features is reported in Table 2, and a more detailed description for each subject is available in Table S2.
Table 2.
Summary of the Clinical Features Occurring in Subjects Carrying de novo HIST1H1E Frameshift Mutations
| Clinical Features | Frequencya |
|---|---|
| DD/ID | 18/18b(100%) |
| motor delay | 12/12 (100%) |
| walking independently, range (mean), months | 15–66 (31) |
| speech delay | 14/14 (100%) |
| hypotonia | 11/15 (73%) |
| autistic features | 5/14 (36%) |
| Craniofacial Features | 19/19 (100%) |
| macrocephaly | 12/19 (63%) |
| scaphocephaly | 6/11 (54%) |
| sparse hair | 6/12 (50%) |
| high anterior hairline | 14/16 (87%) |
| prominent forehead | 13/14 (93%) |
| hypertelorism | 10/11 (91%) |
| downslanted palpebral fissures | 8/12 (67%) |
| full nasal tip | 10/12 (83%) |
| low set ears | 8/14 (57%) |
| Aging Appearence | 20/20 (100%) |
| skin hyperpigmentation | 5/15 (33%) |
| hypotrichosis | 6/20 (30%) |
| skin hyperpigmentation | 5/20 (25%) |
| nail abnormalities | 6/20 (30%) |
| dental problems | 6/20 (30%) |
| advanced bone age | 4/20 (20%) |
| Other | 9/15 (60%) |
| feeding or eating difficulties | 9/15 (60%) |
| hearing loss | 5/14 (36%) |
| strabismus | 8/15 (53%) |
The reported cohort includes 13 presently described subjects and seven previously reported cases.
Subjects showed variable ID: five mild, nine moderate, and two severe. In two cases, the severity of ID was unspecified.
Notably, the oldest individuals of the present cohort (S1 and S3) and two previously reported subjects (S15 and S17) had facial appearances compatible with a much more advanced age. When we evaluated the clinical presentation of the entire cohort of affected individuals, including those previously reported, we noticed an overrepresentation of features that are generally less common for individuals in this age group but more commonly found in elderly individuals. These features included hypotrichosis, ptosis, cutis laxa, hyperkeratosis, skin hyperpigmentation, dry skin, nail abnormalities, hearing loss, cataracts, diabetes mellitus, and osteopenia (Table S3). One girl was diagnosed with childhood hypophosphatasia (osteopenia and advanced bone age that was investigated after she presented with multiple small stress fractures of the lower limbs). Three other children also had advanced bone age. A fifth child had multiple fractures after minor trauma. Several children had dental problems including missing permanent teeth and small, fragile teeth.
Functional Characterization of HIST1H1E Mutations
Based on the unique narrow spectrum of mutations, we hypothesized a specific disruptive impact on HIST1H1E function as a consequence of the frameshift. Haploinsufficiency was considered unlikely because endogenous HIST1H1E mRNA levels from two unrelated affected subjects (S1, aged 49 years, and S2, aged 4 years) were comparable to those of controls, and transient transfection experiments involving Xpress-tagged HIST1H1E constructs in COS-1 cells documented an increased stability of the mutant proteins compared to the wild-type protein (Figure S1). Furthermore, confocal microscopy analysis performed in transiently transfected HeLa cells showed proper nuclear localization and stable binding to chromatin of the tested mutants (Figure S2). These data, along with the finding that a mutant allele expressing the third open reading frame at an equivalent position of the C terminus (this allele does not occur in affected individuals) was characterized by compromised chromatin binding, indicating loss of function (Figure S2), strongly suggested a dominant negative or neomorphic effect as the mechanism of disease.
H1 linker histones are core chromatin components, and they bind to short DNA segments at the entry and exit sites on the surfaces of nucleosomes, stabilizing their structure and regulating chromatin folding.4 The C-terminal tail of H1 histones contains a number of serine/threonine residues that undergo reversible phosphorylation, modulating the dynamics of chromatin compaction; these residues are lost in disease-associated HIST1H1E mutants. Of note, although partial phosphorylation of the C-terminal tail allows chromatin relaxation during interphase, full phosphorylation is required for maximal chromatin condensation during mitosis.4, 12 To assess chromatin compaction, we performed a single-cell gel electrophoresis (SCGE) assay in primary fibroblasts from subjects S1 and S2. Increasing run times allowed DNA loops to extend under the electrophoretic field and showed significantly more nucleoid relaxation in fibroblasts from affected individuals than in control cells; these results were quantified as “tail moment” values (Figure 2A). Because chromatin compaction is associated with a specific pattern of histone modifications, including methylation of histone H3 at specific lysine residues,40 the methylation profile of histone H3 was assessed in the two fibroblast lines. Consistent with the SCGE data, a decrease in H3K4me2, H3K9me3, and H3K27me3 staining was documented by immunofluorescence analyses (Figure 2B). In line with these findings, reduced levels of HP1β, a member of the HP1 family mediating heterochromatinization by binding to methylated H3K9, and whose level is reduced in cells with a defective heterochromatin state,41 was also observed (Figure 2C). Finally, a caliculin-induced premature chromosome condensation assay documented significantly fewer condensed chromosomes in fibroblasts from affected individuals (11.3% of analyzed cells) than in control cells (19.6%) (p < 0.01, χ2 test). Overall, these data indicated a more relaxed state of chromatin in cells expressing the disease-causing HIST1H1E mutants.
Figure 2.

SCGE Assay, Immuno-Fluorescence Studies, and Methylation Profiling Analysis
(A) Increasing electrophoresis run times (20, 40, and 60 min) highlighted significant differences in relaxation of DNA supercoiling between fibroblasts from control and from affected individuals. DNA migration was quantified as Tail moment values, which are defined as the products of the tail length and the fraction of total DNA in the tail (upper panel). Nucleoids of cells from subjects S1 and S2 showed a significantly higher Tail moment value (∗p < 0.05, ∗∗p < 0.01; two-tailed Student’s t test). For each experimental point, at least 75 cells were analyzed. Values are mean ± SEM of three independent experiments. Representative images of nucleoids from fibroblasts from control and affected individuals at each run time are shown (lower panel).
(B) Confocal laser scanning microscopy (CLSM) observations document overall decreased amounts of H3K4me2, H3K9me3, and H3K27me3 staining (green) in S1 cells compared to control cells. Nuclei were stained with DAPI. Images are representative of >200 analyzed cells. Scale bars represent to 8 μm.
(C) Fibroblasts from subject S1 show a decreased amount of HP1β compared to control cells (WT). Cells were stained with antibodies against HP1β (red) and HIST1H1E (H1.4) (green); DNA are DAPI-stained (blue). Scale bars represent 7 μm.
(D) Multidimensional scaling plot of genome-wide methylation analysis using the top 1,000 most variable probes among samples. The plot shows the distinct methylation profiles of pediatric (open circles) and adult (S1, duplicate) (filled circles) affected individuals, compared to healthy controls (filled squares).
(E) DNA damage was induced by 1 or 2 Gy γ-ray irradiation. Tail moment values indicate the amount of radiation-induced DNA damage measured by SCGE assay immediately after treatment. S1 and S2 fibroblasts showed a higher sensitivity to γ-ray irradiation (∗p < 0.02, ∗∗p < 0.001; two-tailed Student’s t test). For each experimental point, at least 75 cells were analyzed. Values are means ± SEM of three independent experiments.
Methylome Profiling
Because altered chromatin condensation is expected to impact DNA methylation,42 we used a genome-wide methylation profiling analysis to investigate perturbations at the epigenome level. To this end, Infinium MethylationEPIC BeadChip profiling was used for comparing a subset of DNA samples obtained from leukocytes of six affected individuals to samples from a control group composed of 35 healthy individuals of European descent. When the entire set of assayed target probes is considered, the analysis did not highlight a substantial change in methylation pattern (data not shown). However, MDS analysis was carried out taking into account the information associated with the 1,000 most differentially methylated probes among all samples (independently from their group classification), and the results indicated clear-cut divergence between the methylation profiles of healthy controls and those of affected individuals, the latter clustering according to the ages of the individuals (Figure 2D). Among these informative probes, 170 differentially methylated CpGs were located within the UCSC CpG islands (from 200 bp to 1500 bp from TSS) (Table S4). Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) cellular component enrichment analyses revealed a significant enrichment of genes linked to pathways mainly related to neurological, immunological, and cell adhesion/membrane function; these represent cellular processes relevant to the clinical phenotype of affected individuals (Table S5). A separate clustering of affected pediatric individuals from controls was also observed through the use of a recently defined episignature set, which successfully profiled 14 developmental disorders (data not shown);43 this further supports the occurrence of a specific methylation profile in people with HIST1H1E frameshift mutations. These findings are in line with those of previous studies performed in cells with defective histone H1 function, which showed a minor impact on global DNA methylation compared to controls. Rather, in these cells, changes involved specific CpGs within regulatory domains of regulated genes,44 indicating that chromatin rearrangement in these cells does not globally impact the methylation status of DNA but instead affects specific subsets of genes and cellular processes.
HIST1H1E Mutations Cause Accelerated Cellular Senescence
On the basis of the occurrence of phenotypic features suggestive of premature aging and the fact that a more relaxed compaction of chromatin has been functionally linked to replicative senescence,45 a feature occurring in progeroid syndromes,46 we hypothesized that the aberrant function of the HIST1H1E mutants might promote accelerated senescence. To test this hypothesis, we evaluated proliferative competence and morphological and biochemical markers of cellular senescence. Trypan blue exclusion assay documented a variable but consistently reduced proliferation rate in cells from affected individuals (Figure 3A). Of note, a complete proliferative arrest was observed in S1 fibroblasts at relatively early passages. Consistent with this finding, flow-cytometry analysis documented a block in cell-cycle transition from G0/G1 to S phase (Figure 3B). Compared to control fibroblasts, those derived from affected individuals were larger and had a flattened and irregular shape. This was apparent at early passages and became more pronounced with passage of cultured cells (Figure S3). Additionally, compared to age- and passage-matched control cells, fibroblasts endogenously carrying the HIST1H1E mutant allele were characterized by a significantly augmented SA-β-gal activity (Figure 4A), which is an established marker of cellular senescence.46 Furthermore, enhanced levels of TP53, whose amount increases during replicative senescence,47 were observed at early passages (Figure 4B). These data demonstrate a direct link between aberrant HIST1H1E function and replicative senescence.
Figure 3.

Proliferation Assay and Cell-Cycle Progression
(A) Cells were seeded at 200,000 cells/well in a six-well plate and incubated a 37°C. Cell numbers (mean of three replicates ± SD) were counted by trypan blue exclusion assay after four and seven days. A significantly decreased proliferation rate was observed in the fibroblast lines from the two unrelated affected individuals (S1 and S2). Cells from subject S1 show a permanent cell growth arrest.
(B) Cell cycle phases of S1/S2’s (right) and control (left) fibroblasts as measured by BrdU incorporation and propidium iodide (PI) flow cytometry analysis. The upper box identifies cells incorporating BrdU (S phase), the lower left box identifies G0/G1 cells and the lower right box represents G2/M cells. One of three independent experiments is reported with the percentage of cells in each cell cycle phase.
Figure 4.

Defective HISTH1E Function Results in Altered SA-β-gal Activity and p53 Expression Level
(A) Representative images (left) and quantification (right) of SA-β-gal activity evaluated on S1 and control (C) fibroblasts at different culture passages. The significance was measured by one-way Anova with Tukey’s multiple comparison test (∗p < 0.01, ∗∗p < 0.0001).
(B) Compared to control cell lysates, fibroblast lysates from individuals S1 and S2 showed enhanced TP53 protein levels at earlier passages. Representative blots (left) and mean ± SD densitometry values (right) of three independent experiments are shown (∗p < 0.05, ∗∗p < 0.002; two-tailed Student’s t test).
In premature-aging disorders, replicative senescence can be caused by multiple events, including DNA double-strand breaks (DSBs), telomere dysfunction and/or accelerated shortening, and lamin defects.48, 49, 50 To evaluate the telomere status, we first performed Q-FISH analysis, which showed no difference in telomere length between control cells and fibroblasts from affected individuals (Figure S4A). Immunofluorescence analysis designed to identify γH2AX foci, markers of DSBs,51 showed an increased number of positive cells and foci per cell among those carrying mutant HIST1H1E compared to control cells (Figure S4B), indicating increased spontaneous DNA damage. ImmunoFISH analysis also documented a higher frequency of co-localization between γH2AX foci and telomeres in cells from affected individuals than in control cells (Figure S4C), highlighting the presence of dysfunctional telomeres in these cells. Fibroblasts carrying the mutated HIST1H1E allele also showed a higher sensitivity to low doses of γ-ray irradiation (Figure 2E), documenting augmented susceptibility to DNA damage. These findings suggest that the proliferative arrest and senescent phenotype of cells expressing the mutant HIST1H1E allele might result, at least in part, from ineffective DNA repair and persistent activation of DNA damage response (DDR) signaling.52, 53 To explore this possibility, the fraction of residual DNA damage was evaluated by SCGE assay post-γ-ray irradiation. Kinetics data documented defective and/or delayed DNA repair of single-strand breaks and DSBs in mutant fibroblasts compared to control cells (Figure S5).
Cells carrying LMNA (MIM: 150330) gene mutations that drive cellular senescence exhibit characteristic morphological nuclear abnormalities due to altered mechanical properties of the lamina,54 a network of structural filaments that interact with chromatin to participate in chromatin remodeling and organization as well as DNA replication and transcription.55, 56, 57, 58 Thus, we explored possible changes in nuclear morphology and architecture. Immunofluorescence analysis showed aberrant lamin A/C morphology in cells that were taken from affected individuals and induced to divide after thymidine/nocodazole treatment (Figure 5A). Aberrant morphology ranged from abnormal nuclear shapes to nuclear blebbing. Of note, an increased number of aberrantly shaped nuclei in fibroblasts carrying the HIST1H1E frameshift was observed at early passages in cells from affected individuals compared to control cells, and this difference became more pronounced as passages increased (p < 0.0000001; two-tails Fisher’s exact test) (Figure 5B). This feature is reminiscent of changes in nuclear shape in progeroid syndromes and confirms the senescent phenotype associated with aberrant HIST1H1E function.
Figure 5.

Defective HISTH1E Function Results in Aberrant Nuclear Morphology That Is Exacerbated over Cell-Culture Passages
(A) CLSM analysis was performed in steady-state (left) and synchronized (right) skin fibroblasts induced to divide after being treated with thymidine/nocodazole and recovered with fresh medium. The panels show an aberrant nuclear morphology in cells from subject S1. Whereas control cells proceed through mitosis (representative metaphases are shown), S1 fibroblasts fail to progress. Experiments were carried out at early passages (passage 3). Cells were stained with an antibody against lamin A/C (red) and DAPI (blue). Images are representative of >200 analyzed cells. Scale bars represent 7 μm.
(B) CLSM analysis was performed on S1 and control fibroblasts seeded at different culture passages. The panels show an aberrant nuclear morphology at early passages in subject S1’s cells compared to control cells, which were seeded at late passages (passage 16). Percentages refer to the number of cells with aberrant nuclear morphology. Cells were stained as above. Images are representative of >200 analyzed cells. Scale bars represent 27 μm.
HIST1H1E Mutations Trigger Nucleolar Instability
Heterochromatin plays a role in maintaining nucleolar stability and controlling rRNA synthesis,59 and decreased heterochromatin levels have been associated with nucleolar instability and upregulated transcription of rRNA genes that, in turn, has been suggested to enhance protein synthesis and contribute to overgrowth and accelerated aging.60, 61 Besides functioning in ribosome biogenesis, the nucleolus is involved in cell-cycle control,62 and linker histones have been reported to interact with multiple nucleolar proteins that are implicated in various nucleolar functions.63 On the basis of these considerations, we explored whether HIST1H1E mutations trigger nucleolar instability. As shown, S1 fibroblasts displayed fragmentation of the nucleolus, as revealed by loss of the nucleolar marker nucleolin (Figure 6A), and these fibroblasts also showed increased 28S rRNA levels and an increased amount of 18S (Figure 6B); these results further document the pleiotropic effect of HIST1H1E mutations. Notably, these findings suggest that besides affecting nucleolar function, dysregulated rRNA synthesis driven by HIST1H1E frameshift mutations might represent the molecular event causally linked to macrosomia, a recurring feature among young children with this class of mutations.19, 20 This hypothesis is in line with other observations correlating tissue plasticity to ribosomal biogenesis, as documented for the skeletal muscle mass modulated by seasonal acclimatization in Cyprinus carpio.64
Figure 6.
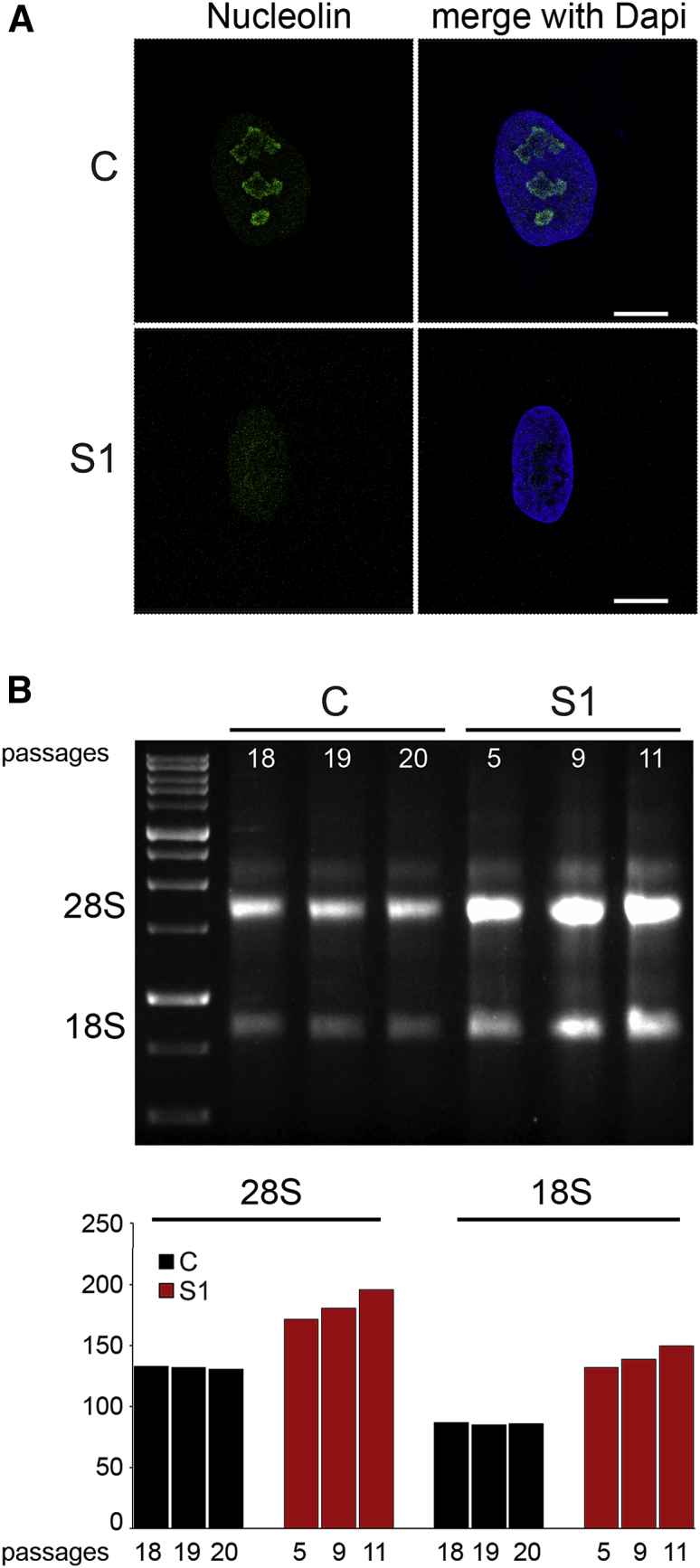
Defective HIST1H1E Function Results in Nucleolar Fragmentation and Increased 18S and 28S rRNA Levels
(A) CLSM observations were performed on S1 and control (C) fibroblasts. Panels show a significant decrease in C3 clone antibody staining in S1 fibroblasts, revealing nucleolar fragmentation. Nuclei were stained with DAPI. Images are representative of >200 analyzed cells. Scale bars represent 9 μm.
(B) Total RNA was extracted from the same amount of S1 and C cells at different cellular passages. Three μl of total RNA was loaded for size separation on 1% agarose gel and stained with ethidium bromide. Increased amounts (compared to those in control cells) of both 28S and 18S rRNA codifying for ribosomal subunits are evident in fibroblasts from subject S1.
Aneuploidy Is a Major Feature of Cells Bearing HIST1H1E Frameshift
Finally, because maximal chromatin compaction is required for proper chromosomal segregation and aneuploidy is a marker of the chromosomal instability which characterizes the senescent phenotype, we expected the occurrence of aneuploidy in cells expressing the disease-causing HIST1H1E mutants. Indeed, direct count of chromosomes in metaphases revealed a remarkably high proportion of aneuploidy in early-passage fibroblasts from S1 (41.2% of analyzed cells), which was significantly higher even when compared to late-passage control fibroblasts (18.9% of cells) (p < 0.01, χ2 test).
Discussion
Here we show that a specific class of dominantly acting frameshift mutations affecting the C-terminal tail of HIST1H1E disrupts chromatin structure and nuclear lamina organization and drives cellular replicative senescence. By assessing the clinical records of a relatively large cohort of individuals carrying these mutations, we also show that this endophenotype is mirrored by features suggestive of accelerated aging.
HIST1H1E is one of the members of the linker histone family, whose members function as structural components of chromatin to control the extent of DNA compaction and contribute to the regulation of gene expression and DNA replication, recombination, and repair.4, 6, 8, 44 Linker histones are encoded by multiple genes in the mammalian genomes; such genes include paralogs characterized by a diverse expression pattern during development and having either a ubiquitous distribution or an expression restricted to specific cell types.9, 65 Among these, HIST1H1E has been reported to be expressed ubiquitously at a high level,65 and similarly to the other replication-dependent linker histones, HIST1H1E is synthetized during the S phase to assemble chromatin with newly replicated DNA.66 As with the other linker histones, the protein is relatively depleted from active promoters and other regulatory regions controlling transcription and enriched in portions of the genome carrying repressive histone marks.6 Notably, previous studies provided evidence that inactivation of any of this class of histones does not significantly perturb murine development, which is compromised only when a concomitant inactivation of multiple subtypes occurs, resulting in an extensive reduction of these proteins.10, 11 Consistent with this functional redundancy, our data do not support haploinsufficiency as the molecular mechanism implicated in pathogenesis (see below) but point to a specific, dominantly acting effect causing a profound perturbation of multiple cellular processes directly and indirectly controlled by chromatin remodeling. Such pleiotropic effects involve genome instability, epigenetic modifications, inefficient DNA repair ability, improper chromosome compaction and segregation, and nucleolar fragmentation, converging toward cellular senescence and replicative impasse.
Similar to what is observed in progeroid disorders (e.g., Hutchinson-Gilford progeria [MIM: 176670], Werner syndrome [MIM: 277700], and lipodystrophy syndromes), the cellular processes affected in the disorder associated with aberrant HIST1H1E function, link cellular senescence to premature aging. The clinical profile of the affected individuals with HIST1H1E mutations, however, differs from those of individuals with progeroid disorders, and this profile is quite peculiar. The main features are a distinctive facies characterized by bitemporal narrowing, macrocephaly, prominent forehead, high anterior hairline, sparse frontotemporal hair, hypertelorism, downslanted palpebral fissures, a broad nasal tip, and low-set and posteriorly rotated ears. DD/ID invariantly occurs, whereas overgrowth, which had been originally reported as a key feature of the disorder, was not observed in any of the subjects included in this work at last assessment; this suggests that enhanced growth might represent a feature characterizing infancy. Although the facial gestalt can help in recognition of the disorder, we noted that no pathognomonic features can be used for a definitive clinical diagnosis. In early childhood, a tentative differential diagnosis should include Pallister-Killian syndrome and mild phenotypes within the spectrum of Weaver syndrome, Werner syndrome, and other progeroid disorders.
Besides documenting the multiple events contributing to cellular senescence, the present findings provide a mechanistic model for the multifaceted impact of this class of HIST1H1E mutations. Our data further document the specific and narrow spectrum of disease causative mutations affecting this gene. The long, lysine-rich C-terminal tail of HIST1H1E encompasses multiple regulatory residues that are substrates for cyclin-dependent kinases, and the extent of phosphorylation controls the proper compaction required for chromosome segregation, as well as local chromatin decondensation needed to promote DNA transcription, replication, and repair. The phosphorylation of this region during the cell cycle has been theorized to contribute to the control of the chromatin state by a twofold process.3, 4, 12, 67 Partial phosphorylation is attained during interphase (G0–S phase), allowing dynamical chromatin relaxation and access to DNA.68, 69, 70 On the other hand, a maximal phosphorylation is attained during mitosis (M phase), which is considered to be required for chromatin condensation and segregation of chromosomes into daughter cells during cell division.71, 72, 73, 74, 75 In this context, the functionally equivalent disease-causing frameshift events are the only mutations that are predicted to not significantly affect the capability of the mutant protein to retain efficient chromatin binding with concomitant loss of the regulatory sites required for modulation of higher-order chromatin architecture. Consistent with this model, the HIST1H1E truncating variants reported in gnomAD affect regions of the protein that do not overlap the mutational hotspot here defined but are much more proximal to the N terminus or to the C terminus (Figure S6). The former are predicted to result in truncated proteins with defective binding to chromatin and/or expected to undergo accelerated degradation, whereas the latter retain the regulatory serine/threonine residues and are not expected to have a dominant negative effect. The complexity by which the C-terminal tail controls HIST1H1E function, the high stability of these mutants, and their ability to bind to chromatin strongly support the possibility of a dominant negative effect of these mutants. However, neomorphism or gain-of-function as alternative mechanism(s) of disease cannot be ruled out a priori, and experimentally testing the different models will require dedicated studies.
Taken together, the present findings provide evidence that dominantly acting functional dysregulation of a linker histone causes a complex cellular phenotype characterized by replicative senescence and results in a neurodevelopmental disorder characterized by accelerated aging.
Declaration of Interests
A.Be. and R.P. declare no additional conflicts of interest beyond their employment affiliation. All of the other authors declare no competing interests.
Acknowledgments
We thank the patients and their families for their participation in this study, and Serenella Venanzi (Istituto Superiore di Sanità, Rome) for technical support. This project was supported, in part, by Fondazione Bambino Gesù (Vite Coraggiose to M.T.), the Italian Ministry of Health (Ricerca Corrente 2017 and 2018 to A.C. and M.T.), Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG21390 to G.M. and IG21614 to M.T.) and Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) Dipartimenti di Eccellenza (miur)(Project D15D18000410001) to the Department of Medical Sciences, University of Turin, Italy. M.T. acknowledges Consorsio Interuniversitario per il Calcolo Automatico (CINECA) for the computational resources. The Broad Center for Mendelian Genomics (UM1 HG008900) is funded by the National Human Genome Research Institute with supplemental funding provided by the National Heart, Lung, and Blood Institute under the Trans-Omics for Precision Medicine (TOPMed) program and the National Eye Institute. M.H.W. is supported by National Institutes of Health (NIH) grant T32GM007748. R.F.K. and A.V.D. are supported by grants from the European Research Area Network (ERA-NET) NEURON through the Research Foundation—Flanders -(FWO). K.Õ., S.P., and K.R. are supported by Estonian Research Council grants PRG471 and PUTJD827.
Published: August 22, 2019
Footnotes
Supplemental Data can be found online at https://doi.org/10.1016/j.ajhg.2019.07.007.
Contributor Information
R. Frank Kooy, Email: frank.kooy@uantwerpen.be.
Marco Tartaglia, Email: marco.tartaglia@opbg.net.
Accession Numbers
All mutations identified in this work have been submitted to ClinVar (submission IDs: SCV000925231 to SCV000925238).
Web Resources
Combined Annotation Dependent Depletion (CADD), https://cadd.gs.washington.edu/
Database for nonsynonymous SNPs’ functional predictions (dbNSFP), https://sites.google.com/site/jpopgen/dbNSFP
ExAC database, http://exac.broadinstitute.org/
Gene SeT AnaLysis, http://www.webgestalt.org/option.php
Online Mendelian Inheritance in Man (OMIM), https://www.omim.org/
Supplemental Data
References
- 1.Wolffe A. Academic Press; London: 1998. Chromatin: Structure and function. [Google Scholar]
- 2.Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 3.Harshman S.W., Young N.L., Parthun M.R., Freitas M.A. H1 histones: current perspectives and challenges. Nucleic Acids Res. 2013;41:9593–9609. doi: 10.1093/nar/gkt700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hergeth S.P., Schneider R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015;16:1439–1453. doi: 10.15252/embr.201540749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crane-Robinson C. Linker histones: History and current perspectives. Biochim. Biophys. Acta. 2016;1859:431–435. doi: 10.1016/j.bbagrm.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Izzo A., Kamieniarz-Gdula K., Ramírez F., Noureen N., Kind J., Manke T., van Steensel B., Schneider R. The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep. 2013;3:2142–2154. doi: 10.1016/j.celrep.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Geeven G., Zhu Y., Kim B.J., Bartholdy B.A., Yang S.M., Macfarlan T.S., Gifford W.D., Pfaff S.L., Verstegen M.J., Pinto H. Local compartment changes and regulatory landscape alterations in histone H1-depleted cells. Genome Biol. 2015;16:289. doi: 10.1186/s13059-015-0857-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayona-Feliu A., Casas-Lamesa A., Reina O., Bernués J., Azorín F. Linker histone H1 prevents R-loop accumulation and genome instability in heterochromatin. Nat. Commun. 2017;8:283. doi: 10.1038/s41467-017-00338-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan C., Fan Y. Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim. Biophys. Acta. 2016;1859:496–509. doi: 10.1016/j.bbagrm.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirotkin A.M., Edelmann W., Cheng G., Klein-Szanto A., Kucherlapati R., Skoultchi A.I. Mice develop normally without the H1(0) linker histone. Proc. Natl. Acad. Sci. USA. 1995;92:6434–6438. doi: 10.1073/pnas.92.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Y., Sirotkin A., Russell R.G., Ayala J., Skoultchi A.I. Individual somatic H1 subtypes are dispensable for mouse development even in mice lacking the H1(0) replacement subtype. Mol. Cell. Biol. 2001;21:7933–7943. doi: 10.1128/MCB.21.23.7933-7943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao R., Mizzen C.A. Interphase H1 phosphorylation: Regulation and functions in chromatin. Biochim. Biophys. Acta. 2016;1859:476–485. doi: 10.1016/j.bbagrm.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Kurotaki N., Imaizumi K., Harada N., Masuno M., Kondoh T., Nagai T., Ohashi H., Naritomi K., Tsukahara M., Makita Y. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 14.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi H.H., Sarkissian M., Hu G.Q., Wang Z., Bhattacharjee A., Gordon D.B., Gonzales M., Lan F., Ongusaha P.P., Huarte M. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuen B.T., Knoepfler P.S. Histone H3.3 mutations: A variant path to cancer. Cancer Cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maze I., Noh K.M., Soshnev A.A., Allis C.D. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014;15:259–271. doi: 10.1038/nrg3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tessadori F., Giltay J.C., Hurst J.A., Massink M.P., Duran K., Vos H.R., van Es R.M., Deciphering Developmental Disorders Study. Scott R.H., van Gassen K.L.I., Bakkers J., van Haaften G. Germline mutations affecting the histone H4 core cause a developmental syndrome by altering DNA damage response and cell cycle control. Nat. Genet. 2017;49:1642–1646. doi: 10.1038/ng.3956. [DOI] [PubMed] [Google Scholar]
- 19.Tatton-Brown K., Loveday C., Yost S., Clarke M., Ramsay E., Zachariou A., Elliott A., Wylie H., Ardissone A., Rittinger O. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am. J. Hum. Genet. 2017;100:725–736. doi: 10.1016/j.ajhg.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takenouchi T., Uehara T., Kosaki K., Mizuno S. Growth pattern of Rahman syndrome. Am. J. Med. Genet. A. 2018;176:712–714. doi: 10.1002/ajmg.a.38616. [DOI] [PubMed] [Google Scholar]
- 21.Flex E., Niceta M., Cecchetti S., Thiffault I., Au M.G., Capuano A., Piermarini E., Ivanova A.A., Francis J.W., Chillemi G. Biallelic mutations in TBCD, encoding the tubulin folding cofactor d, perturb microtubule dynamics and cause early-onset encephalopathy. Am. J. Hum. Genet. 2016;99:962–973. doi: 10.1016/j.ajhg.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauer C.K., Calligari P., Radio F.C., Caputo V., Dentici M.L., Falah N., High F., Pantaleoni F., Barresi S., Ciolfi A. Mutations in KCNK4 that affect gating cause a recognizable neurodevelopmental syndrome. Am. J. Hum. Genet. 2018;103:621–630. doi: 10.1016/j.ajhg.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorboz I., Aiello C., Simons C., Stone R.T., Niceta M., Elmaleh M., Abuawad M., Doummar D., Bruselles A., Wolf N.I. Biallelic mutations in the homeodomain of NKX6-2 underlie a severe hypomyelinating leukodystrophy. Brain. 2017;140:2550–2556. doi: 10.1093/brain/awx207. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y., Muzny D.M., Xia F., Niu Z., Person R., Ding Y., Ward P., Braxton A., Wang M., Buhay C. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandeweyer G., Van Laer L., Loeys B., Van den Bulcke T., Kooy R.F. VariantDB: A flexible annotation and filtering portal for next generation sequencing data. Genome Med. 2014;6:74. doi: 10.1186/s13073-014-0074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Retterer K., Juusola J., Cho M.T., Vitazka P., Millan F., Gibellini F., Vertino-Bell A., Smaoui N., Neidich J., Monaghan K.G. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016;18:696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 28.Andreoli C., Leopardi P., Crebelli R. Detection of DNA damage in human lymphocytes by alkaline single cell gel electrophoresis after exposure to benzene or benzene metabolites. Mutat. Res. 1997;377:95–104. doi: 10.1016/s0027-5107(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 29.Catanzaro G., Besharat Z.M., Miele E., Chiacchiarini M., Po A., Carai A., Marras C.E., Antonelli M., Badiali M., Raso A. The miR-139-5p regulates proliferation of supratentorial paediatric low-grade gliomas by targeting the PI3K/AKT/mTORC1 signalling. Neuropathol. Appl. Neurobiol. 2018;44:687–706. doi: 10.1111/nan.12479. [DOI] [PubMed] [Google Scholar]
- 30.Zhou C., Cunningham L., Marcus A.I., Li Y., Kahn R.A. Arl2 and Arl3 regulate different microtubule-dependent processes. Mol. Biol. Cell. 2006;17:2476–2487. doi: 10.1091/mbc.E05-10-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cordeddu V., Redeker B., Stellacci E., Jongejan A., Fragale A., Bradley T.E., Anselmi M., Ciolfi A., Cecchetti S., Muto V. Mutations in ZBTB20 cause Primrose syndrome. Nat. Genet. 2014;46:815–817. doi: 10.1038/ng.3035. [DOI] [PubMed] [Google Scholar]
- 32.Berardinelli F., Antoccia A., Buonsante R., Gerardi S., Cherubini R., De Nadal V., Tanzarella C., Sgura A. The role of telomere length modulation in delayed chromosome instability induced by ionizing radiation in human primary fibroblasts. Environ. Mol. Mutagen. 2013;54:172–179. doi: 10.1002/em.21761. [DOI] [PubMed] [Google Scholar]
- 33.Perner S., Brüderlein S., Hasel C., Waibel I., Holdenried A., Ciloglu N., Chopurian H., Nielsen K.V., Plesch A., Högel J., Möller P. Quantifying telomere lengths of human individual chromosome arms by centromere-calibrated fluorescence in situ hybridization and digital imaging. Am. J. Pathol. 2003;163:1751–1756. doi: 10.1016/S0002-9440(10)63534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris T.J., Butcher L.M., Feber A., Teschendorff A.E., Chakravarthy A.R., Wojdacz T.K., Beck S. Champ: 450k chip analysis methylation pipeline. Bioinformatics. 2014;30:428–430. doi: 10.1093/bioinformatics/btt684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guarrera S., Fiorito G., Onland-Moret N.C., Russo A., Agnoli C., Allione A., Di Gaetano C., Mattiello A., Ricceri F., Chiodini P. Gene-specific DNA methylation profiles and LINE-1 hypomethylation are associated with myocardial infarction risk. Clin. Epigenetics. 2015;7:133. doi: 10.1186/s13148-015-0164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:130–137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duffney L.J., Valdez P., Tremblay M.W., Cao X., Montgomery S., McConkie-Rosell A., Jiang Y.H. Epigenetics and autism spectrum disorder: A report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2018;177:426–433. doi: 10.1002/ajmg.b.32631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helsmoortel C., Vandeweyer G., Ordoukhanian P., Van Nieuwerburgh F., Van der Aa N., Kooy R.F. Challenges and opportunities in the investigation of unexplained intellectual disability using family-based whole-exome sequencing. Clin. Genet. 2015;88:140–148. doi: 10.1111/cge.12470. [DOI] [PubMed] [Google Scholar]
- 40.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Daujat S., Zeissler U., Waldmann T., Happel N., Schneider R. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J. Biol. Chem. 2005;280:38090–38095. doi: 10.1074/jbc.C500229200. [DOI] [PubMed] [Google Scholar]
- 42.Allis C.D., Jenuwein T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 43.Aref-Eshghi E., Bend E.G., Colaiacovo S., Caudle M., Chakrabarti R., Napier M., Brick L., Brady L., Carere D.A., Levy M.A. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am. J. Hum. Genet. 2019;104:685–700. doi: 10.1016/j.ajhg.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan Y., Nikitina T., Zhao J., Fleury T.J., Bhattacharyya R., Bouhassira E.E., Stein A., Woodcock C.L., Skoultchi A.I. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199–1212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 45.Criscione S.W., Teo Y.V., Neretti N. The chromatin landscape of cellular senescence. Trends Genet. 2016;32:751–761. doi: 10.1016/j.tig.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulju K.S., Lehman J.M. Increased p53 protein associated with aging in human diploid fibroblasts. Exp. Cell Res. 1995;217:336–345. doi: 10.1006/excr.1995.1095. [DOI] [PubMed] [Google Scholar]
- 48.Cau P., Navarro C., Harhouri K., Roll P., Sigaudy S., Kaspi E., Perrin S., De Sandre-Giovannoli A., Lévy N. Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin. Cell Dev. Biol. 2014;29:125–147. doi: 10.1016/j.semcdb.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 49.Rossiello F., Herbig U., Longhese M.P., Fumagalli M., d’Adda di Fagagna F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 2014;26:89–95. doi: 10.1016/j.gde.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lidzbarsky G., Gutman D., Shekhidem H.A., Sharvit L., Atzmon G. Genomic instabilities, cellular senescence, and aging: In vitro, in vivo and aging-like human syndromes. Front. Med. 2018;5:104. doi: 10.3389/fmed.2018.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sedelnikova O.A., Bonner W.M. GammaH2AX in cancer cells: A potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle. 2006;5:2909–2913. doi: 10.4161/cc.5.24.3569. [DOI] [PubMed] [Google Scholar]
- 52.Downey M., Durocher D. GammaH2AX as a checkpoint maintenance signal. Cell Cycle. 2006;5:1376–1381. doi: 10.4161/cc.5.13.2899. [DOI] [PubMed] [Google Scholar]
- 53.Fillingham J., Keogh M.C., Krogan N.J. GammaH2AX and its role in DNA double-strand break repair. Biochem. Cell Biol. 2006;84:568–577. doi: 10.1139/o06-072. [DOI] [PubMed] [Google Scholar]
- 54.Dahl K.N., Scaffidi P., Islam M.F., Yodh A.G., Wilson K.L., Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 2006;103:10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Broers J.L., Ramaekers F.C., Bonne G., Yaou R.B., Hutchison C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 56.Gruenbaum Y., Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015;84:131–164. doi: 10.1146/annurev-biochem-060614-034115. [DOI] [PubMed] [Google Scholar]
- 57.Shumaker D.K., Dechat T., Kohlmaier A., Adam S.A., Bozovsky M.R., Erdos M.R., Eriksson M., Goldman A.E., Khuon S., Collins F.S. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gonzalez-Suarez I., Gonzalo S. Nurturing the genome: A-type lamins preserve genomic stability. Nucleus. 2010;1:129–135. doi: 10.4161/nucl.1.2.10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Padeken J., Heun P. Nucleolus and nuclear periphery: velcro for heterochromatin. Curr. Opin. Cell Biol. 2014;28:54–60. doi: 10.1016/j.ceb.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Larson K., Yan S.J., Tsurumi A., Liu J., Zhou J., Gaur K., Guo D., Eickbush T.H., Li W.X. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012;8:e1002473. doi: 10.1371/journal.pgen.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsekrekou M., Stratigi K., Chatzinikolaou G. The nucleolus: In genome maintenance and repair. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18071411. pii: E1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pederson T. The nucleolus. Cold Spring Harb. Perspect. Biol. 2011;3:a000638. doi: 10.1101/cshperspect.a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalashnikova A.A., Rogge R.A., Hansen J.D. Linker histone H1 and protein-protein interactions. Biochim. Biophys. Acta. 2016;1859:455–461. doi: 10.1016/j.bbagrm.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fuentes E.N., Zuloaga R., Valdes J.A., Molina A., Alvarez M. Skeletal muscle plasticity induced by seasonal acclimatization involves IGF1 signaling: implications in ribosomal biogenesis and protein synthesis. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2014;176:48–57. doi: 10.1016/j.cbpb.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Parseghian M.H., Hamkalo B.A. A compendium of the histone H1 family of somatic subtypes: an elusive cast of characters and their characteristics. Biochem. Cell Biol. 2001;79:289–304. [PubMed] [Google Scholar]
- 66.Happel N., Warneboldt J., Hänecke K., Haller F., Doenecke D. H1 subtype expression during cell proliferation and growth arrest. Cell Cycle. 2009;8:2226–2232. doi: 10.4161/cc.8.14.8982. [DOI] [PubMed] [Google Scholar]
- 67.Roque A., Ponte I., Suau P. Interplay between histone H1 structure and function. Biochim. Biophys. Acta. 2016;1859:444–454. doi: 10.1016/j.bbagrm.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 68.Chadee D.N., Taylor W.R., Hurta R.A., Allis C.D., Wright J.A., Davie J.R. Increased phosphorylation of histone H1 in mouse fibroblasts transformed with oncogenes or constitutively active mitogen-activated protein kinase. J. Biol. Chem. 1995;270:20098–20105. doi: 10.1074/jbc.270.34.20098. [DOI] [PubMed] [Google Scholar]
- 69.Herrera R.E., Chen F., Weinberg R.A. Increased histone H1 phosphorylation and relaxed chromatin structure in Rb-deficient fibroblasts. Proc. Natl. Acad. Sci. USA. 1996;93:11510–11515. doi: 10.1073/pnas.93.21.11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chadee D.N., Allis C.D., Wright J.A., Davie J.R. Histone H1b phosphorylation is dependent upon ongoing transcription and replication in normal and ras-transformed mouse fibroblasts. J. Biol. Chem. 1997;272:8113–8116. doi: 10.1074/jbc.272.13.8113. [DOI] [PubMed] [Google Scholar]
- 71.Gurley L.R., Walters R.A., Tobey R.A. Sequential phosphorylation of histone subfractions in the Chinese hamster cell cycle. J. Biol. Chem. 1975;250:3936–3944. [PubMed] [Google Scholar]
- 72.Hohmann P., Tobey R.A., Gurley L.R. Phosphorylation of distinct regions of f1 histone. Relationship to the cell cycle. J. Biol. Chem. 1976;251:3685–3692. [PubMed] [Google Scholar]
- 73.Gurley L.R., D’Anna J.A., Barham S.S., Deaven L.L., Tobey R.A. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur. J. Biochem. 1978;84:1–15. doi: 10.1111/j.1432-1033.1978.tb12135.x. [DOI] [PubMed] [Google Scholar]
- 74.Matsumoto Y., Yasuda H., Mita S., Marunouchi T., Yamada M. Evidence for the involvement of H1 histone phosphorylation in chromosome condensation. Nature. 1980;284:181–183. doi: 10.1038/284181a0. [DOI] [PubMed] [Google Scholar]
- 75.Ajiro K., Borun T.W., Cohen L.H. Phosphorylation states of different histone 1 subtypes and their relationship to chromatin functions during the HeLa S-3 cell cycle. Biochemistry. 1981;20:1445–1454. doi: 10.1021/bi00509a007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.