

Abstract
Diabetic nephropathy (DN) is a common complication of diabetes that is the dominant cause of end-stage renal disease. However, the pathological mechanism of DN is yet to be elucidated. Serum and glucocorticoid induced kinase (SGK) 1, a ubiquitously expressed kinase, was employed in the current study to assess its effect on DN in vivo and in vitro. Male BALB/C mice and a human tubular epithelial cell line (HK-2) were utilized for experimentation. Male BALB/C mice and a human tubular epithelial cell line (HK-2) were utilized for experimentation. Pathological changes were measured via HE and staining and immunohistochemistry was performed to measure the expression of SGK 1. An SGK1 inhibitor, GSK650394, was applied to analyze the role of SGK1 in HK-2 cell epithelial-mesenchymal transition (EMT). Associated protein expressions were assessed via western blotting. In addition, migration was measured using a scratch wound healing assay. 3-methyladenine (3-MA), an autophagy inhibitor, was used to determine the variation of autophagy following SGK1 inhibition. The expression of autophagy proteins were analyzed. Furthermore, the expression of PI3K, AKT, mTOR and their levels of phosphorylation were measured. The results revealed that the ultrastructure of renal tissue suffered damage and that the expression of SGK1 was markedly increased. After SGK1 inhibition, HK-2 cell EMT was suppressed and cell migration was attenuated. Furthermore, the autophagy of HK-2 cells was promoted, an increased expression of Beclin-1 and LC3 II was detected, and a decreased expression of p62 was observed. Additionally, the phosphorylation of PI3K, AKT and mTOR were markedly upregulated. The results indicated that blocking autophagy signaling via 3-MA muted SGK1-protected against HG-evoked cell injury. Our study demonstrated that SGK1 inhibition promoted autophagy and suppressed renal tubular epithelial cell EMT in DN, indicating that SGK1 may serve as a potential therapeutic target of DN.
Keywords: Autophagy, diabetic nephropathy, serum-and glucocorticoid induced kinase, epithelial mesenchymal transition
Introduction
Diabetic nephropathy (DN) is an important microvascular complication of diabetes that is one of the leading causes of end-stage kidney disease, which is also primarily attributed to the increasing prevalence of type 2 diabetes [1,2]. With an increasing number of new diagnoses and a 5-year survival rate of ~20%, DN has attracted much attention [3]. The main pathological features of DN include glomerular hypertrophy, mesangial proliferation, thickening of the glomerular basement membrane and the accumulation of extracellular matrix [4,5]. Emerging evidence has indicated that proximal tubule injury is involved in renal injury and serves a crucial role in the early stages of DN [6].
Renal tubular epithelial cell epithelial-mesenchymal transition (EMT) is the main pathological process that leads to end-stage renal disease in various kidney diseases, including DN [7,8]. It has been well documented that EMT is a process from which epithelial cells lose their polarity and are converted into a mesenchymal phenotype epithelial cells [9]. Serum-and glucocorticoid induced kinase (SGK) 1, a ubiquitously expressed kinase, acts as an anti-oncogene in many different types of cancer, including colon, gastric and endometrial cancer [10-12]. A previous study reported that the inhibition of SGK1 in prostate cancer epithelial cells suppressed cell invasion, migration and EMT via autophagy [13]. Furthermore, SGK1 is involved in the mediation of obstructive nephropathy-associated fibrosis by attenuating EMT progression [14].
Autophagy is an intracellular catabolic process in which the lysosome participates in ageing and degrades damaged organelles and proteins. It is essential for maintaining cellular homeostasis [15,16]. Accumulating studies have revealed that autophagy is implicated in the pathogenesis of DN and may serve as a novel therapeutic target [17,18]. The different phases of autophagy are regulated by a number of autophagy proteins. A well-known previous study determined that Beclin-1 is a subunit of the class III phosphoinositide-3-kinase (PI3K) complex, which is critical for the initiation of autophagosome formation [19]. The PI3K/AKT signaling serves a crucial role in regulation of cell growth and metabolism [20]. In addition, Mammalian target of rapamycin (mTOR) is considered as an intracellular repressor of autophagy, which could be regulated by PI3K/AKT signaling [21].
In our study, we investigated the role of SGK1 in DN and to elucidate its underlying mechanism. The current study demonstrated that SGK1 inhibition promoted autophagy and suppressed renal tubular cell EMT in DN by activating the PI3K/AKT/mTOR signaling. It was concluded that SGK1 may serve as a potential therapeutic agent and provide a new therapeutic direction for the clinical treatment of DN.
Material and methods
Animals and treatment
Male BALB/C mice were obtained from Shanghai SLAC Laboratory Animal Company Ltd. All animals were raised in a suitable environment with a temperature of 21 ± 3°C under a 12 h light/dark cycle. Mice were given free access to food and water and were acclimatized to the environment for at least 1 week prior to experimentation. All study protocols were approved by the Ethics Committee on Animal Experiments of Bengbu Medical College. Mice were divided into two groups arbitrary: A normal control group (n = 10) and a model group (n = 10). Body weight was measured before the experiment began and animals were starved for 12 h. Subsequently, 55 mg/kg Streptozotocin (STZ; Sigma-Aldrich; Merck KGaA) was administered to model mice via an intraperitoneal injection to induce DN. Control mice were injected with the same volume of natrium citricum buffer solution (0.1 mol/l; pH 4.5). Fasting blood glucose levels were then measured from blood obtained via the caudal vein in triplicate, 3 days after injection. When the concentration of glucose was ≥ 16.7 mmol/l, the diabetic model was deemed to be successfully established. The serum urea protein and urine volume in 24 h of metabolic cage exceeded 50% of that before the model meaning the successful DN model was made. All mice were sacrificed at 12 weeks following anesthesia, after which the kidneys were collected and preserved for the further investigation.
Cell culture and treatment
The human tubular epithelial cell line HK-2 was obtained from The Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences. Cells were cultured in DMEM-F12 (3:1) containing 10% fetal bovine serumin 5% CO2 at 37°C. When cells reached a confluence of 80%, they were exposed to 30 mM glucose to induce DN. GSK650394 (GSK; Selleck Chemicals), an inhibitor of SGK1, was used to block the expression of SGK1. Additionally, 3-methyladenine (3-MA; Selleck chemicals), an autophagy inhibitor, was utilized to assess changes in autophagy following SGK1 inhibition.
Hematoxylin and eosin (HE) staining and Masson staining
Harvested kidney tissues were conventionally fixed in 4% paraformaldehyde overnight at 4°C. Paraffin-embedded posterior segments, which contained the optic disc, were then sectioned into 5 μm slices. The sections were deparaffinized with graded ethanol together with xylene. After HE staining, sections were dehydrated with graded ethanol and xylene. The remaining paraffin-embedded kidneys were cut into 2 mm sections and stained with Masson’s trichrome to assess pathological changes. Images were subsequently obtained using an OLYMPUS DP73 microscope (Olympus Corporation).
Immunohistochemical assay (IHC)
ICH was performed using 5 μm paraffin-embedded kidney sections. After deparaffinization and rehydration, slides were placed in citric acid buffer and heated for antigen retrieval. Subsequently, all kidney slides were blocked with 5% bovine serum albumin for 60 min at 37°C. Sections were then incubated with primary anti-rabbit antibodies against SGK1 (1:1000; cat. no. ab59337; Abcam) overnight at 4°C in a wet box. Slides were incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies and observed under a light microscope. Semi-quantitative analysis was subsequently performed using Image-Pro Plus version 6.0 software.
Immunofluorescence assay
Slides containing HK-2 cells were soaked in PBS three times in the culture plate. Cells were subsequently fixed in 4% paraformaldehyde solution at 4°C for 24 h and treated with 0.1% Triton X-100 in PBS for blocking at room temperature for 2 h. Cells were then incubated with anti-LC3 II (1:1000; cat. no. ab48394; Abcam) at 4°C for 24 h. After washing in triplicate with PBS, HK-2 cells were incubated with DyLight™ 488-conjugated secondary antibodies (Thermo Fisher Scientific, Inc.) for 2 h. Immunofluorescence was detected via fluorescence microscopy (Olympus Corporation).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from renal tissues or HK-2 cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently, RNA was reverse-transcribed to cDNA using the QuantiTect RT kit (Qiagen GmbH) in accordance with the manufacturer’s protocol. Real-time PCR was performed using a System 7500 instrument (Applied Biosystems; Thermo Fisher Scientific, Inc.). The primer sequences utilized for qPCR were as follows: SGK1 forward, 5’-GCAGAAGAAGTGTTCTATGCAGT-3’ and reverse, 5’-CCGCTCCGACATAATATGCTT-3’; GAPDH forward, 5’-GGAGCGAGATCCCTCCAAAAT-3’ and reverse, 5’-GGCTGTTGTCATACTTCTCATGG-3’. The relative expression of target genes were normalized to GAPDH, which served as the endogenous control. Expressions were analyzed using the 2-ΔΔCq method [22].
Western blotting
Proteins were extracted from renal tissues or HK-2 cells using RIPA lysis buffer (Beyotime Institute of Biotechnology). A BCA protein assay kit (Bio-rad Laboratories, Inc.) was utilized to determine protein concentration. Subsequently, 40 μg were loaded for SDS-PAGE analysis and transferred to PVDF membranes. Membranes were blocked with 5% skimmed milk. Samples were then incubated with primary antibodies, and secondary antibodies were added. Immunoreactive bands were measured using ECL (Thermo Fisher Scientific, Inc.). The expression of GAPDH was used as an endogenic control in all samples. The following antibodies were obtained from Abcam. Anti-SGK1 (cat. no. 59337) and anti-p62 (cat. no. 8690T). Anti-E-cadherin (cat. no. 3195T), anti-N-cadherin (cat. no. 13116T), anti-Vimentin (cat. no. 5741T), anti-Beclin-1 (cat. no. 3495T), anti-PI3K (cat. no. 4249T), anti-AKT (cat. no. 9272S), anti-mTOR (cat. no. 2972S), anti-phosphorylated (p)-PI3K (cat. no. 4228S), anti-p-AKT (cat. no. 4060S), anti-p-mTOR (cat. no. 5536S) and anti-GAPDH (cat. no. 5174S) antibodies were purchased from Cell Signaling Technology, Inc.
Cell migration assay
A scratch wound healing assay was performed to measure HK-2 cell migration. HK-2 cells were plated into 6-well plates for 24 h. Subsequently formed monolayers were scratched using a pipette tip and floating cells were removed by washing with PBS. Cells were incubated in serum-free DMEM medium for 48 h and photographed with a camera connected to a microscope. The percentage of wound healing was analyzed using ImageJ software (National Institutes of Health).
Statistical analysis
All results were confirmed in at least three independent experiments. All experimental results were expressed as mean ± SD. Statistical analysis was performed using SPSS software 16.0 (SPSS, Inc.). Statistical comparisons were made via one-way ANOVA followed by a post-hoc Dunnett’s test. A significance level of P < 0.05 was adopted for all analyses.
Results
Diabetic nephropathy is successfully established in model mice
Fasting blood glucose levels were monitored after mice were fasted for 12 h following STZ injections for 72 h. As exhibited in Figure 1A, murine fasting blood glucose levels were > 16.7 mmol/l in the model group after treatment with STZ. Furthermore, relative urine volume and urine protein levels were markedly decreased at 24 h (Figure 1B and 1C), indicating that the diabetic model was successfully established. Subsequently, HE and Masson staining were performed to analyze the associated changes in renal tissue. The structure of the glomerulus exhibited certain abnormalities, including an increased glomerular volume and significantly increased mesangial cell proliferation when compared with the control group (Figure 1D and 1E). These data demonstrated that the murine model of DN was successfully established.
Figure 1.
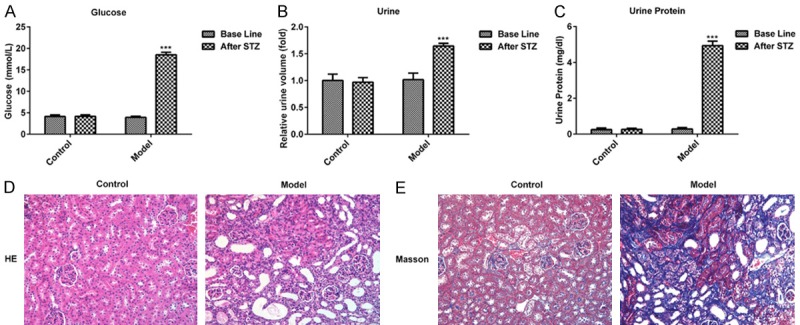
Physiological changes of mice after injection with STZ for 72 h. (A) The levels of glucose, (B) urine volume and (C) urine protein in control and STZ treated mice are presented. The pathologic changes of renal tissues were measured via (D) hematoxylin and eosin and (E) Masson staining. (magnification, ×100). ***P < 0.001 vs. the control. STZ, streptozotocin.
Expression of SGK1 and transforming growth factor (TGF)-β1 increases in DN mice
The expressions of SGK1 and TGF-β1 in the renal tissue of DN mice were measured via western blotting and RT-qPCR. As presented in Figure 2A and 2B, the expressions of SGK1 and TGF-β1 were significantly upregulated. Furthermore, the positively marked cells of SGK1 were increased in the DN group compared with the control group (Figure 2C). The results indicated that SGK 1 was overexpressed in DN mice.
Figure 2.
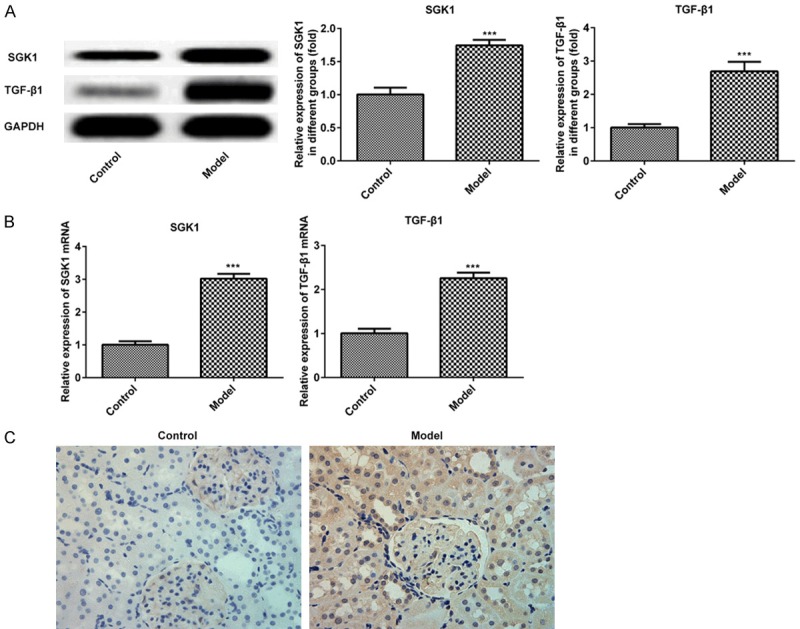
Expression of SGK1 and TGF-β1 in murine renal tissue following treatment with STZ. The expression of SGK1 and TGF-β1 were assessed via (A) western blotting and (B) reverse transcription-quantitative PCR. (C) The expression of SGK1 was determined via immunohistochemistry. (magnification, ×200). ***P < 0.001 vs. the control. SGK1, serum and glucocorticoid induced kinase; TGF-β1, transforming growth factor; STZ, streptozotocin.
Inhibition of SGK1 suppresses EMT progression in high glucose induced HK-2 cells
To investigate the role of SGK1 in vitro, HK-2 cells were exposed to 30 mM glucose to induce DN cell modelling. The results revealed that protein and mRNA levels were augmented (Figure 3A and 3B). GSK, as an inhibitor of SGK1, was then utilized in the present study. As presented in Figure 3C, the expression of E-cadherin was decreased, and the expression of N-cadherin and Vimentin were increased in the Glucose+GSK group. In TGF-β1 and high glucose treatment groups, the protein expression of E-cadherin was downregulated, which was accompanied with an upregulated expression of N-cadherin and Vimentin compared with the Glucose+GSK group. After treatment with TGF-β1 in combination with GSK, the expression of E-cadherin was augmented. Furthermore, N-cadherin and Vimentin levels were augmented. These results indicated that the inhibition of SGK1 suppressed EMT progression.
Figure 3.

Expression of SGK1, E-cadherin, N-cadherin and Vimentin following the treatment of high glucose, TGF-β1 or GSK in HK-2 cells. The expression of SGK1 was measured via (A) western blotting and (B) reverse transcription-quantitative PCR. (C) The expression of E-cadherin, N-cadherin and Vimentin was determined via western blotting. ***P < 0.001 vs. the control; #P < 0.05, ##P < 0.01 and ###P < 0.001 vs. the Glucose group; ΔΔP < 0.01 and ΔΔΔP < 0.001 vs. the Glucose+TGF-β1 group. SGK1, serum and glucocorticoid induced kinase; TGF-β1, transforming growth factor; GSK, GSK650394.
Inhibition of SGK1 reduced the migration of high glucose-induced HK-2 cells
Cell migration was assessed via scratch wound healing and Transwell assays. As presented in Figure 4A-D, cell migration was reduced in the Glucose+GSK group. Subsequently, TGF-β1 was administered to treat high glucose induced HK-2 cells. The results revealed an increased cell migration when compared with the Glucose+GSK group. Additionally, treatment with TGF-β1 in combination with GSK in high glucose culture conditions markedly attenuated cell migration when compared with the Glucose+TGF-β1 group. The aforementioned data indicated that the inhibition of SGK1 suppressed the migration of high induced HK-2 cells.
Figure 4.
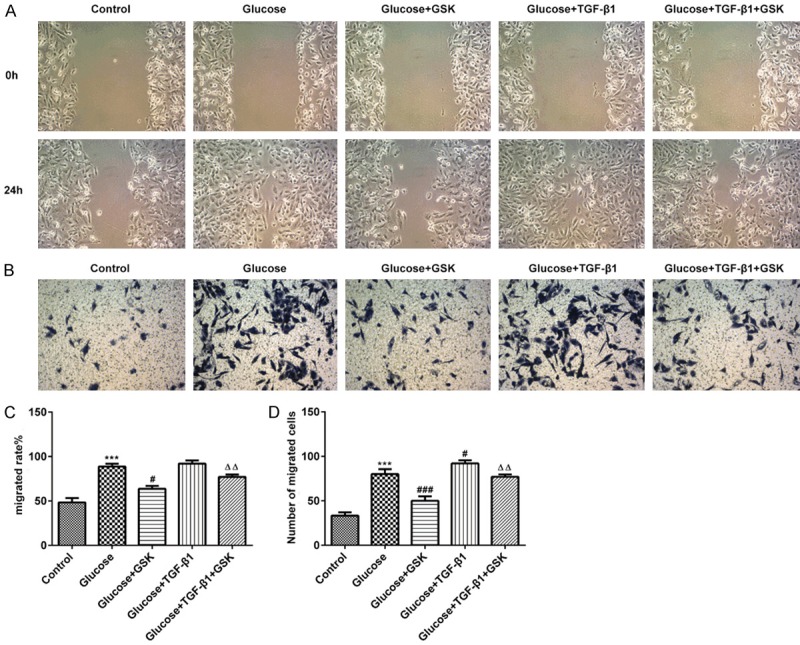
HK-2 cell migration following treatment with high glucose, TGF-β1 or GSK. The migratory activity of HK-2 cells was assessed via (A and C) scratch and (B and D) Transwell assays (magnification, ×20). ***P < 0.001 vs. the control; #P < 0.05 and ###P < 0.001 vs. the Glucose group; ΔΔP < 0.01 vs. the Glucose+TGF-β1 group. TGF-β1, transforming growth factor; GSK, GSK650394.
SGK1 inhibition promotes high glucose induced HK-2 cell autophagy
To assess the effect of SGK1 on high glucose-induced HK-2 cell autophagy, the expression of Beclin-1 and p62 were detected via western blotting. As presented in Figure 5A, the expression of Beclin-1 and p62 were down- and upregulated, respectively. Furthermore, levels of Beclin-1 and p62 were increased and decreased respectively following treatment with Glucose and GSK. After blocking with 3-MA, the aforementioned effects on Beclin-1 and p62 were reversed. To further confirm the level of autophagy, immunofluorescence was performed to measure the level of LC3. As presented in Figure 5B and 5C, the fluorescence intensity of the autophagy marker protein, LC3, was significantly lower than that of the control group, indicating that autophagy was decreased. After treatment with high glucose and GSK, the fluorescence intensity of LC3 expression was high. However, a decreased expression of LC3 was demonstrated in HK-2 cells following treatment with 3-MA, which indicated that SGK1 inhibition promoted high glucose-induced HK-2 cell autophagy.
Figure 5.
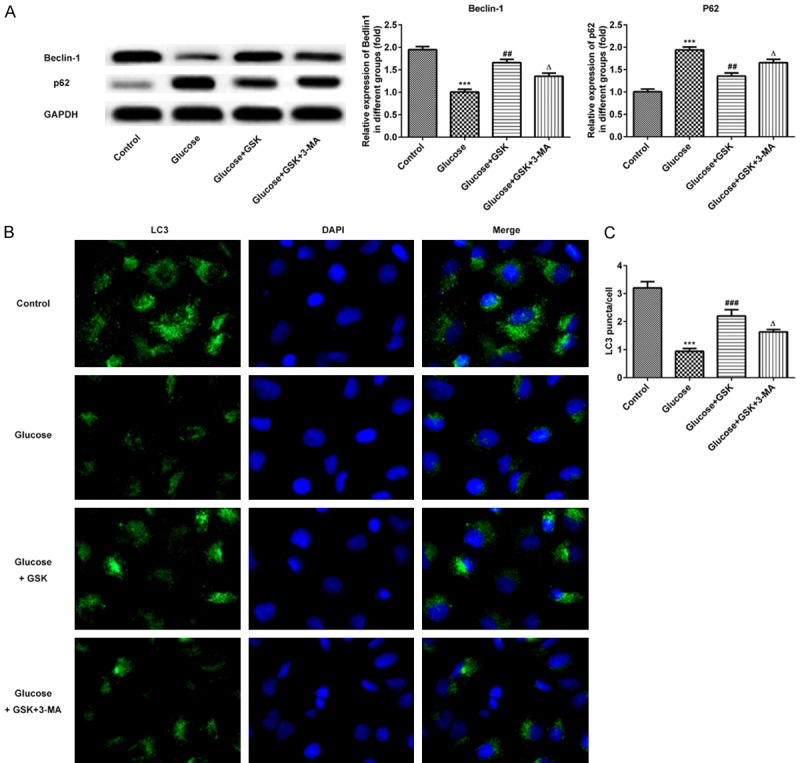
Expression of Beclin-1, p62 and LC3 following HK-2 cell treatment with high glucose, GSK or 3-MA. A. The expressions of Beclin-1 and p62 were measured via western blotting. B. Levels of LC3 were determined via immunofluorescence. (magnification, ×200). C. Quantitative analysis for immunofluorescence. ***P < 0.001 vs. the control; ##P < 0.01 and ###P < 0.001 vs. the Glucose group; ΔP < 0.05 vs. the Glucose+TGF-β1 group. GSK, GSK650394’ 3-MA, 3-methyladenine.
Inhibition of SGK1 activates the PI3K/AKT/mTOR signaling pathway in high glucose-induced HK-2 cells
The expressions of PI3K/AKT/mTOR were measured via western blotting. As presented in Figure 6, the expression of p-PI3K, p-AKT and p-mTOR were downregulated in high glucose induced HK-2 cells compared with control. However, these proteins were upregulated in the glucose and GSK group. Following treatment with GSK in combination with 3-MA, the expressions of p-PI3K, p-AKT and p-mTOR were decreased compared with the glucose and GSK group. These results indicated that SGK1 inhibition promoted autophagy by activating the PI3K/AKT/mTOR signaling pathway.
Figure 6.
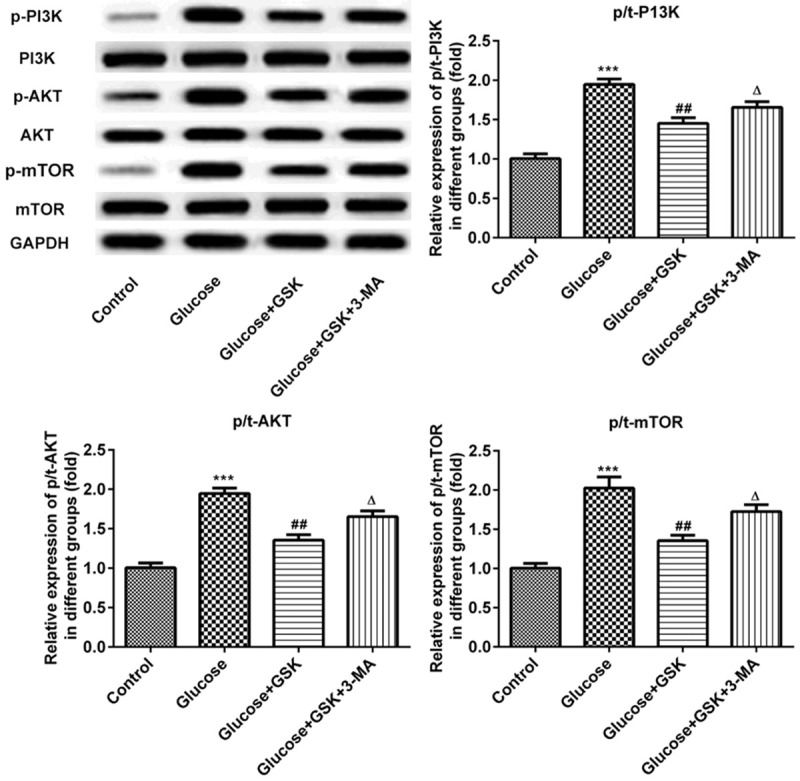
Expression of p-PI3K, p-AKT and p-mTOR following HK-2 cell treatment with high glucose, GSK or 3-MA. The expressions of p-PI3K, p-AKT and p-mTOR were measured via western blotting. ***P < 0.001 vs. the control; ##P < 0.01 vs. the Glucose group; ΔP < 0.05 vs. the Glucose+TGF-β1 group. p, phosphorylated; GSK, GSK650394’ 3-MA, 3-methyladenine.
Discussion
The rapidly increasing prevalence of diabetes is resulting in a concomitant increase in the prevalence of DN [23]. In recent years, accumulating evidence has focused on elucidating the molecular mechanisms that underlie the initiation and progression of DN, in the hope of developing novel therapeutic methods. However, DN remains a worldwide health problem and as such, an alternative novel therapeutic agent for the treatment of DN is urgently required [24]. The present study demonstrated for the first time that the inhibition of SGK1 ameliorates DN by promoting autophagy and suppressing EMT via the PI3K/AKT/mTOR signaling pathway in renal tubular epithelial cells.
EMT is characteristic for the transformation of primer epithelial cells to mesenchymal cells, which contributes to cell movement during development and facilitates the pathogenesis of disease [25,26]. EMT serves an important role in the progression of DN and previous studies have supported the notion that the amelioration of high glucose-induced renal tubular EMT exerts protective effects on DN [27,28]. It has been well documented that high glucose or TGF-β1 treatment induces human renal tubular cell EMT, resulting in the decreased expression of E-cadherin accompanied with the increased expression of N-cadherin and Vimentin [29]. In the current study, GSK, an inhibitor of SGK1, was used to determine the role of SGK1 in high glucose-induced HK-2 cells. Previous studies have demonstrated that high glucose treatment increases SGK1 protein and mRNA expression, leading to EMT by increasing the effects exerted by TGF-β1 in HK-2 cells [30,31]. The results of the present study were consistent with those of the aforementioned research. After treatment with TGF-β1 in combination with GSK, the expression of E-cadherin, N-cadherin and Vimentin were reversed when compared with those after TGF-β1 treatment alone. In addition, the results of the migration assay revealed the same changes as described above.
Autophagy is an evolutionarily conserved cellular process that degrades protein aggregates, damaged organelles and other macromolecules in the cytoplasm [31]. It is therefore considered to serve important roles in cellular homeostasis [32]. Levels of autophagy are decreased in individuals with DN and several studies have reported that autophagy exerts a defensive role against kidney damage caused by hyperglycemia. This indicates that the induction of autophagy may provide protection against DN [33,34]. Beclin-1, a key regulator of autophagy and the mammalian ortholog of yeast Atg6, has been reported to induce autophagy by regulating PI3K [35-37]. LC3 and Beclin-1 are effective biomarkers for monitoring autophagy. p62 is well-known for its role in linking polyubiquitinated protein aggregates in autophagic machinery. Furthermore, its upregulated protein expression indicates the inhibition of autophagy [38]. The present results revealed an upregulated expression of Beclin-1 and LC3-II/I, and a decreased expression of p62 in high glucose and GSK treated HK-2 cells compared with high glucose treatment alone, which demonstrated that SGK1 inhibition induced autophagy. After exposure to 3-MA, levels of Beclin-1 and LC3-II/I in HK-2 cells were decreased, and the expression of p62 was increased. These results further indicated that the induction of autophagy induced by the inhibition of SGK1 protects against DN.
Previous studies have suggested that the PI3K/AKT/mTOR signaling participats in the control of autophagy [39,40]. Emerging evidence supports that inhibition of PI3K/AKT/mTOR signaling initiates autophagy and protects against kidney tubular epithelial injury [41]. In addition, KCa3.1 mediates the dysfunction of tubular autophagy in DN through PI3K/AKT/mTOR pathway [42]. The present results demonstrated that glucose suppresses autophagy and upregulates the expression of p-PI3K, p-AKT and p-mTOR. After GSK was used to block SGK1, autophagy was induced and the decreased phosphorylation of PI3K, AKT and mTOR was observed. In contrast, the above mentioned protein expressions were increased after treatment with 3-MA. The results indicated that SGK1 inhibition promotes renal tubular epithelial cell autophagy in DN through PI3K/AKT/mTOR signaling.
Conclusion
The results of our study demonstrated that SGK1 inhibition promotes autophagy and suppresses the EMT of renal tubular epithelial cells in DN via the PI3K/AKT/mTOR signaling pathway. On the basis of these results, SGK1 inhibition may serve as a potential therapeutic target of DN.
Acknowledgements
The present study was supported by the Natural Science Foundation of Anhui Province (Grant No. 1908085QH319).
Disclosure of conflict of interest
None.
References
- 1.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–2340. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badal SS, Danesh FR. New insights into molecular mechanisms of diabetic kidney disease. Am J Kidney Dis. 2014;63:S63–83. doi: 10.1053/j.ajkd.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magee C, Grieve DJ, Watson CJ, Brazil DP. Diabetic nephropathy: a tangled web to unweave. Cardiovasc Drugs Ther. 2017;31:579–592. doi: 10.1007/s10557-017-6755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han Q, Zhu H, Chen X, Liu Z. Non-genetic mechanisms of diabetic nephropathy. Front Med. 2017;11:319–332. doi: 10.1007/s11684-017-0569-9. [DOI] [PubMed] [Google Scholar]
- 5.Palsamy P, Subramanian S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochim Biophys Acta. 2011;1812:719–731. doi: 10.1016/j.bbadis.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, Hosoya K, Komatsu M, Kaneko Y, Kanda T, Kubota E, Tokuyama H, Hayashi K, Guarente L, Itoh H. Renal tubular sirt1 attenuates diabetic albuminuria by epigenetically suppressing claudin-1 overexpression in podocytes. Nat Med. 2013;19:1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boffa JJ, Dussaule JC, Ronco P, Chatziantoniou C. Chronic kidney disease, new therapeutic approaches. Rev Prat. 2012;62:72–75. [PubMed] [Google Scholar]
- 8.Wagnew F, Eshetie S, Kibret GD, Zegeye A, Dessie G, Mulugeta H, Alemu A. Diabetic nephropathy and hypertension in diabetes patients of sub-saharan countries: a systematic review and meta-analysis. BMC Res Notes. 2018;11:565. doi: 10.1186/s13104-018-3670-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling L, Tan Z, Zhang C, Gui S, Hu Y, Chen L. Long noncoding RNA ENSRNOG00000037522 is involved in the podocyte epithelialmesenchymal transition in diabetic rats. Int J Mol Med. 2018;41:2704–2714. doi: 10.3892/ijmm.2018.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu T, Yu T, Hu H, He K. Knockdown of the long non-coding RNA HOTTIP inhibits colorectal cancer cell proliferation and migration and induces apoptosis by targeting SGK1. Biomed Pharmacother. 2018;98:286–296. doi: 10.1016/j.biopha.2017.12.064. [DOI] [PubMed] [Google Scholar]
- 11.Yao Y, Jiang Q, Jiang L, Wu J, Zhang Q, Wang J, Feng H, Zang P. Lnc-SGK1 induced by Helicobacter pylori infection and highsalt diet promote Th2 and Th17 differentiation in human gastric cancer by SGK1/Jun B signaling. Oncotarget. 2016;7:20549–20560. doi: 10.18632/oncotarget.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conza D, Mirra P, Cali G, Tortora T, Insabato L, Fiory F, Schenone S, Amato R, Beguinot F, Perrotti N, Ulianich L. The SGK1 inhibitor SI113 induces autophagy, apoptosis, and endoplasmic reticulum stress in endometrial cancer cells. J Cell Physiol. 2017;232:3735–3743. doi: 10.1002/jcp.25850. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Wang X, Wang Y, Dai Y, Xie Y, Ping Y, Yin B, Yu P, Liu Z, Duan X, Liao Z, Chen Y, Liu C, Li X, Tao Z. SGK1 inhibition-induced autophagy impairs prostate cancer metastasis by reversing EMT. J Exp Clin Cancer Res. 2018;37:73. doi: 10.1186/s13046-018-0743-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng J, Truong LD, Wu X, Kuhl D, Lang F, Du J. Serum- and glucocorticoid-regulated kinase 1 is upregulated following unilateral ureteral obstruction causing epithelial-mesenchymal transition. Kidney Int. 2010;78:668–678. doi: 10.1038/ki.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol. 2013;9:713–723. doi: 10.1038/nrendo.2013.184. [DOI] [PubMed] [Google Scholar]
- 17.Wei W, An XR, Jin SJ, Li XX, Xu M. Inhibition of insulin resistance by PGE1 via autophagy-dependent FGF21 pathway in diabetic nephropathy. Sci Rep. 2018;8:9. doi: 10.1038/s41598-017-18427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, Chen S, Xu J, Zhu Q, Ye X, Ding D, Yao W, Lu Y. Dysregulation of lncRNAs GM5524 and GM15645 involved in highglucoseinduced podocyte apoptosis and autophagy in diabetic nephropathy. Mol Med Rep. 2018;18:3657–3664. doi: 10.3892/mmr.2018.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deshpande S, Abdollahi M, Wang M, Lanting L, Kato M, Natarajan R. Reduced autophagy by a microRNA-mediated signaling cascade in diabetes-induced renal glomerular hypertrophy. Sci Rep. 2018;8:6954. doi: 10.1038/s41598-018-25295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3-kinase signalling-which way to target? Trends Pharmacol Sci. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- 21.Shao X, Lai D, Zhang L, Xu H. Induction of autophagy and apoptosis via PI3K/AKT/TOR pathways by azadirachtin a in spodoptera litura cells. Sci Rep. 2016;6:35482. doi: 10.1038/srep35482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol. 2016;12:73–81. doi: 10.1038/nrneph.2015.173. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka Y, Kume S, Kitada M, Kanasaki K, Uzu T, Maegawa H, Koya D. Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res. 2012;2012:628978. doi: 10.1155/2012/628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kothari AN, Mi Z, Zapf M, Kuo PC. Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin Transl Med. 2014;3:35. doi: 10.1186/s40169-014-0035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srivastava SP, Koya D, Kanasaki K. MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. Biomed Res Int. 2013;2013:125469. doi: 10.1155/2013/125469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv ZM, Wang Q, Wan Q, Lin JG, Hu MS, Liu YX, Wang R. The role of the p38 MAPK signaling pathway in high glucose-induced epithelial-mesenchymal transition of cultured human renal tubular epithelial cells. PLoS One. 2011;6:e22806. doi: 10.1371/journal.pone.0022806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He T, Guan X, Wang S, Xiao T, Yang K, Xu X, Wang J, Zhao J. Resveratrol prevents high glucose-induced epithelial-mesenchymal transition in renal tubular epithelial cells by inhibiting NADPH oxidase/ROS/ERK pathway. Mol Cell Endocrinol. 2015;402:13–20. doi: 10.1016/j.mce.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Song S, Qiu D, Luo F, Wei J, Wu M, Wu H, Du C, Du Y, Ren Y, Chen N, Duan H, Shi Y. Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. J Mol Endocrinol. 2018;61:101–113. doi: 10.1530/JME-18-0069. [DOI] [PubMed] [Google Scholar]
- 30.Chen CM, Juan SH, Chou HC. Hyperglycemia activates the renin-angiotensin system and induces epithelial-mesenchymal transition in streptozotocin-induced diabetic kidneys. J Renin Angiotensin Aldosterone Syst. 2018;19:1470320318803009. doi: 10.1177/1470320318803009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sayarlioglu H, Okuyucu A, Bedir A, Salis O, Yenen E, Bekfilavioglu G, Kaya C. Is there any role of epithelial to mesenchymal transition in the pathogenesis of contrast nephropathy? Ren Fail. 2016;38:1249–1255. doi: 10.1080/0886022X.2016.1209381. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebrahim N, Ahmed IA, Hussien NI, Dessouky AA, Farid AS, Elshazly AM, Mostafa O, Gazzar WBE, Sorour SM, Seleem Y, Hussein AM, Sabry D. Mesenchymal stem cell-derived exosomes ameliorated diabetic nephropathy by autophagy induction through the mTOR signaling pathway. Cells. 2018;7 doi: 10.3390/cells7120226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Yang L, Chang B, Zhang J, Guo Y, Yang X. The protective effects of rapamycin on cell autophagy in the renal tissues of rats with diabetic nephropathy via mTOR-S6K1-LC3II signaling pathway. Ren Fail. 2018;40:492–497. doi: 10.1080/0886022X.2018.1489287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glover K, Li Y, Mukhopadhyay S, Leuthner Z, Chakravarthy S, Colbert CL, Sinha SC. Structural transitions in conserved, ordered beclin 1 domains essential to regulating autophagy. J Biol Chem. 2017;292:16235–16248. doi: 10.1074/jbc.M117.804195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mei Y, Glover K, Su M, Sinha SC. Conformational flexibility of BECN1: essential to its key role in autophagy and beyond. Protein Sci. 2016;25:1767–1785. doi: 10.1002/pro.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 39.Deng F, Ma YX, Liang L, Zhang P, Feng J. The pro-apoptosis effect of sinomenine in renal carcinoma via inducing autophagy through inactivating PI3K/AKT/mTOR pathway. Biomed Pharmacother. 2018;97:1269–1274. doi: 10.1016/j.biopha.2017.11.064. [DOI] [PubMed] [Google Scholar]
- 40.Inoki K. mTOR signaling in autophagy regulation in the kidney. Semin Nephrol. 2014;34:2–8. doi: 10.1016/j.semnephrol.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du C, Zhang T, Xiao X, Shi Y, Duan H, Ren Y. Protease-activated receptor-2 promotes kidney tubular epithelial inflammation by inhibiting autophagy via the PI3K/Akt/mTOR signalling pathway. Biochem J. 2017;474:2733–2747. doi: 10.1042/BCJ20170272. [DOI] [PubMed] [Google Scholar]
- 42.Huang C, Lin MZ, Cheng D, Braet F, Pollock CA, Chen XM. KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways. Sci Rep. 2016;6:23884. doi: 10.1038/srep23884. [DOI] [PMC free article] [PubMed] [Google Scholar]