

Abstract
Babaodan capsule (BBD), a traditional Chinese (TCM) formula, has been widely used as an alternative remedy for multiple types of malignancies, clinically. However, the underlying mechanisms behind the efficacy of BBD remain poorly understood, particularly in regard to lung cancer. Herein, we demonstrate that BBD induced autophagic death in A549 and A549DDP cells without apoptosis. Treatment with autophagic inhibitor 3-MA, Baf-A1 and PI3K agonist, IGF-1, fully proved our conclusion, as well as uncovered the potential downregulated signaling pathway, PI3K/AKT/mTOR. The study additionally found that BBD could downregulate the expression of MDR1 and increase the chemosensitivity of cisplatin. Collectively, our results, both in vivo and in vitro, demonstrate that BBD leads to autophagic cell death through downregulating the PI3K/AKT/mTOR signaling pathway and improved the antitumor effects of cisplatin in non-small cell lung cancer (NSCLC).
Keywords: Babaodan capsule (BBD), non-small cell lung cancer (NSCLC), autophagy, PI3K/AKT/mTOR pathway
Introduction
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer related death worldwide, with approximately 2 million new cases diagnosed in 2018 [1]. Non-small cell lung cancer (NSCLC), accounts for nearly 80-90% of lung cancer, and patients without typical mutation still rely on the platinum-based double chemotherapy [2]. However, the majority of patients suffer chemotherapeutic failure due to developed multidrug resistance (MDR) and intolerable toxicity induced by high doses of these agents [3,4]. To circumvent these problems, exploration of new effective strategies is now in the forefront of cancer research. Rational therapeutic drug combinations, such as natural products together with chemotherapeutic drugs, may provide novel therapeutic options for the treatment of this disease.
Traditional Chinese medicine (TCM) is associated with minimal side effects, and has been widely applied clinically for a range of diseases, including cancer. For instance, Artemisinin (ART), which was isolated from a traditional Chinese herb, has miraculous effects on malaria and parasitic diseases [5,6]. Yu Ping Feng San (YPFS), a TCM formula, reverses cisplatin-induced multi-drug resistance in NSCLC cells through the regulation of drug transporters and p62/TRAF6 signaling [7]. A series of studies also suggested that an increasing number of TCMs play important roles on anti-NSCLC activities via modulating pro-death autophagy, and even affect the acquired MDR [8-13].
Autophagy is a conserved cellular catabolic process in which organelles and cytoplasmic proteins are degraded and recycled through a autophagosome-lysosome manner [14]. It is known to keep cellular homeostasis and protect against various diseases [15,16]. However, the involvement of autophagy in cancer remains incompletely understood [17,18]. Many reports have emphasized that autophagy was a “double-edge sword” for tumor development [19-21]. Even so, there are still some agents that focus on neoplasms by modulating autophagic death, such as etoposide, rapamycin, and temozolomide [22], as well as the above mentioned active ingredients extracted from Chinese herbs or plants.
Babaodan capsule (BBD) is an ancient TCM formula, which has been widely used as a folk remedy since the Ming Dynasty. It is composed of Moschus, Snake Gall, Pearl, antelope’s horn, Calculus Bovis and Radix Notoginseng, with properties of heat-clearing, detoxification, jaundice-eliminating and promotion of blood circulation. Traditionally, BBD is prescribed for the treatment of viral hepatitis, acute urinary tract infection and other inflammation-related diseases. However, in recent years, we have recognized in the clinic that patients with primary liver and lung cancer who received treatment of BBD combined with chemotherapeutic drugs, had a better prognosis and relatively mild side effects [23]. Thus, we hypothesized that BBD could have a measure of antitumor effect, and may increase the sensitivity of chemotherapeutic agents.
In our study, we investigated the anti-cancer activity of BBD on lung cancer cell lines A549 and A549DDP, and attempted to identify the underlying molecular mechanism, aiming to explore its potential value of therapeutic effects on malignancy.
Material and methods
Materials and reagents
RPMI-1640 medium, phosphate buffered saline (PBS), and foetal bovine serum were purchased from Gibco (BRL Life Technologies, Grand Island, NY, USA), The cell-counting Kit-8 (CCK-8) assay was obtained from Dojin-do (Dojin-do, Japan), 3-Methyladenine (3-MA, an early-stage autophagy inhibitor) and Bafilomycin A1 (Baf-A1, a late-stage autophagy inhibitor) were purchased from Selleck (Selleck Chemicals, CA, USA). IGF-1 (a PI3K activator) was bought from Cell Sciences, (MA, USA). Antibodies against Bcl-2, Bax, cleaved-PARP, P-gp, PI3K, phospho-PI3K (p-PI3K), AKT, phospho-AKT (p-AKT), mTOR, phospho-mTOR (p-mTOR) were purchased from Cell Signaling Technology (Danvers, MA, USA), anti-LC3 was purchased from Novus, and antibody against GAPDH from Bioworld Technology, USA.
Cells and cell culture
Human NSCLC cell lines A549 were acquired from the American Type Culture Collection (ATCC). The cisplatin-resistant subline, A549DDP, was established in our laboratory by culturing A549 cells with increasing concentrations of cisplatin. Both A549 and A549DDP cells were cultured in RPMI-1640 medium containing 10% foetal bovine serum in a humidified incubator with 5% CO2, at 37°C.
Preparation of BBD
BBD was obtained from and authenticated by the manufacturer Xiamen Traditional Chinese Medicine Company Limited. The powder was dissolved with phosphate-buffered saline (PBS) to make a storage concentration of 20 mg/ml. Working concentrations were made by diluting the stock solution with culture medium.
CCK8 assay
Cells were seeded in 96-well plates at a density of 5×103/well, treated with various concentrations of BBD for 24 h or 48 h, respectively, after which the medium was replaced with CCK8, and incubated for 1-3 h at 37°C. The absorbance was analysed at 450 nm with a microplate reader (Biotex, USA). We also pretreated the cells with 3-MA, and BBD treatment, and after one hour, the levels of inhibition were calculated.
To determine the effect of BBD on the sensitivity of cisplatin, different doses of cisplatin with or without BBD were added to the adherent cells for 48 h. IC50 values were then calculated and compared with the single cisplatin group. Each experiment was performed in triplicate.
Western blotting
Cells were collected, then washed twice with PBS and lysed with cold RIPA buffer containing protease inhibitors. Equal amounts of protein samples were subjected to SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% BSA for 2 h at RT and incubated with primary antibodies at 4°C overnight. Subsequently, membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1-2 h at RT. Membranes were then washed and examined by the enhanced chemiluminescence solution and visualized with a ChemiDoc XRS + system (BioRad, USA).
RNA isolation and qRT-PCR
Total RNA was extracted using Trizol (Takara, Japan) and detected the RNA concentration with a NanoDrop2000 spectrophotometer (Thermo Fisher, USA). The cDNA was synthesized using a PrimeScrpt RT reagent kit (Takara, Japan). QRT-PCR was performed with SYBR premix (Takara, Japan) and a CFX96 PCR detection system (Bio-Rad, USA). The mRNA levels were normalized to human GAPDH, and all reactions were conducted in triplicate. Results were calculated by 2-ΔΔCt method. The primer pairs used were as follows: MDR1: 5’-GCCTGGTCATCTTGTGCTTCTTCC-3’ and 5’-AAGTGCTTCAATGAACCGCCTCTC-3’. GAPDH: 5’-GCTCATGACCACAGTCCATG-3’ and 5’-TCAGCTCTGGGATGACCTTG-3’.
Colony formation assay
A549 and A549DDP cells were pretreated for 24 h with cisplatin alone or BBD-cisplatin combination, then counted and seeded again at 500-600 cells/well. Control untreated cells were also processed in parallel. After 12-14 days incubation, the cells were fixed with 4% paraformaldehyde for 30 mins and stained with 0.1% crystal violet. The number of clones were then counted in each group. Each experiment was performed in triplicate.
Cell invasion and migration assay
For the migration assay, cells were seeded at 3×105 cells/well in a 6-well plate and treated with cisplatin alone (1.25 µg/ml for A549, 2.5 µg/ml for A549DDP) or BBD-cisplatin combination (0.4 mg/ml-1.25 µg/ml for A549, 0.4 mg/ml-2.5 µg/ml for A549DDP) for 24 h. When cell confluence reached approximately 90%, a scratch was made using a 200 µI micropipette tip. The migration area was observed at 0 h, 24 h, and 48 h, and photos were obtained using an optical microscopy (Leica, Germany). The wound area was analysed with ImageJ software.
Cell invasion was evaluated with 24-well transwell cell culture chambers coated with Matrigel matrix (BD, USA) in the upper chamber. Then, as in the colony formation assay, the pretreated cells were placed in the upper chamber without FBS, while 500 µI of 10% FBS RPMI1640 was added to the bottom chamber. After cell invasion occurred for 48 h, the cells in fields on the lower side of the membrane were photographed and counted with an upright metallurgical microscopy (Leica, Germany). Each assay was performed at least three times.
Transmission electron microscopy
Cells were seeded on 6-well plates and incubated in different treatment groups according to the experimental design. Samples were fixed with 2.5% glutaraldehyde, then dehydrated and embedded in araldite resin. Representative areas were chosen to cut into ultrathin sections and stained with uranyl acetate and lead citrate. Finally, samples were examined by electron microscopy.
Tumor xenograft experiments
This study was performed in accordance with institutional guidelines of Guangdong Province. Female nude BALB/c mice aged 4-6 weeks were purchased from the Experimental Animal Center of Southern Medical University (Guangzhou, China). BBD was dissolved by PBS, and A549 cells were also suspended in sterile PBS at a concentration of 1×107 cells/0.2 ml and injected subcutaneously into the right flank of the mice. When tumors reached about 50-100 mm3, mice were randomly divided into four groups (four mice per each group): (a) control (PBS injection once every three days and PBS gavage once daily); (b) BBD intragastric administration (0.25 g/kg, once daily for BBD and PBS injection once every three days); (c) cisplatin injection (3 mg/kg, once every three days for cisplatin and PBS gavage once daily); (d) BBD intragastric administration and cisplatin injection (3 mg/kg, once every three days for cisplatin; 0.25 g/kg, once daily for BBD). Tumor size was monitored every 4 days. After ~4 weeks, mice were euthanized, and tumors were removed and photographed.
Statistical analysis
Data are expressed as means ± SD of at least three independent experiments. Differences of Independent samples were compared by Student’s t-test, while one-way ANOVA was used to analyze the differences between groups. All comparisons for which P-value < 0.05 were considered statistically significant.
Results
Babaodan capsule (BBD) inhibits cell growth in a dose-dependent manner in A549 and A549DDP cells
To evaluate the potential therapeutic effect of BBD for NSCLC, we selected the NSCLC cell line, A549, and its cisplatin (DDP)-resistant cell line, A549DDP, for which the IC50 of DDP was 7.65 μg /ml. Such an IC50 was ~4-fold higher than that of A549, which was 1.84 μg/ml (Figure 1A). We treated cells with various concentrations (0-2 mg/ml) of BBD, and results revealed that BBD inhibited cell growth dose-dependently, and cell viability was decreased when exposed to BBD for 48 h (Figure 1B and 1C). Moreover, cell viability exhibited no obvious differences between A549DDP and its parental A549 cells (Figure 1D). Moreover, with increasing concentrations of BBD, the colony formation capacity of both cells decreased significantly, as compared to controls (Figure 1E and 1F). These findings suggested that BBD similarly inhibited cell growth of A549 and A549DDP in a dose- and time-dependent manner.
Figure 1.
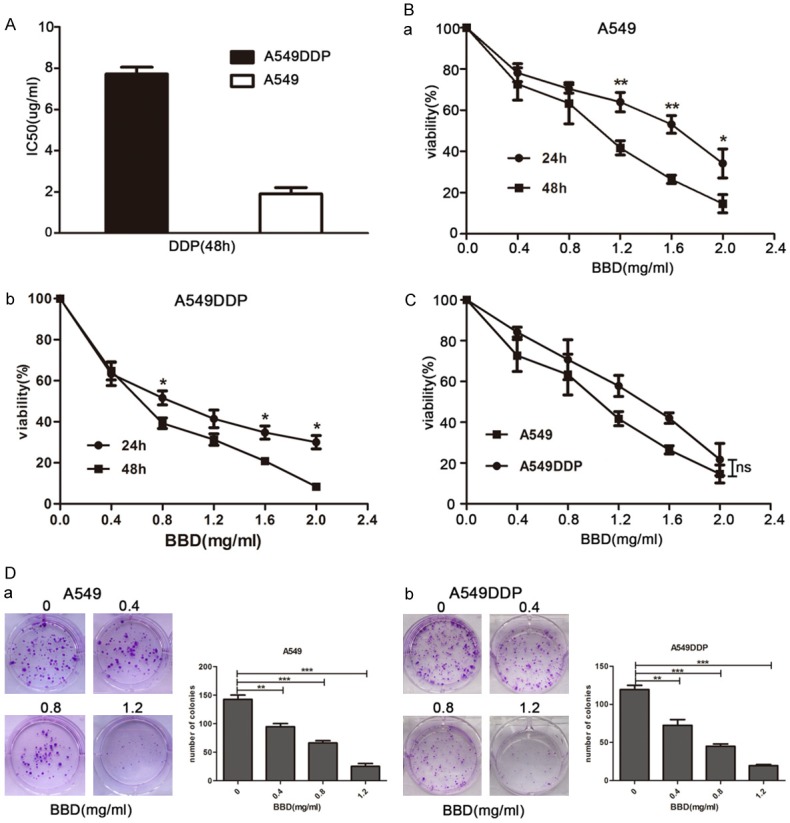
BBD inhibited cell growth in a dose-dependent manner in A549 and A549DDP cells. A. IC50 value of cisplatin was determined by CCK8 assay. B. Cell viability after BBD treatment for 24 hours or 48 hours was determined through CCK8 assay. C. Cells were incubated in the same concentrations of BBD for similar 24 hours, cell viability was detected and compared. D. Photographs of crystal violet-stained colonies of BBD-treated both cells growing in 6-well plates for 12-14 days after infection, and the number of cells in each colony was counted. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
BBD induces cytotoxicity in A549 and A549DDP primarily by autophagy
The data above demonstrated the growth-inhibitory efficacy of BBD. Intriguingly, western blot analysis indicated that the protein levels of Bax/Bcl-2, and caspase-cleaved PARP remained almost unchanged, compared to controls with different concentrations of BBD added (Figure 2A and 2B). To further determine the reason, we pretreated the cells with 3-methyladenine (3-MA), an early-stage autophagy inhibitor, following subsequent BBD treatment, and surprisingly found the drugs exhibited mild toxic effects than the single BBD group (Figure 2C and 2D). Thus, to examine whether autophagy contributed to the cell death, we checked the expression of LC3-II in BBD-treated cells. As expected, LC3-II expression rose dramatically when cells were exposed to BBD (Figure 2E and 2F).
Figure 2.
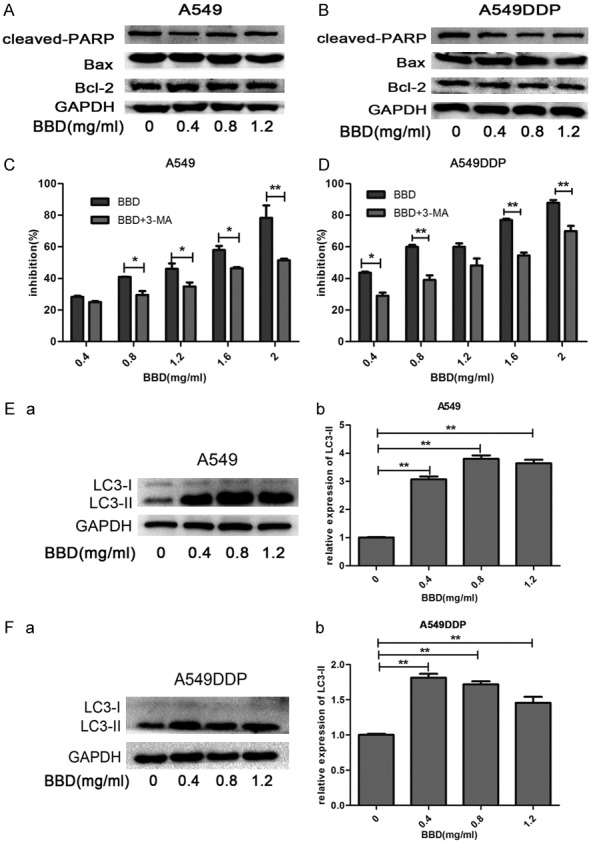
BBD induced cytotoxicity in A549 and A549DDP primarily by autophagy. A549 (A) and A549DDP (B) cells were treated with different concentrations of BBD, the cellular proteins Bcl-2, Bax, caspase-cleaved PARP were determined by Western-blot analysis. A549 (C) and A549DDP (D) cells were pretreated with 3-MA, and then treated for another 24 hours with BBD, cell viability was then determined by CCK8 assay and compared to the BBD treatment alone. (E, F) Western blot analysis of LC3II expression after BBD administration for 24 hours in both cells. The corresponding expression levels are shown as bar graphs. *, P < 0.05; **, P < 0.01.
However, the increased LC3-II level can be correlated to increased autophagic flux and the decreased degradation of autophagosomes. To distinguish between these two possibilities and to detect autophagic flux, we used bafilomycin A1 (Baf-A1), a late-stage autophagic inhibitor that blocks the fusion of autophagosomes with lysosomes [24]. The results showed that co-incubation of cells with BBD and Baf-A1 resulted in increased LC3-II levels compared with each drug alone (Figure 3A and 3B). Autophagosomes were evaluated by transmission electron microscopy (TEM), and we not only found autophagosomes in BBD-treated A549 cells, but also an increased number in the combination treatment group (Figure 3C). Moreover, western blot analysis showed 3-MA reversed the augmented LC3-II level produced by BBD (Figure 3D and 3E), indicating that it may elevate autophagic flux. All results demonstrated that BBD exerted a cytotoxic effect on A549 and A549DDP through enhancing the autophagic flux, rather than triggering apoptosis.
Figure 3.

BBD increased autophagic flux. A549 and A549DDP cells were treated with Baf-A1, BBD, BBD + Baf-A1, as well as untreated control group. (A, B) Western blot analysis revealed the expression of LC3II in both cells. (C) TEM evaluated autophagy in A549 cells. A549 (D) and A549DDP (E) cells were treated with 3-MA, BBD, BBD + 3-MA, as well as untreated control group. Similarly the LC3II status was determined by Western blot. The corresponding expression levels are shown as bar graphs. **, P < 0.01.
BBD induces autophagy via downregulating PI3K/AKT/mTOR signaling pathway
To determine if BBD regulated autophagy through the PI3K/AKT/mTOR signaling pathway [25]. We performed a western blot and observed that increasing doses of BBD markedly decreased the protein expression levels of p-PI3K, p-AKT, p-mTOR in both cells, while the total proteins remained unaltered (Figure 4A). Insulin-like growth factor-1 (IGF-1) is known to bind to the IGF-1 receptor, which can activate the PI3K/AKT signaling pathway [26], and thus, weaken autophagy (Figure 4B). Together with IGF-1 and BBD, the relevant downregulated status of LC3-II by IGF-1 was overturned to some extent (Figure 4C and 4D). Hence, we concluded that BBD induced autophagy in A549 and A549DDP via deactivation of the PI3K/AKT/mTOR signaling pathway.
Figure 4.

BBD induced autophagy via downregulating the PI3K/AKT/mTOR signaling pathway. The expression of PI3K, p-PI3K, AKT, p-AKT, mTOR, p-mTOR were examined by Western blot after BBD (A) or IGF-1 (B) treated for 24 hours. A549 (C) and A549DDP (D) cells were treated with IGF-1, BBD, BBD + IGF-1, as well as untreated control group, LC3II expression was determined by Western blot. **, P < 0.01.
BBD potentiates the chemosensitivity of cisplatin in A549 and A549DDP cells
To further prove whether BBD pretreatment conditioned lung adenocarcinoma cells to become increasingly susceptible to cisplatin, we first administered 24-hour exposure to 0.4 mg/ml BBD, a concentration which exhibited about 20% growth inhibition, prior to cisplatin incubation, and observed it synergized the cell death effect of cisplatin. The IC50 values of cisplatin alone were 3.16 µg/ml (A549) and 7.9 µg/ml (A549DDP), whereas the BBD-pretreated groups were 1.76 µg/ml (A549) and 4.85 µg/ml (A549DDP) (Figure 5A). Furthermore, a wound healing assay (Figure 5B) showed an additional decrease in motility of both cells after BBD pre-treated, and the transwell assay (Figure 5C) in parallel indicated that combination-treated cells invaded little compared to that cisplatin group or untreated control. Additionally, we performed qRT-PCR and western blot to evaluate the expression of MDR1, and the results showed that BBD dose-dependently reduced the mRNA and protein level of MDR1 (Figure 5D and 5E). These data suggest that BBD indeed improved chemosensitivity of cisplatin in A549 and A549DDP, and that the reduction of MDR1 may play a pivotal role.
Figure 5.
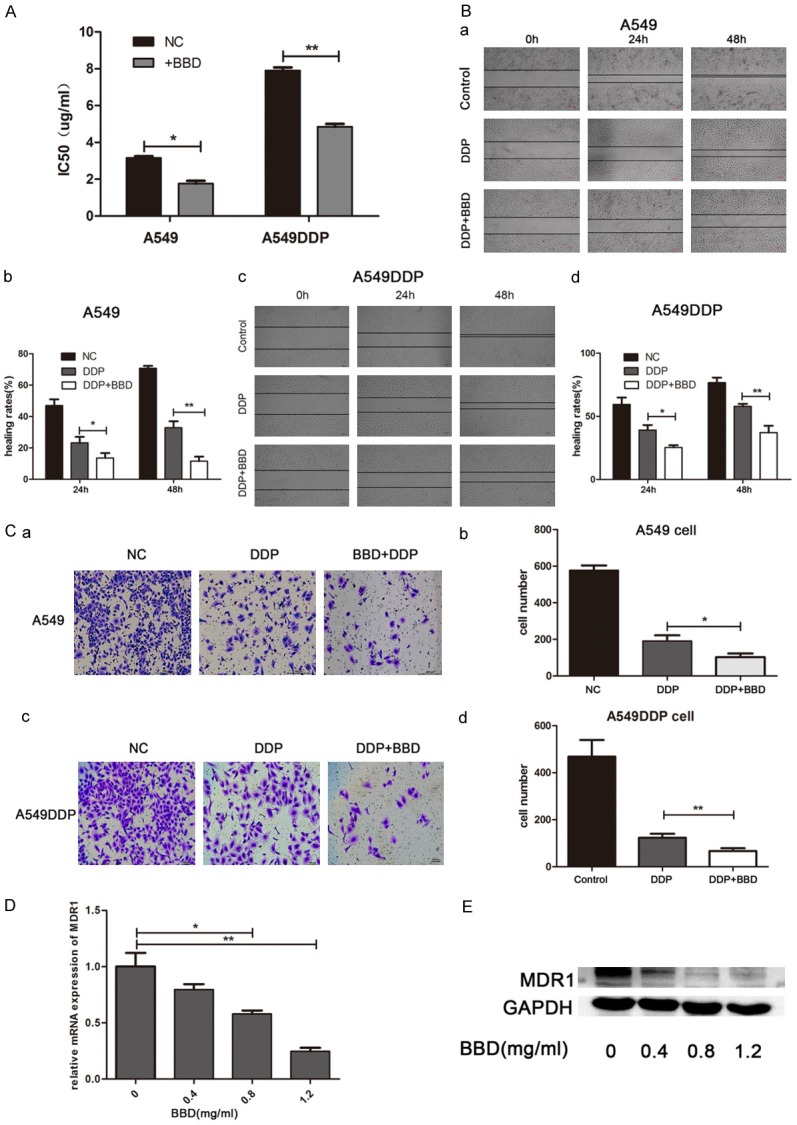
BBD potentiated the chemosensitivity of cisplatin in A549 and A549DDP. (A) IC50 value of cisplatin with or without BBD-pretreatment. (B) Wound healing assay exhibited the migration ability of BBD + DDP treated cells, compared with DDP alone group. (C) Transwell assay revealed the invasion ability of BBD + DDP treated cells, compared with DDP alone group. qRT-PCR assay (D) and Western blot assay (E) determined the expression of MDR1. *, P < 0.05; **, P < 0.01.
BBD exerts anti-tumor activity in A549 xenografts
To further test the hypothesis, we performed a second in vivo study with A549 xenograft experiments. In agreement with our prior results, BBD inhibited tumor growth. More importantly, combination treatment resulted in significantly decreased tumor volumes, compared with BBD or cisplatin alone (Figure 6A and 6B). Moreover, there were no significant alterations to body weight of all mice, suggesting that BBD is nontoxic.
Figure 6.
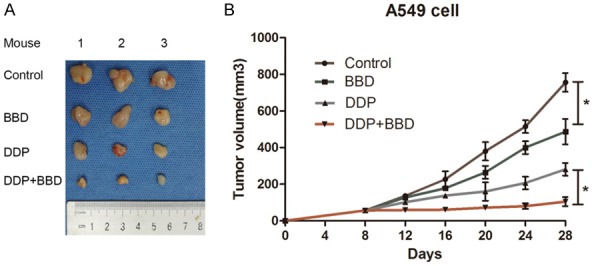
BBD exerted anti-tumor activity in A549 xenografts. A549 cells were injected into the nude mice, and BBD, DDP, and control PBS administered as indicated. A. Representative images of tumors from mice in each group. B. Tumor volume was measured every 4 days. *, P < 0.05.
Discussion
BBD, as a TCM formula, has been widely used for cancer therapy because of the properties of multi-components and multi-target. However, there are still relatively few studies focused on the antitumor effects of BBD, particularly in the case of lung cancer. Here, we demonstrated for the first time that BBD inhibited tumor growth by triggering autophagy through downregulation of PI3K/AKT/mTOR signaling pathway in lung adenocarcinoma.
We first examined the cytotoxic effects of BBD on A549 and A549DDP cells, and surprisingly found it significantly suppressed the cell viability and inhibited the colony-forming activity. However, it failed to generate apoptosis in these cells, with no changes observed in the protein level of Bax/Bcl-2, and caspase-cleaved PARP with BBD treatment. Similar to apoptosis, there is another programmed cell death pathway-autophagy. Though the correlation between apoptosis and autophagy is complicated and controversial, accumulating evidence has indicated that autophagic death indeed plays an indispensable role in apoptosis-resistant cancer cells [27,28].
To verify whether BBD exerts its therapeutic effects via modulating autophagy, we first tested the conversion of LC3-I to LC3-II and found that the protein level of LC3-II was distinctly elevated when cells were exposed to BBD. To further discriminate the increased level of LC3-II either from the activated autophagic flux or impaired degradation of autophagolysosome, we analyzed the level of LC3-II in the presence of BBD, combined with Bafilomycin A1 (Baf-A1) pre-exposure. The results showed that co-incubation of cells with BBD and Baf-A1 showed higher level of LC3-II, compared with Baf-A1 alone, indicating that BBD could facilitate autophagosome formation. In parallel, observation of increased autophagosomes and autolysosomes by TEM corroborated this finding. Furthermore, pretreatment of the cells with 3-MA caused weaker inhibition of cell viability. Western blot analysis also indicated the BBD-induced high level of LC3-II was rescued by 3-MA pretreatment. All data suggested that BBD induced cytotoxic effects on A549 and A549DDP cells very likely by triggering nonprotective autophagy.
To further understand the biological function of BBD to lung adenocarcinoma cells, the PI3K/AKT/mTOR signaling pathway, which is a classic autophagic pathway [25], was investigated. We surprisingly found that all the phosphorylated products mentioned above in the pathway, like p-PI3K, p-AKT, p-mTOR were reduced by BBD. We additionally used the PI3K/AKT agonist, insulin-like growth factor-1 (IGF-1), to clarify the relationship between the activation of the PI3K/AKT/mTOR pathway and BBD-induced autophagy. The results showed the effect of IGF-1 on LC3-II impairment was partially rescued by BBD addition. Mechanistically, the data indicated that BBD augmented cell autophagy through downregulation of PI3K/AKT/mTOR signaling pathway.
Intrinsic or acquired MDR is a severe challenge for nearly all chemotherapeutic agents [29]. This is due to the reduction of intracellular drug accumulation. Drug efflux pumps play an important role in extruding therapeutic compounds from the cell, among which P-gp is the most widely studied [30-32]. Our study manifested that BBD appreciably decreased the mRNA and protein expression of MDR1 in a drug sequence-dependent manner. Increased suppression of cell invasion and migration is a feature of cellular response to combined BBD and cisplatin treatment, which certified as the synergetic antitumor effect on cells. Consistent with our results in vitro, the nude mouse model also suggested that BBD inhibited tumor growth and elevated sensitivity of cisplatin in NSCLC cells.
In summary, our data experimentally proved for the first time that BBD could inhibit tumor growth and increase the chemosensitivity of cisplatin in NSCLC in vivo and in vitro. Furthermore, we determined the molecular mechanism underlying the phenotype, the inhibition of PI3K/AKT/mTOR pathway. Given that this was a preliminary study, and inevitably has some defects, it provides a solid theoretical foundation for the wide pharmacological effects of BBD as a valuable therapeutic adjuvant. It will also be necessary for further research to explore which active ingredient in BBD is responsible for such functions.
Acknowledgements
This study was supported by the manufacturer Xiamen Traditional Chinese Medicine Company Limited.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, Cheney RT, Chirieac LR, D’Amico TA, Demmy TL, Dilling TJ, Dobelbower MC, Govindan R, Grannis FJ, Horn L, Jahan TM, Komaki R, Krug LM, Lackner RP, Lanuti M, Lilenbaum R, Lin J, Loo BJ, Martins R, Otterson GA, Patel JD, Pisters KM, Reckamp K, Riely GJ, Rohren E, Schild SE, Shapiro TA, Swanson SJ, Tauer K, Yang SC, Gregory K, Hughes M. Non-small cell lung cancer, version 6.2015. J Natl Compr Canc Netw. 2015;13:515–524. doi: 10.6004/jnccn.2015.0071. [DOI] [PubMed] [Google Scholar]
- 3.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunn PJ, Kelly K. New chemotherapeutic agents prolong survival and improve quality of life in non-small cell lung cancer: a review of the literature and future directions. Clin Cancer Res. 1998;4:1087–1100. [PubMed] [Google Scholar]
- 5.Tu Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat Med. 2011;17:1217–1220. doi: 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- 6.van Agtmael MA, Eggelte TA, van Boxtel CJ. Artemisinin drugs in the treatment of malaria: from medicinal herb to registered medication. Trends Pharmacol Sci. 1999;20:199–205. doi: 10.1016/s0165-6147(99)01302-4. [DOI] [PubMed] [Google Scholar]
- 7.Lou JS, Yan L, Bi CW, Chan GK, Wu QY, Liu YL, Huang Y, Yao P, Du CY, Dong TT, Tsim KW. Yu Ping Feng San reverses cisplatin-induced multi-drug resistance in lung cancer cells via regulating drug transporters and p62/TRAF6 signalling. Sci Rep. 2016;6:31926. doi: 10.1038/srep31926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong M, Ye T, Bi Y, Wang Q, Kuerban K, Li J, Feng M, Wang K, Chen Y, Ye L. A novel hybrid of 3-benzyl coumarin seco-B-ring derivative and phenylsulfonylfuroxan induces apoptosis and autophagy in non-small-cell lung cancer. Phytomedicine. 2019;52:79–88. doi: 10.1016/j.phymed.2018.09.216. [DOI] [PubMed] [Google Scholar]
- 9.Poornima P, Weng CF, Padma VV. Neferine from Nelumbo nucifera induces autophagy through the inhibition of PI3K/Akt/mTOR pathway and ROS hyper generation in A549 cells. Food Chem. 2013;141:3598–3605. doi: 10.1016/j.foodchem.2013.05.138. [DOI] [PubMed] [Google Scholar]
- 10.Fu T, Wang L, Jin XN, Sui HJ, Liu Z, Jin Y. Hyperoside induces both autophagy and apoptosis in non-small cell lung cancer cells in vitro. Acta Pharmacol Sin. 2016;37:505–518. doi: 10.1038/aps.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun B, Gao L, Ahsan A, Chu P, Song Y, Li H, Zhang Z, Lin Y, Peng J, Song Z, Wang S, Tang Z. Anticancer effect of SZC015 on lung cancer cells through ROS-dependent apoptosis and autophagy induction mechanisms in vitro. Int Immunopharmacol. 2016;40:400–409. doi: 10.1016/j.intimp.2016.09.026. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh MJ, Chen MK, Yu YY, Sheu GT, Chiou HL. Psoralen reverses docetaxel-induced multidrug resistance in A549/D16 human lung cancer cells lines. Phytomedicine. 2014;21:970–977. doi: 10.1016/j.phymed.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Wu J, Fan M, Shen C, Dai W, Bao Y, Liu J, Yu B. Novel dihydroartemisinin derivative DHA-37 induces autophagic cell death through upregulation of HMGB1 in A549 cells. Cell Death Dis. 2018;9:1048. doi: 10.1038/s41419-018-1006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–20. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 17.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mokarram P, Albokashy M, Zarghooni M, Moosavi MA, Sepehri Z, Chen QM, Hudecki A, Sargazi A, Alizadeh J, Moghadam AR, Hashemi M, Movassagh H, Klonisch T, Owji AA, Los MJ, Ghavami S. New frontiers in the treatment of colorectal cancer: autophagy and the unfolded protein response as promising targets. Autophagy. 2017;13:781–819. doi: 10.1080/15548627.2017.1290751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wasik AM, Grabarek J, Pantovic A, Cieslar-Pobuda A, Asgari HR, Bundgaard-Nielsen C, Rafat M, Dixon IM, Ghavami S, Los MJ. Reprogramming and carcinogenesis--parallels and distinctions. Int Rev Cell Mol Biol. 2014;308:167–203. doi: 10.1016/B978-0-12-800097-7.00005-1. [DOI] [PubMed] [Google Scholar]
- 20.Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226:255–273. doi: 10.1002/path.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao Q, Yu C, Xue R, Hsueh W, Pan P, Chen Z, Wang S, McNutt M, Gu J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int J Biochem Cell Biol. 2008;40:272–283. doi: 10.1016/j.biocel.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 22.Feng ZQ, Wu LC, Shen MH, Shu QJ, Deng YC, Shao GL. Clinical observe report of babao pellet compounded of TACE treating the heat toxic and blood stagnation type patients with primary hepatocellular carcinoma. Chinese Archives of Traditional Chinese Medicine. 2007 [Google Scholar]
- 23.Bowman EJ, Graham LA, Stevens TH, Bowman BJ. The bafilomycin/concanamycin binding site in subunit c of the V-ATPases from neurospora crassa and saccharomyces cerevisiae. J Biol Chem. 2004;279:33131–33138. doi: 10.1074/jbc.M404638200. [DOI] [PubMed] [Google Scholar]
- 24.Nagelkerke A, Sweep FC, Geurts-Moespot A, Bussink J, Span PN. Therapeutic targeting of autophagy in cancer. Part I: molecular pathways controlling autophagy. Semin Cancer Biol. 2015;31:89–98. doi: 10.1016/j.semcancer.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Nurwidya F, Andarini S, Takahashi F, Syahruddin E, Takahashi K. Implications of insulin-like growth factor 1 receptor activation in lung cancer. Malays J Med Sci. 2016;23:9–21. [PMC free article] [PubMed] [Google Scholar]
- 26.Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu B, Pan S, Dong X, Tan G, Wei Z, Qiao H, Jiang H, Sun X. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13:1589–1598. doi: 10.1158/1535-7163.MCT-13-1043. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu S, Yoshida T, Tsujioka M, Arakawa S. Autophagic cell death and cancer. Int J Mol Sci. 2014;15:3145–3153. doi: 10.3390/ijms15023145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 29.Fojo T, Bates S. Strategies for reversing drug resistance. Oncogene. 2003;22:7512–7523. doi: 10.1038/sj.onc.1206951. [DOI] [PubMed] [Google Scholar]
- 30.Johnstone RW, Ruefli AA, Smyth MJ. Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem Sci. 2000;25:1–6. doi: 10.1016/s0968-0004(99)01493-0. [DOI] [PubMed] [Google Scholar]
- 31.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 32.Stavrovskaya AA. Cellular mechanisms of multidrug resistance of tumor cells. Biochemistry (Mosc) 2000;65:95–106. [PubMed] [Google Scholar]