

Abstract
Atherosclerosis is a lipid-driven, chronic inflammatory disease that leads to plaque formation at specific sites of the arterial tree. Being the common cause of many cardiovascular disorders, atherosclerosis makes a tremendous impact on morbidity and mortality rates of cardiovascular diseases (CVDs) in countries with higher income. Animal models of atherosclerosis are utilized as useful tools for studying the aetiology, pathogenesis and complications of atherosclerosis, thus, providing a valuable platform for the efficacy testing of different pharmacological therapies and validation of imaging techniques. To date, a large variety of models is available. Pathophysiological changes can be induced in animals by either an atherogenic diet or genetic manipulations. The discussion of advantages and disadvantages of some murine, rabbit and porcine genetic models currently available for the atherosclerosis research is the scope of the following review.
Keywords: Atherosclerosis, genetic animal models of human atherosclerosis, murine models of human atherosclerosis, rabbit models of human atherosclerosis, porcine models of human atherosclerosis
Introduction
In industrialized countries, atherosclerosis is the most significant contributor to CVDs that are accountable for over 50% of total mortality in these countries [1]. Atherosclerosis is the age-dependent disease, which can start at a young age and its prevalence and extent progress with years. Importantly, atherosclerotic lesion development can have a long asymptomatic phase [2,3], and, in many cases, its first clinical manifestations emerge as lesions of the advanced stages that may cause the significant arterial occlusion with severe life-threatening consequences. In association with the presence of the atherosclerosis risk factors, many studies described the subclinical form of atherosclerosis in a large population of young adults [4-8]. Moreover, the high prevalence (up to 100%) of coronary atherosclerosis in asymptomatic teenagers and young adults was reported [9]. There are several different risk factors of atherosclerotic disease, including hyperlipidemia, hypertension, smoking, male gender, genetic disposition and diabetes mellitus. According to the current understanding, these conditions can cause damage to the vascular endothelium allowing lipid penetration into the vascular wall. Particularly, the increased plasma low-density lipoprotein (LDL) level is the most significant risk factor for the development of atherosclerosis and subsequent cardiovascular disease (CVD). Randomized clinical trials of lipid-lowering therapy revealed up to 30% decrease in main coronary events, thus, confirming the significance of hyperlipidemia as a major contributing risk factor of atherosclerosis [10]. Besides, hypercholesterolemia was reported to be the chief population attributable risk factor for coronary heart disease (CHD) [11]. Furthermore, the high content of blood cholesterol and LDL was found to predominantly contribute to the atherogenesis in both humans and animal experimental models [12,13], hence, exposing hypercholesterolemia as an independent risk factor for the atherosclerotic disease. In addition, hypercholesterolemia is a monogenetic cause of familial hypercholesterolemia (FH), an autosomal dominant genetic disorder, in which premature atherosclerosis with subsequent CVD are inevitably developed [14].
It was established that development of the atherosclerotic lesion begins with the accumulation of the circulating modified LDL-cholesterol (LDL-C) in the subendothelial space of the arterial wall, therefore, the subendothelial lipid retention is the major process initiating the atherosclerotic plaque growth [15,16]. Lipid accumulation is proportional to the plasma levels of circulating modified LDL-C and leads to the formation of foam cells. Further accumulation of foam cells in the arterial intimal cells results in the development of primary lesions and consequential fatty streaks, the early-stage lesions in the proatherogenic progression. Thus, LDL-C retention and its accumulation by foam cells in the arterial wall are the key processes leading to the development and progression of the atherosclerotic plaque. Moreover, the intracellular LDL-C retention is accompanied by the migration and accelerated proliferative activity of smooth muscle cells (SMCs), macrophages, lymphocytes, neutrophils and dendritic cells, and the increased synthesis of the components of the extracellular matrix by subendothelial cells [17,18]. Additionally, in response to hyperlipidemia, both the innate and adaptive immune systems are intimately involved in the development of atherosclerotic plaque [19]. Atherosclerotic plaques develop primarily in the walls of large and medium-sized arteries causing thickening of the arterial wall that may lead to the significant narrowing of the arterial lumen and disturbed arterial vessel haemodynamics [20]. Also, for unknown reasons, the atherosclerotic plaques can rupture, often when they are small in size, and lead to the occlusive thrombosis resulting in myocardial infarction (MI) or stroke [21].
The high incidence of atherosclerosis encourages the investigation of its risk factors, causes, and pathomechanisms. In that regards, numerous animal models were explored. Thus, rabbits were the first species used to model atherosclerosis in the study dating back to the beginning of the 20th century that established the direct relationship between the high-cholesterol diet and atherosclerosis [22]. The application of the first models was limited due to the usage of high-fat diets with the noxious side effects. Since then, a number of species, such as mice, rats, guinea pigs, hamsters, birds, dogs, pigs and non-human primates were used for the modelling of human atherosclerosis. In general, animal modelling of atherosclerosis can be based on the accelerated plaque formation due to a cholesterol-rich/Western-type diet, manipulation of genes involved in the cholesterol metabolism, and the introduction of additional atherosclerosis risk factors. Genetically modified animal models were developed to produce spontaneous atherosclerotic lesions and nowadays are preferentially used to study the mechanisms of plaque formation and stability, as well as the development of therapeutic interventions and imaging techniques. None of the models is known to be ideal but each has its own advantages and disadvantages, in terms of precise mimicking of human atherosclerosis and the potential use for the translational research.
In this review, we will discuss the utility of different animal models of human atherosclerosis created by genetic engineering, noting their strengths and weaknesses that will help to increase the understanding of this chronic inflammatory disease and provide the robust foundation for the translational research.
Murine models of human atherosclerosis
Mice become the predominant species to study the experimental atherosclerosis for the following reasons: (i) rapid reproduction, (ii) large proportion of genes is in homology to humans and the ease of genetic manipulation, (iii) ability to monitor atherogenesis in a reasonable time frame, (iv) low cost, and (v) simple in-house maintenance [23,24]. Despite, none of the animal models, including murine models, would replicate human atherosclerotic disease exactly, mice proven to be capable of developing lesion formation processes of close similarity. Like humans, mice spontaneously develop atherosclerotic lesions, in response to the cholesterol-rich diet [23]. Moreover, in humans, the formation of atherosclerotic plaques takes place at the specific arterial regions (vessel bifurcations), where the low and oscillatory endothelial shear stress occurs but varies considerably over a small distance [25,26]. Mice develop atherosclerotic lesions in a similar manner [27].
However, several differences attributed to the developmental process of mouse atherosclerotic lesions should be considered, as they may limit the aptness of murine models to study human atherosclerosis. Thus, the manipulation of the atherogenic process and lipoprotein profile can be difficult in mice due to the substantial genetic differences in the lipid metabolism between mice and man. Cholesterol present in murine plasma is mostly carried out in the atheroprotective high-density lipoprotein (HDL) fraction [28], and atherogenic LDL and its precursors including very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) are rapidly cleared of the blood [29]. The most common lipoprotein pattern of humans suffering from coronary artery disease consists of elevated plasma levels of LDL-C and decreased HDL-C, with or without increases in plasma IDL and VLDL [30]. Moreover, mice and humans exhibit different HDL subclasses that can be indicative of the different levels of atheroprotection [31]. Additionally, mice do not express cholesteryl ester transfer protein (CETP), a plasma protein that attracted much attention as a possible target for atheroprotection in humans [23,32]. Notably, CETR transfers cholesteryl ester from HDL to the apolipoprotein B (ApoB)-containing lipoproteins in exchange for triglycerides. Thus, the lipid profile observed in wild-type mice makes them more resistant to a dietary-induced elevation of LDL and development of atherosclerotic lesions, therefore, the metabolic murine models of human atherosclerosis were not in favour until the recent advances in gene-manipulating technologies. Besides, mice and humans differ in some other parameters that may influence atherogenesis. For example, the heart rate (over 300 bpm in mice, and 70-100 bpm in man), and atherosclerosis generation time (months in genetically modified mice and years in humans) [23].
Human and murine atherosclerosis differ in other features, including topography and morphology of lesions that also may limit the application of murine models. In humans, lesions develop more often in the coronary arteries, carotids and peripheral vessels, such as the iliac artery. Whereas, mice typically do not develop coronary atherosclerosis, except the most proximal regions. Their atherosclerotic lesions are more frequently observed in the aortic root, aortic arch and innominate artery [23]. There is the ingrowth of neo-vessels from vasa vasora into the base of lesions, which is a path for the inflammatory cell infiltration, and is important for the development of local inflammation and core necrosis in humans but such vessels are rarely present in murine lesions [23]. Additionally, taking into account that atherosclerosis development is commonly dependent on local inflammation, it is important to consider the differences in pathomechanisms of both adaptive and innate immune responses in mice and humans [33].
The aspects related to the development of advanced and complicated lesions are also dissimilar in mice models and humans [34] making difficult to study human advanced and complicated atherosclerosis, as well as plaque-stabilizing drugs. The most of murine models do not exhibit the development of unstable atherosclerotic plaque with overlying thrombosis that is the most often associated with clinically significant acute cardiovascular episodes in humans [35]. Also, the lesions acquiring in mice do not typically develop the thick fibrous cap that is frequently attributed to the chronic human atherosclerosis [23].
Another limitation associated with the utility of murine models is their size. The small size of mice can be limiting for some practical investigation procedures, such as visualization of coronary arteries, blood sample collection, and dissection of small arteries [36,37]. Besides, the size constraints limit the stent interventional studies in mice. Stenting is proven to be a safe and effective approach for the management of acute cardiovascular events in humans, for example, ischemic stroke [38]. However, a successful model of murine in situ stenting demonstrating reduction in in-stent restenosis using interleukin-1 receptor-1 (IL-1R1)-deficient mice was described [39]. Taken together, to what extent mice can serve as accurate models of the human atherosclerotic disease is still the subject of discussion [40].
Genetically-modified murine models of human atherosclerosis (Table 1)
Table 1.
Genetic murine models available for the atherosclerosis research
| Model type | Advantages | Disadvantages | Utility | References |
|---|---|---|---|---|
| ApoE-/- mice | Spontaneously develop hyperlipidemia and extensive atherosclerosis of all stages morphologically identical to humans on the low-fat diet | The infrequency of crucial for humans plaque ruptures accompanied by mural thrombosis | Suitable to study atherogenesis evaluating the mechanisms underlying atherosclerosis progression | [139-141] |
| The low extent of coronary lesions | ||||
| LDLR-/- mice | Develop site-specific lesions in a time-dependent manner | The requirement for the long-term high-fat diet | Can be used to facilitate lesion analysis at specific locations, particularly, to study the initiation of the fatty streak, inhibition of lesion progression and regression of advanced plaques | [142] |
| The low extent of coronary atherosclerosis | ||||
| ApoE/LDLR double KO mice | Develop advanced atherosclerosis on the low-fat diet | Differences to humans in lipid profile | Suitable to study the anti-atherosclerotic effects of possible treatments, without the need of an atherogenic diet | [143-145] |
| ApoE*3-Leiden mice | Development of accelerated atherosclerosis and human-like lipoprotein profile | Lack of plaque rupture, thrombus formation, and/or haemorrhage | An accurate model for studying lipoprotein remnant metabolism and development of atherosclerotic plaques | [80,145] |
| Suitable for anti-atherosclerotic drug development | ||||
| Tg ApoB100+/+/LDLR KO mice | Develop accelerated atherosclerosis on a chow diet and human-like lipoprotein profile | Absence of spontaneous plaque ruptures | Suitable to study atherosclerosis and tests of effects of new therapies | [78,79] |
| LDLR/Apobec-1 double KO mice | Develop severe cholesterolemia and spontaneous atherosclerosis diet which progressively worsens with age on a normal chow diet | Absence of spontaneous plaque ruptures | Superior model to study the progression of atheroma | [83] |
| PCSK9-rAAV mice | Develop severe and persistent hypercholesterolemia and advanced atherosclerosis, including calcification of plaque | Absence of spontaneous plaque ruptures | Provide a flexible model of dyslipidemia and atherosclerosis | [93,146,147] |
| Induction of atherosclerosis in mice without germline genetic engineering, the rapid, easy and cost-effective approach | ||||
| ApoE-/-Fbn1C1039G+/- mice | Develop exacerbated atherosclerosis with plaque instability and spontaneous plaque ruptures | Required toxic Western-type diet | The perfect model for studying end-stage atherosclerosis investigating the role of major factors involved in plaque destabilization and rupture, as well as potential molecular targets for novel therapeutic interventions and the development of innovative molecular imaging strategies for vulnerable plaques | [104,106,107,110] |
Note: ApoE-/- - apolipoprotein E deficient mice; LDLR-/- mice - low-density lipoprotein receptor deficient mice; Tg ApoB100+/+/LDLR-/- KO mice - transgenic apolipoprotein B-100 positive/low-density lipoprotein receptor knock out mice; PCSK9-rAAV mice - proprotein convertase subtilisin/kexin type 9-recombinant adeno-associated virus mice; ApoE-/-Fbn1C1039G+/- apolipoprotein E-deficient Fibrillin-1 mutant mice.
Regardless of the limitations, mice remain the favoured animals for the atherosclerosis modelling, since the simplicity of the genetic manipulation using gene knockout, tissue-specific conditional gene expression, and gene knock-in techniques enables identification of genes contributing to the development of atherosclerosis and creating of transgenic models. Moreover, the relatively easy breeding of mice allowing the simultaneous manipulation of several genes in a single animal model represents an important advantage for the atherosclerosis research. The study showed that a combined interruption of genes influencing the development of the inflammatory component of the atherosclerotic process in apolipoprotein E deficient (ApoE-/-) hypercholesterolemic mice resulted in the inhibition of lesion development [41]. To date, most of the current genetic manipulations in mice applied for the atherosclerosis research rely on the disruption of normal lipoprotein regulation and metabolism generating non-HDL-based hypercholesterolemia. This can be the most readily achieved by the genetic knockout of ApoE or the LDL receptor (LDLR). Thus, ApoE-/- mice and low-density lipoprotein receptor-deficient mice (LDLR-/- mice) become the most commonly used genetic murine models of human atherosclerosis, which are going to be described in the next two subsections.
ApoE deficient mice
Apolipoprotein E (ApoE) is a lipoprotein that plays an important atheroprotective role in atherosclerosis development. Many of its anti-atherogenic effects that directly and indirectly involved in the lipoprotein metabolism were described [42]. ApoE is synthesized mainly in the liver, but it can be produced in some other tissues, such as brain, spleen, lung, kidney, arterial wall, and is present in high concentrations in the interstitial fluid [43,44]. Except for LDL, ApoE is a structural component of all lipoprotein particles, as well as it is a high-affinity ligand for both LDLR and LDLR-related proteins. Its interaction with these receptors provides the transport of cholesterol and other lipids between various body cells. In particular, ApoE is a critical ligand for the effective hepatic clearance of plasma lipoproteins (diet-derived chylomicrons and liver-derived VLDL remnants) mediated by LDLR and LDLR-related proteins [45,46]. It should be noted that ApoE functional roles within the LDL particles remain unclear.
ApoE gene deficiency in humans and animal models leads to the development of atherosclerosis, providing the evidence that the ApoE role in the mouse body is identical to that in humans. Various genetic mutations of functional LDLR pathway seen in patients with familial hypercholesterolemia (FH) lead to the impaired hepatic uptake and degradation of LDL, and, in turn, to a dramatic rise in plasma LDL, and an increased incidence of atherosclerosis and premature CVD [47]. Similarly, even on the standard chow diet, ApoE knockout (ApoE KO) mice develop a significant increase in plasma cholesterol level, with proatherogenic VLDL, as the most abundant circulating lipoprotein followed by the development of atherosclerosis [48].
ApoE KO mouse model was the first genetically modified murine model developed to study atherosclerosis [48]. The benefits of the use of this model are long-established. The early studies demonstrated that on the low-fat diet ApoE-/- mice rapidly develop atherosclerotic plaques, compared to wild-type mice, and, moreover, they can develop the morphologically identical lesions of all stages to human atherosclerosis [49,50]. Since the ApoE-/- mice develop atherosclerotic lesion even on the low-fat feed, the highly toxic diet can be avoided [51]. Moreover, in the ApoE-/- mice, the progression from early to advanced lesion occurs in the similar fashion to that in humans: atherosclerotic lesions frequently develop at vascular branch points and grow rapidly into foam cells with fibrous plaques and necrotic lipid cores [52]. The spontaneous plague development was identified in several vascular beds, predominantly in the aortic root, aortic arch and different branch points along the aorta in these mice [50]. The extensive atherosclerosis was seen in ApoE-/- mice at the age of 2-3 months [49]. Monocyte attachment to endothelial cells was detected from 6 weeks of age, foam cell lesions were developed after 8 weeks, and after 15 weeks, advanced lesions (fibrous plaques) were observed [50]. In 20 weeks, fibrous plaques containing SMCs, extracellular matrix, and an overlying fibrous cap were evident [50]. This time period can be accelerated in these mice by the feeding of the Western-type diet (0.15% cholesterol and 21% fat derived from milk fat), with the development of more advanced lesions. Accordingly, ApoE-/- mice fed with the Western-type diet developed extreme hypercholesterolemia accelerating the development and progression of atherosclerotic lesions comprising observable cholesterol crystals, necrotic core, and calcification [49]. The occurrence of the plaque rupture in these mice remains to be a subject of debate for a past decade [53-56]. Some authors indicated that sporadic plaque ruptures may be observed, but they occur after a long period of time, and consequently, clinical events such as MI or ischemic stroke are almost never seen in these models [28]. Though, a plaque rupture may be stimulated in this model by the placement of perivascular collar or cuff [57]. The rarity of crucial for humans plaque rupture is a disadvantage of this model.
The major limitation to the use of this model is linked to the transfer of bone marrow from a mouse expressing ApoE into an ApoE deficient recipient that significantly reduces plasma lipid levels and atherosclerosis [58]. Moreover, the extrapolation of data obtained in ApoE KO model to humans is difficult due to the differences in lipid metabolism, as most of the plasma cholesterol in these animals is in the form of VLDL but not LDL [59]. Besides, apart from the lipoprotein clearance, the presence of additional independent ApoE activities, such as anti-oxidative, anti-proliferative and anti-inflammatory that may lead to the development of atherosclerotic lesions irrespective of the plasma lipid level [44] can make an impact on the applicability of the results obtained in mice to humans. To date, considering the advantages and the limitations attributed to this model, it is widely used for studies of various pathways of atherosclerosis pathogenesis [60,61] and drug discovery [62,63].
LDLR deficient mice
LDLR-/- murine models were developed, in the attempt to overcome the limitations of ApoE-/- models, and they have revealed some advantages. The first advantage of LDLR-/- mouse model is based on the fact that LDLR does not have the variety of functions, therefore, the effects of its deficiency can be more straightforwardly attributed to the lipoprotein homeostasis. In this respect, the study showed that LDLR deficiency predominantly affecting lipoprotein uptake and clearance, resulted in a high prevalence of plasma LDL, as the main cholesterol-carrying lipoprotein in mice on the chow diet [64]. Noteworthy, the LDLR is a membrane receptor that mediates the endocytosis of cholesterol-rich LDL and maintains its plasma level. Besides, it facilitates the cellular uptake of ApoB- and E-containing lipoproteins. Second, LDLR-/- mice have the ability to model human-like plaques commonly observed in human FH, including the lesions in aortic valves and the aortic root [65]. That is advantageous to study atherosclerosis accompanying FH, an autosomal dominant disorder caused by mutations in the LDLR gene [66]. Third, LDLR deficient mice were found to be useful for studying the relationship between diabetes and atherosclerosis, which are often co-exist, as compared to ApoE-/- mice, they are more susceptible to obesity and insulin resistance [67]. In addition, together with ApoE KO model, LDLR-/- mouse model may be useful to study mechanisms of the atherosclerosis regression.
Nevertheless, in case of resistance to an injury-induced neointimal formation, ApoE KO mice were found to be more useful, in comparison with LDLR-/- mice, for studying mechanisms of restenosis following angioplasty [68,69]. Moreover, some reviewers indicated that, compared to ApoE deficient mice, when placed on a normal chow diet, LDLR-/- mice delay development of atherosclerosis, and higher cholesterol intake is required to accelerate the pathological process [70]. Variations in dietary cholesterol intake may lead to a problem of LDLR-/- mouse model standardization across different laboratories.
Interestingly, together with the transfer to a chow diet, reintroduction of ApoE and LDLR genes into ApoE-/- or LDLR-/- mice respectively, resulted in a sharp decrease in plasma cholesterol and a regression of established atherosclerosis [71]. The understanding of the atherosclerotic plaque regression is therapeutically relevant because the most cardiovascular disease patients will be treated after advanced plaques become established. Overall, the introduction of ApoE-and LDLR-deficient mouse models of atherosclerosis has transformed the understanding of the atherogenic process, and, hitherto, the mouse instantly became the most popular mammalian model of human atherosclerosis.
ApoE/LDLR double knock out mice
ApoE/LDLR double knock out (ApoE/LDLR-DKO) mice represent a model that develops both more severe hyperlipidemia and atherosclerosis than ApoE or LDLR single knockouts [72]. ApoE/LDLR-DKO mice demonstrated the more pronounced progression of atherosclerosis even on a regular chow diet [73]. They displayed high levels of VLDL and LDL and the marked elevations in both ApoB-48, ApoB-100 fractions [74]. In that regards, this mouse model can be useful to study atherosclerosis and, consequently, the anti-atherosclerotic drugs, without the need of an atherogenic diet.
However, this double KO model does not closely reflect the lipid profiles of the disease in humans. This is partially due to the presence of an Apobec-1, RNA-specific cytidine deaminase enzyme in the mouse liver. According to the current understanding, this enzyme is a catalytic component of endosomes switching the ApoB-100-encoding mRNA to an mRNA coding for ApoB-48. Incorporation of ApoB-48 into VLDL inducts its rapid clearance by scavenger receptors, prior to conversion of VLDL into LDL particles. The study showed that even moderate Apobec-1 expression leads to aberrant hyper-editing [75]. Such editing results in the resistance of plasma LDL elevation in mice bringing out the limitation to the use of this model. It is possible to overcome the Apobec-1 activity by applying further gene manipulation strategies, such as transgene expression of human ApoB-100 or disruption of mouse Apobec-1 gene, therefore, creating other useful genetic murine models focused on the elevation of ApoB-100 levels that will be described below [76,77]. The creation of the different gene knockout and transgenic mice that do not require dietary manipulations has enhanced the understanding of the mechanisms regulating plasma lipoprotein levels.
Transgenic ApoB100+/+/LDLR-/- knock out mice
Transgenic ApoB100+/+/LDLR-/- knock out (TgApoB+/+/LDLR-/- KO) mice are able to exhibit accelerated atherosclerosis on a chow diet, thus, they provide an excellent model for the atherosclerosis research. These mice showed atherogenic lipid profile of close resemblance to that in human atherosclerosis: dramatically elevated cholesterol and triglyceride plasma levels contained mainly in the IDL/LDL fraction, and significantly reduced HDL-C plasma levels [78]. It was reported that the lesion development does not require a high-fat, high-cholesterol diet (HFHC) intake in these mice, and they were able to develop complex and extensive atherosclerotic lesions involving approximately 15-20% of the aortic intimal surface (abdominal and terminal segments) [78]. This model was successfully used to study the anti-inflammatory effects of statins on the atherosclerotic plaque inflammation [79] and diet interventions on metabolic syndrome [80]. In addition, the hypercholesterolemia of TgApoB+/+/LDLR-/- KO mice is associated with a clear locomotor deficit and impairment of the episodic-like memory, so these mice can also serve as a model for a cognitive and a psycho-motor decline [81].
LDLR/Apobec-1 double knock out mice
LDLR/Apobec-1 double knock out (LDLR/Apobec-1 DKO) mice are deficient in the ability to convert ApoB-100 to ApoB-48 in the liver, and, also in LDL clearance. They exhibit high levels of ApoB-100-LDL-C, closely reproducing the plasma lipid profiles of human Type II FH, the most frequent type of FH observed in humans [82]. The model is characterized by the development of severe spontaneous atherosclerosis on a normal chow diet and its thorough analysis of the plaque development and progression was described [83]. Thus, the lesions that spontaneously develop in LDLR/Apobec-1 DKO mice begin as fatty streaks (stage I, American Heart Association classification) in the proximal aortic regions, as early as 12 weeks, and progressively worsen with age. These lesions spread to distal regions and, by 72 weeks of age, can occupy over 60% of the entire arterial tree. These models can replicate histologically different stages of atheroma beginning with fatty streaks, progressing to a clear human stage IV atheroma and then to a thin cap fibrous atheroma. Cap rupture is unusual for this model but there is a piece of evidence for the cap erosion [83]. Accordingly, LDLR/Apobec-1 DKO is a potentially available important model to study the evolution of atheroma.
Apolipoprotein ApoE*3-Leiden mice
Apolipoprotein ApoE*3-Leiden mutation is associated with one of the genetic forms of hyperlipidemia [84]. Transgenic mice were generated using a genomic DNA construct containing the mutant ApoE and ApoC1 genes with all regulatory elements isolated from the APOE*3-Leiden proband [85]. The primary effect of the dominant ApoE*3-Leiden mutation is an impaired clearance of triglyceride-rich lipoproteins (chylomicron- and VLDL-remnants) caused by a reduced affinity of the apolipoprotein ApoE*3Leiden for the LDLR3. The ApoE*3Leiden transgenic mice can develop diet-dependent hyperlipidemia and are highly susceptible to diet-induced atherosclerosis. In these mice, when fed a mildly HFHC diet, early fatty-streak formation was observed in the aortic arch after 3 months, and late complex atherosclerotic lesions consisting of plaques with a necrotic core and a fibrous cap (stages IV and V) was registered after 3 to 6 months of feeding a severe HFHC diet [86]. Also, they showed dramatically elevated total plasma cholesterol and triglyceride levels attributed to an increase in VLDL/LDL particles [87].
One of the advantages to the use of this model is the presence of the functional ApoE, so the effects of hyperlipidemia can be studied independently of other ApoE activities. Another advantage is that the introduction of human CETP gene into ApoE*3-Leiden mice results in more human-like lipoprotein metabolism in ApoE*3Leiden. CETP mice [88]. These mice showed a human-like response to the clinically used lipid-modulating pharmacological interventions, such as statins, fibrates, ezetimibe, and niacin [89-91]. Based on numerous features in common with human atherosclerotic lesions and the similar diet-induced lipid metabolism, transgenic ApoE*3-Leiden mice were considered as one of the most accurate animal models of human atherosclerosis [92] posing a great value to study lipoprotein remnant metabolism and accelerated atherosclerosis. However, the disadvantage of this model is that ApoE3-Leiden mice lack plaque rupture, thrombus formation, and/or haemorrhage, the atherosclerotic events of profound importance for humans.
Proprotein convertase subtilisin/kexin type 9-recombinant adeno-associated virus mice
The construction of proprotein convertase subtilisin/kexin type 9-recombinant adeno-associated virus (PCSK9-rAAV) mouse model is a rapid, easy and cost-effective approach to study atherosclerosis [93]. Noteworthy, PCSK9, is a novel subtilase serine protease highly expressed in liver and intestine. It binds to hepatic LDLR on the cell surface promoting LDL lysosomal degradation that increases LDL plasma levels in humans and mice [94,95]. The intracellular binding of PCSK9 was also described [96]. It was reported that PCSK9 gene regulates cholesterol homeostasis exclusively through the LDLR [97]. FH with severe hypercholesterolemia and early CVD can be associated with “gain-of-function” mutations in the PCSK9 gene [98]. In mice, AAV transduction with human or murine “gain-of-function” PCSK9 mutant gene resulted in doubling of serum cholesterol on the normal diet, compared to controls, and this effect was stable and preserved after 12 months post-transduction [28]. The Western-type diet exacerbated hyperlipidemia in these mice [28]. The lipoprotein profile of PCSK9DY-AAV mice fed with the Western-type diet revealed an equal distribution between VLDL and LDL particles [99].
Moreover, the atherosclerosis progression in PCSK9DY transgenic mice was observed in a dose-dependent manner of feeding [28]. Lesions resembling those of LDLR-/- mice occurred throughout the vasculature progressing to the fibro-atheromatous stage [100], and vascular calcification occurred within the time frame of 15-20 weeks [101]. In that regards, this model makes possible to conduct studies on vascular calcification of atherosclerotic plaques and therapeutic interventions by avoiding laborious and costly mouse colony generation allowing to model human atherosclerosis in animals with different genetic backgrounds. In addition, similarly to other described above murine models, the main limitation of this model is the absence of spontaneous plaque ruptures, the leading cause of acute events in humans.
Apolipoprotein E-deficient fibrillin-1 mutant (ApoE-/-Fbn1C1039G+/-) mice
Fibrillin-1 (Fbn1) is a component of microfibrils associated with elastic-fibres that provide structural support to a vessel wall [102]. Notably, mutations in the Fbn1 gene are the cause of the Marfan syndrome, a genetic disorder characterized by fragmentation of elastic fibres [103]. On a Western-type diet, ApoE deficient Fibrillin-1 mutant (ApoE-/-Fbn1C1039G+/-) mice develop fragmentation of the elastic fibers and large atherosclerotic plaques with prominent features of plaque instability, including an enlarged necrotic core occupying about 30% of total plaque area, strongly diminished collagen content (a thin fibrous cap with an important loss of collagen fibres), high level of inflammatory cytokines and matrix metalloproteinases, T-cell infiltration, SMC apoptosis, and numerous concealed caps, not only at the level of aortic valves but also in the brachiocephalic artery, and in different areas of the aorta [104-106]. Also, the elastin loss would result in fibrous cap exposure to increased biomechanical stress [106]. Moreover, similar to human pathology, elastin fragmentation in combination with a Western-type diet in these mice are the essentials for the development of atherosclerotic plaque neovascularization, intra-plaque haemorrhage, and plaque rupture resulting in MI, stroke and sudden death [104,107]. These features are uncommon for murine atherosclerosis models but are known to favourably affect plaque progression and vulnerability in humans [108]. Spontaneous plaque ruptures were observed in this model [107,109]. Several reviewers indicated that sudden death in ApoE-/-Fbn1C1039G+/- mice fed with a Western-type diet can be observed in the time period between 16 and 23 weeks, with 50% mortality rate after 20 weeks [28]. Mice that died suddenly exhibited a significantly higher frequency of coronary stenosis, compared to survivors [107], suggesting that the plaque development in coronary artery plays an important role in cardiac death.
Taken together, ApoE-/-Fbn1C1039G+/- mice can model human end-stage atherosclerosis, therefore, offer an opportunity to investigate the role of key factors involved in plaque destabilization and potential molecular targets for therapeutic interventions. Importantly, among all murine models, these mice are the best suited for spontaneous plaque rupture studies. They can be also used for the development of innovative molecular imaging strategies for vulnerable plaques [110].
Rabbit models of human atherosclerosis
Rabbits were the first animals to model atherosclerosis but, since the year 2000, there is a downward trend of using rabbit models, and that is probably due to the wide availability of murine models. However, rabbits have the same advantages as mice for modelling of human atherosclerosis, including ease of maintenance and availability, low economical cost, and are more appropriate for catheter-based procedures and non-invasive imaging. Moreover, rabbit models are considered to be the best models to study hyperlipidemia and, in turn, atherosclerosis because they possess several unique features of lipoprotein metabolism that are identical to humans and, therefore, advantageous for the atherosclerosis research. The features of lipid metabolism attributed to rabbits are the following: (i) alike humans, rabbits abundantly express plasma CETP, an important regulator of the cholesterol metabolism [111]; (ii) rabbit ApoB-containing lipoproteins are similar to those of humans in their chemical composition and apoprotein content (VLDL and LDL) [111]; (iii) ApoB mRNA editing does not occur in a rabbit’s liver, hence, ApoB-100-containing VLDL is produced and that is similar to humans [111]; and (iv) finally, rabbits are very susceptible to diet-induced atherosclerosis [112]. In that regards, two strains, namely Watanabe Heritable Hyperlipidaemic (WHHL) and St. Thomas’ Hospital (STH) rabbits were reported as relevant models for human hyperlipidemia and atherosclerosis (Figure 1) [113,114]. Noteworthy, naturally defective in LDLR, WHHL rabbits exhibit lipid metabolism pattern identical to that seen in human FH [115]. The recent advances in gene technology enabled the generation of a variety of transgenic rabbits, therefore, providing a unique system to study the properties important for human atherosclerosis. One example, ApoB-100 transgenic rabbits were created, which are capable to manifest combined hyperlipidemia with reduced HDL-cholesterol concentrations allowing to study of ApoB metabolism, therefore, dietary manipulation and drug intervention studies in these animals should be more relevant to humans [116]. Other transgenic rabbit models of human atherosclerosis highlighting the effects of individual genes on lipoprotein metabolism and atherosclerosis susceptibility were also described [117].
Figure 1.
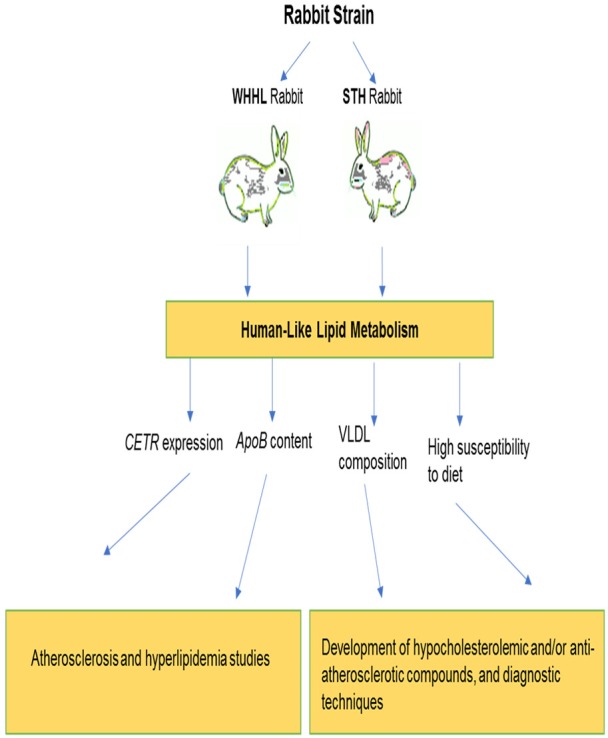
The utility of genetic rabbit models of human atherosclerosis. Note: WHHL - Watanabe Heritable Hyperlipidaemic; STH - St. Thomas’ Hospital; CETR - cholesteryl ester transfer protein; ApoB - apolipoprotein B; VLDL - very low-density lipoprotein.
Porcine models of human atherosclerosis (Table 2)
Table 2.
Genetic porcine models available for the atherosclerosis research
| Model type | Advantages | Disadvantages | Utility | References |
|---|---|---|---|---|
| Tg D374Y-PCSK9 minipigs | Develop moderate hypercholesterolemia and human-like atherosclerosis on the standard low-fat diet | The high-fat diet required to develop severe hypercholesterolemia and advanced atherosclerosis | Useful for testing different imaging techniques from 12 months of age. | [12,122,130-132] |
| Acquire reduced hepatic LDLR levels | Multiple expression of the mutant transgene may limit the utility for treatments designed to increase LDLR expression or reduce PCSK9 activity | |||
| Impaired LDL clearance | ||||
| LDLR-/- minipigs | Develop advanced human-like atherosclerosis, including coronary lesions | The high-fat diet required | Suitable for coronary atherosclerosis research, development of plaque stabilization drugs and percutaneous diagnostic and interventional devices | [130,133,148] |
| ApoE-/- minipigs | Develop moderate cholesterolemia and atherosclerosis on the low-fat diet | The high-fat diet required to develop prominent human-like dyslipidemia and progressive atherosclerosis | Suitable for translational studies of atherosclerosis | [136,137] |
| ApoE /LDLR double KO minipigs | Develop atherosclerosis-related lipid metabolism | Validation of atherosclerosis phenotype is limited possibly due to the fact that in-frame mutations in both the ApoE and LDLR alleles may give rise to truncated ApoE and LDLR proteins with the partially retained function | Useful for studies determining the function of genes involved in the progression of human atherosclerosis | [138] |
Note: Tg D374Y-PCSK9 minipigs - Transgenic D374Y-PCSK9 minipigs; LDLR - low-density lipoprotein receptor; LDL - low-density lipoprotein; LDLR-/- - low-density lipoprotein receptor deficient; ApoE-/- - apolipoprotein E deficient; ApoE/LDLR double KO minipigs - apolipoprotein E and low-density lipoprotein receptor double knockout minipigs.
Pigs, particularly minipigs, offer large-animal models that have the human-like size and cardiovascular anatomy of stronger genetic resemblance to humans. Having a large animal model capable of developing human-like atherosclerosis is crucial for translational research.
In terms of atherosclerosis developmental process, pigs have a number of important differences to humans, such as lack of CETP, the requirement for high dietary cholesterol intake typically combined with cholic acid, and lesion progression is restricted to only foam cell formation stage within a reasonable time frame [12]. These differences caused some limitations to the use of porcine models but genetic engineering of minipigs helped to overcome the disadvantages. Cloning by the somatic cell nuclear transfer become the method of choice for developing of both transgenic and gene-edited animals. Most of the published genetically modified pig models were developed by gene editing in porcine somatic cells followed by the animal cloning [118-121].
It was found that atherosclerotic lesions developing in pigs exhibit overall morphology and several specific pathological features that are common for human lesions but not seen in mice. Thus, lesions grow in the abdominal aorta, iliofemoral arteries, and proximal segments of the coronary arteries; initial foam cell lesions evolve in an intima that also contains connective tissue matrix and SMCs [12]. Plaque neovascularization, as well as calcification, are also present [122]. Importantly, as in murine models, plaque ruptures are rare events in pigs and that limits the suitability of porcine models to study thrombotic complications [12]. Moreover, the development of spontaneous atherosclerosis in pigs is rare, but pigs on HFHC diet can develop advanced atherosclerotic lesions similar in type and location to those that occur in humans [123]. However, HFHC diets are very expensive and require a long time to manifest the disease, thus, making many studies unaffordable.
Because of the closer phylogenetic relationship [124], anatomy, physiology, body weight, lifespan, and atherosclerosis pathology to humans [125], the efficacy of drug testing in porcine models may be more predictive than using murine models. Though, the absence of CETP makes them unsuitable to study CETP inhibitors or other lipid-altering interventions that may require CETP for modelling the full effects of lipids on atherosclerosis [126]. The predictive value of porcine models for drug discovery studies is currently unknown due to the scarcity of the data. The close match of the porcine cardiovascular system to humans, including pig heart size, blood supply, coronary system function, the anatomy of the aorta [127], and the relevant morphology of porcine coronary lesions allow validation of intravascular imaging tools in vivo. Pigs offer an opportunity to perform examinations of atherosclerotic lesions in conjunction with the evaluation of clinical imaging end-points, hence, to decide which of the end-points is most sensitive for the specific pathology [128,129]. In this respect, they are considered promising models bridging preclinical and human clinical studies. Thus, the pig is an important animal for the research of atherosclerosis and CVDs. Overall, limitations to porcine atherosclerotic models include the necessity for well-developed infrastructure to support animal maintenance, a long-time period to create and monitor lesions and substantial financial investment.
D374Y-PCSK9 transgenic minipigs
Yucatan or D374Y-PCSK9 transgenic minipig model was created by the liver-specific expression of the “gain-of-function” mutant D374Y in the PCSK9 gene that can cause a severe form of hypercholesterolemia and, ultimately, atherosclerosis [12]. They exhibited moderate hypercholesterolemia on standard feed and more severe hypercholesterolemia and human-like atherosclerosis were observed on HFHC diet [12]. Moreover, these animals have severely reduced hepatic LDLR levels, thus, augmented concentrations of LDL-C in combination with HFHC diet are sufficient conditions to induce atherosclerosis. The lesions were detected in the thoracic and abdominal aorta, the iliofemoral arteries, and the coronary arteries at 12 months of age [12]. At this age, Yucatan minipigs reach near-human weight making this model useful for testing different imaging techniques without modifications, such as clinical scanners and intravascular devices, in order to advance them into clinics.
Furthermore, the study reported that atherosclerotic lesions in these transgene minipigs recapitulated several histologic features of human atherosclerosis, such as the development of complex lesions with SMCs, extracellular matrix, and inflammatory infiltrate, in addition to macrophage-derived foam cells [122]. Atheromas demonstrated characteristics of advanced human plaques, including necrotic cores, fibrous tissue, calcification, plaque angiogenesis, and intraplaque haemorrhage [122]. However, with the mutant transgene being permanently overexpressed at nearly five hundred times over the normal level, these pigs may have limited utility for treatments designed to increase LDLR expression or reduce PCSK9 activity [130]. Potential benefits of this model include their human-like size, their well-described background, and the ability to induce progressive atherosclerotic lesion without the use of cholic acid, which has several unwanted side-effects. In addition, a few studies reported the suitability of this model for studies of the advanced coronary plaque formation mechanisms [131] and in vivo validation of coronary optical coherence tomography for plaque classification [132].
LDLR deficient minipigs
LDLR KO minipig model was generated using AAV-mediated delivery of gene targeting vector and somatic cell nuclear transfer in Yucatan minipigs with subsequent breeding of heterozygous [130]. Accordingly, in comparison to PCSK9-D374Y transgenic pigs, a more severe and more rapidly developing atherosclerosis occurs in this model, thus, decreasing the duration and costs of studies. Feeding HFHC diet, they develop advanced atherosclerotic lesions in the coronary arteries and abdominal aorta that closely resemble those in humans [130]. Thus, LDLR KO minipigs can be used for the development of human-like advanced coronary atherosclerosis, and, in turn, for translational research, particularly, for the development of plaque stabilization drugs and percutaneous diagnostic and interventional devices [133]. However, prolonged trials with the expensive HFHC diet makes this model cost-prohibitive for the broad application.
This model was reproduced using pig of different genetic background, such as domestic pigs that allowed to decrease the time of lesion development to four months on HFHC diet, and, moreover, to get more complicated human-like advanced plaques containing the necrotic core, hemorrhage, and calcification [133]. Besides, even more accelerated coronary plaque development can be achieved in these pigs with the use of coronary artery balloon injury. For instance, coronary plaques were detected eight weeks after the coronary artery injury [134]. Coronary angioplasty is an established procedure in domestic swine with normal cholesterol level but accelerated plaque development poorly resembles the natural course of human atherosclerosis, i.e. the lesions have abundant fibrotic tissue with little necrotic core formation [135]. This model can be useful for studies of specific atherosclerosis-related processes and imaging studies detecting a specifically localized plaque.
ApoE deficient minipigs
In order to establish an improved large animal model of FH and atherosclerosis, ApoE-/- minipigs were produced using the clustered regularly interspaced short palindromic repeat-associated protein 9 system (CRISPR/Cas9) to disrupt the ApoE gene in Bama miniature pigs [136]. The recent study demonstrated that similarly to PCSK9 transgenic pigs and LDLR KO pigs, ApoE-/- pigs showed moderately increased plasma cholesterol levels on a low-fat diet resembling the human FH phenotype [136]. Moreover, when fed HFHC diet, these pigs showed severe hypercholesterolemia and developed progressive atherosclerotic lesions [136]. Likewise, another study showed that targeted gene KO of ApoE in Yucatan minipigs on the low-fat diet can cause remnant lipoproteinemia closely resembling the human familial dysbetalipoproteinemia, which on HFHC feed was significantly accentuated, and also, these minipigs displayed accelerated progressive atherosclerosis [137]. In that regards, ApoE-/- minipigs could be beneficial for the elucidation of ApoE gene functions and translational studies of atherosclerosis because their serum lipid profile and atherosclerotic lesions can sufficiently recapitulate those in human disease.
ApoE and LDLR double knockout minipigs
ApoE/LDLR double knockout pigs were generated via CRISPR/Cas9 gene-editing approach targeting ApoE gene and LDLR simultaneously in Bama minipig embryonic fibroblasts, then they were used as nuclear donors to produce the double KO animals [138]. The gene-modified swine showed an abnormal lipid metabolism related to atherosclerosis from an early age. As compared to wild-type pig, at 2 months of age serum LDL-C was elevated by approximately 41%; there was also 57% elevation of serum total cholesterol and 120% elevation of serum triglycerides [138]. Atherosclerosis phenotype was not described in these pigs, possibly due to the fact that in-frame mutations in both the ApoE and LDLR alleles may give rise to truncated ApoE and LDLR proteins with the partially retained function [138]. Since ApoE and LDLR gene mutations play an important role in the atherosclerotic disease progression, this model can aid in determining the function of genes, thus, establish a valuable tool to study human atherosclerosis.
Conclusion and comments for future
Modelling atherosclerosis in genetically modified animals is currently mainstream in atherosclerotic research. The genetic animal models available to date can simulate all stages of plaque development providing an opportunity to study the pathogenesis of lesion formation, mechanisms of plaque vulnerability and regression and, therefore, evaluation of new therapeutics and imaging techniques. Each model differs in its potentials and possibilities of application. A good understanding of the similarities and differences between the animal model and human disease is important for the effective extrapolation of data for the translational application.
All described models of atherosclerosis rely on the hypercholesterinemia, as a major triggering factor, and this is one of the critical limitations because the real-life disease is usually multifactorial. In future studies, the inclusion of other risk factors of atherosclerosis may lead to better models of this complex disease with higher translational validity.
In addition, targeting LDL-lowering therapy to individuals at risk remains challenging, due to the lack of sensitive and specific diagnostic methods that can detect silent atherosclerosis prior to life-threatening clinical events. Further studies are required to solve this problem.
Acknowledgements
We acknowledge that this work was supported by the Russian Science Foundation (Grant # 17-75-20249).
Disclosure of conflict of interest
None.
References
- 1.Orekhov AN, Ivanova EA. Introduction of the special issue “Atherosclerosis and Related Diseases”. Vessel Plus. 2017;1:163–5. [Google Scholar]
- 2.Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. 2002;15:439. doi: 10.1111/j.1540-8183.2002.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 3.Mohiaddin RH, Burman ED, Prasad SK, Varghese A, Tan RS, Collins SA, Hughes RL, Gatehouse PD, Jhooti P, Longmore DB, Yang GZ, Firmin DN, Pennell DJ. Glagov remodelling of the atherosclerotic aorta demonstrated by cardiovascular magnetic resonance: the CORDA asymptomatic subject plaque assessment research (CASPAR) project. J Cardiovasc Magn Reson. 2004;6:517–25. doi: 10.1081/jcmr-120030576. [DOI] [PubMed] [Google Scholar]
- 4.McMahan CA, Gidding SS, Fayad ZA, Zieske AW, Malcom GT, Tracy RE, Strong JP, McGill HC. Risk scores predict atherosclerotic lesions in young people. Arch Intern Med. 2005;165:883–90. doi: 10.1001/archinte.165.8.883. [DOI] [PubMed] [Google Scholar]
- 5.Tzou WS, Douglas PS, Srinivasan SR, Bond MG, Tang R, Chen W, Berenson GS, Stein JH. Increased subclinical atherosclerosis in young adults with metabolic syndrome: the Bogalusa heart study. J Am Coll Cardiol. 2005;46:457–63. doi: 10.1016/j.jacc.2005.04.046. [DOI] [PubMed] [Google Scholar]
- 6.Insull W Jr. The pathology of atherosclerosis: plaque development and plaque responses to medical treatment. Am J Med. 2009;122:S3–14. doi: 10.1016/j.amjmed.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Spring B, Moller AC, Colangelo LA, Siddique J, Roehrig M, Daviglus ML, Polak JF, Reis JP, Sidney S, Liu K. Healthy lifestyle change and subclinical atherosclerosis in young adults: coronary artery risk development in young adults (CARDIA) study. Circ. 2014;130:10–7. doi: 10.1161/CIRCULATIONAHA.113.005445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eikendal AL, Groenewegen KA, Bots ML, Peters SA, Uiterwaal CS, den Ruijter HM. Relation between adolescent cardiovascular risk factors and carotid intima-media echogenicity in healthy young adults: the atherosclerosis risk in young adults (ARYA) study. J Am Heart Assoc. 2016;5:e002941. doi: 10.1161/JAHA.115.002941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuzcu EM, Kapadia SR, Tutar E, Ziada KM, Hobbs RE, McCarthy PM, Young JB, Nissen SE. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circ. 2001;103:2705–10. doi: 10.1161/01.cir.103.22.2705. [DOI] [PubMed] [Google Scholar]
- 10.Nissen SE, Tuzcu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J, Orazem J, Magorien RD, O’Shaughnessy C, Ganz P. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med. 2005;352:29–38. doi: 10.1056/NEJMoa042000. [DOI] [PubMed] [Google Scholar]
- 11.Wadhera RK, Steen DL, Khan I, Giugliano RP, Foody JM. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J Clin Lipidol. 2016;10:472–89. doi: 10.1016/j.jacl.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 12.Shim J, Al-Mashhadi RH, Sørensen CB, Bentzon JF. Large animal models of atherosclerosis-new tools for persistent problems in cardiovascular medicine. J Pathol. 2016;238:257–66. doi: 10.1002/path.4646. [DOI] [PubMed] [Google Scholar]
- 13.Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013;24:129–36. doi: 10.1016/j.tem.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–90. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circ. 2007;116:1832–44. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 16.Sukhorukov VN, Karagodin VP, Orekhov AN. Atherogenic modification of low-density lipoproteins. Biomed Khim. 2016;62:391–402. doi: 10.18097/PBMC20166204391. [DOI] [PubMed] [Google Scholar]
- 17.Orekhov AN, Tertov VV, Kudryashov SA, Smirnov VN. Trigger-like stimulation of cholesterol accumulation and DNA and extracellular matrix synthesis induced by atherogenic serum or low-density lipoprotein in cultured cells. Circ Res. 1990;66:311–20. doi: 10.1161/01.res.66.2.311. [DOI] [PubMed] [Google Scholar]
- 18.Alipov VI, Sukhorukov VN, Karagodin VP, Grechko AV, Orekhov AN. Chemical composition of circulating native and desialylated low-density lipoprotein: what is the difference? Vessel Plus. 2017;1:107–15. [Google Scholar]
- 19.Hartvigsen K, Chou MY, Hansen LF, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. The role of innate immunity in atherogenesis. J Lipid Res. 2009;50(Suppl):S388–93. doi: 10.1194/jlr.R800100-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orekhov AN, Ivanova EA. Cellular models of atherosclerosis and their implication for testing natural substances with anti-atherosclerotic potential. Phytomedicine. 2016;23:1190–7. doi: 10.1016/j.phymed.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, Mehran R, McPherson J, Farhat N, Marso SP, Parise H, Templin B, White R, Zhang Z, Serruys P. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–35. doi: 10.1056/NEJMoa1002358. [DOI] [PubMed] [Google Scholar]
- 22.Gu W, Xie Y, Xu Q. The ESC Textbook of Vascular Biology. Oxford, UK: Oxford University Press; 2017. Animal models to study pathophysiology of the vasculature. [Google Scholar]
- 23.Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, Blanco-Colio L, Lavin B, Mallavia B, Egido J. Animal models of cardiovascular diseases. Biomed Res Int. 2011;2011:497841. doi: 10.1155/2011/497841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caro CG. Discovery of the role of wall shear in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:158–161. doi: 10.1161/ATVBAHA.108.166736. [DOI] [PubMed] [Google Scholar]
- 26.Chatzizisis YS, Coskun AU, Jonas M, Edelman EA, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodelling. J Am Coll Cardiol. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 27.Huo Y, Guo X, Kassab GS. The flow field along the entire length of mouse aorta and primary branches. Ann Biomed Eng. 2008;36:685–699. doi: 10.1007/s10439-008-9473-4. [DOI] [PubMed] [Google Scholar]
- 28.Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, De Meyer GRY. Animal models of atherosclerosis. Eur J Pharmacol. 2017;816:3–13. doi: 10.1016/j.ejphar.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Miyajima C. Characterization of atherosclerosis formation in a murine model of type IIa human familial hypercholesterolemia. Biomed Res Int. 2018;2018:1878964. doi: 10.1155/2018/1878964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiner Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat Rev Cardiol. 2017;14:401. doi: 10.1038/nrcardio.2017.31. [DOI] [PubMed] [Google Scholar]
- 31.Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–6. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson MH. HDL and CETP inhibition: will this DEFINE the future? Curr Treat Options Cardiovasc Med. 2012;14:384–390. doi: 10.1007/s11936-012-0191-8. [DOI] [PubMed] [Google Scholar]
- 33.Mestas J, Hughes CC. Of mice and not men: Differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 34.Chen YC, Bui AV, Diesch J, Manasseh R, Hausding C, Rivera J, Hagemeyer CE, Hannan RD, Bobik A, Peter K. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res. 2013;113:252–265. doi: 10.1161/CIRCRESAHA.113.301562. [DOI] [PubMed] [Google Scholar]
- 35.Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;21:434–440. doi: 10.1097/MOL.0b013e32833ded6a. [DOI] [PubMed] [Google Scholar]
- 36.Parasuraman S, Raveendran R, Kesavan R. Blood sample collection in small laboratory animals. J Pharmacol Pharmacother. 2010;1:87–93. doi: 10.4103/0976-500X.72350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee YT, Lin HY, Chan YW, Li KH, To OT, Yan BP, Liu T, Li G, Wong WT, Keung W, Tse G. Mouse models of atherosclerosis: a historical perspective and recent advances. Lipids Health Dis. 2017;16:12. doi: 10.1186/s12944-016-0402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu F, Bracard S, Anxionnat R, Derelle AL, Tonnelet R, Liao L, Mione G, Humbertjean L, Lacour JC, Hossu G, Anadani M, Richard S, Gory B. Impact of emergent cervical carotid stenting in tandem occlusion strokes treated by thrombectomy: a review of the TITAN collaboration. Front Neurol. 2019;10:206. doi: 10.3389/fneur.2019.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chamberlain J, Wheatcroft M, Arnold N, Lupton H, Crossman DC, Gunn J, Francis S. A novel mouse model of in situ stenting. Cardiovasc Res. 2009;85:38–44. doi: 10.1093/cvr/cvp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;5:434–440. doi: 10.1097/MOL.0b013e32833ded6a. [DOI] [PubMed] [Google Scholar]
- 41.Cambadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedqui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Lys6C(hi) and Ly6C(lo) monocytosis and almost abolished atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 42.Greenow K, Pearce NJ, Ramji DP. The key role of apolipoprotein E in atherosclerosis. J Mol Med (Berl) 2005;83:329–342. doi: 10.1007/s00109-004-0631-3. [DOI] [PubMed] [Google Scholar]
- 43.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 44.Getz GS, Reardon CA. Apoprotein E as a lipid transport and signalling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50(Suppl):S156–S161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 46.Raffai RL, Hasty AH, Wang Y, Mettler SE, Sanan DA, Linton MF, Weisgraber KH. Hepatocyte-derived ApoE is more effective than non-hepatocyte-derived ApoE in remnant lipoprotein clearance. J Biol Chem. 2003;278:11670–11675. doi: 10.1074/jbc.M212873200. [DOI] [PubMed] [Google Scholar]
- 47.Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Rev Cardiol. 2007;4:214. doi: 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- 48.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–53. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 49.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–7. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 50.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–40. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 51.Plump AS, Breslow JL. Apolipoprotein E and the apolipoprotein E-deficient mouse. Annu Rev Nutr. 1995;15:495–518. doi: 10.1146/annurev.nu.15.070195.002431. [DOI] [PubMed] [Google Scholar]
- 52.Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24:1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–713. doi: 10.1161/01.ATV.0000261709.34878.20. [DOI] [PubMed] [Google Scholar]
- 54.Jackson CL. Defining and defending murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:973–977. doi: 10.1161/01.ATV.0000261545.53586.f0. [DOI] [PubMed] [Google Scholar]
- 55.Jawien J. The role of an experimental model of atherosclerosis: apoE-knockout mice in developing new drugs against atherogenesis. Curr Pharm Biotechnol. 2012;13:2435–2439. [PubMed] [Google Scholar]
- 56.Falk E, Schwartz SM, Galis ZS, Rosenfeld ME. Putative murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:969–972. doi: 10.1161/01.ATV.0000261572.33474.e0. [DOI] [PubMed] [Google Scholar]
- 57.Sasaki T, Kuzuya M, Nakamura K, Cheng XW, Shibata T, Sato K, Iguchi A. A simple method of plaque rupture induction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1304–9. doi: 10.1161/01.ATV.0000219687.71607.f7. [DOI] [PubMed] [Google Scholar]
- 58.Linton MF, Fazio S. Macrophages, lipoprotein metabolism, and atherosclerosis: insights from murine bone marrow transplantation studies. Curr Opin Lipidol. 1999;10:97–105. doi: 10.1097/00041433-199904000-00003. [DOI] [PubMed] [Google Scholar]
- 59.Pendse AA, Arbones-Mainar JM, Johnson LA, Altenburg MK, Maeda N. Apolipoprotein E knock-out and knock-in mice: atherosclerosis, metabolic syndrome, and beyond. J Lipid Res. 2009;50(Suppl):S178–82. doi: 10.1194/jlr.R800070-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marzolla V. Induction of atherosclerotic plaques through activation of mineralocorticoid receptors in apolipoprotein E-deficient mice. J Vis Exp. 2018 doi: 10.3791/58303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li HY, Leu YL, Wu YC, Wang SH. Melatonin inhibits in vitro smooth muscle cell inflammation and proliferation and atherosclerosis in apolipoprotein E-deficient mice. J Agric Food Chem. 2019;67:1889–1901. doi: 10.1021/acs.jafc.8b06217. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H, Zhou W, Cao C, Zhang W, Liu G, Zhang J. Amelioration of atherosclerosis in apolipoprotein E-deficient mice by combined RNA interference of lipoprotein-associated phospholipase A2 and YKL-40. PLoS One. 2018;13:e0202797. doi: 10.1371/journal.pone.0202797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rakipovski G. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE-/- and LDLr-/- mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci. 2018;3:844–857. doi: 10.1016/j.jacbts.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teupser D, Persky AD, Breslow JL. Induction of atherosclerosis by low-fat, semisynthetic diets in Ldl receptor-deficient C57bl/6j and Fvb/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement) Arterioscler Thromb Vasc Biol. 2003;23:1907–13. doi: 10.1161/01.ATV.0000090126.34881.B1. [DOI] [PubMed] [Google Scholar]
- 65.VanderLaan PA, Reardon CA, Thisted RA, Getz GS. VLDL best predicts aortic root atherosclerosis in LDL receptor-deficient mice. J Lipid Res. 2009;50:376–85. doi: 10.1194/jlr.M800284-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merat S, Casanada F, Sutphin M, Palinski W, Reaven PD. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose-rich diet. Arterioscler Thromb Vasc Biol. 1999;19:1223–1230. doi: 10.1161/01.atv.19.5.1223. [DOI] [PubMed] [Google Scholar]
- 68.Ali ZA, Alp NJ, Lupton H, Arnold N, Bannister T, Hu Y, Mussa S, Wheatcroft M, Greaves DR, Gunn J, Channon KM. Increased in-stent stenosis in ApoE knockout mice: insights from a novel mouse model of balloon angioplasty and stenting. Arterioscler Thromb Vasc Biol. 2007;27:833–40. doi: 10.1161/01.ATV.0000257135.39571.5b. [DOI] [PubMed] [Google Scholar]
- 69.Jin R, Xiao AY, Song Z, Yu S, Li J, Cui MZ, Li G. platelet cd40 mediates leukocyte recruitment and neointima formation after arterial denudation injury in atherosclerosis-prone mice. Am J Pathol. 2018;188:252–263. doi: 10.1016/j.ajpath.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan J, Karere GM, Cox LA, VandeBerg JL. Animal models of diet-induced hypercholesterolemia. In Hyper-cholesterolemia. 2015 IntechOpen. [Google Scholar]
- 71.Burke AC. Regression of atherosclerosis: lessons learned from genetically modified mouse models. Curr Opin Lipidol. 2018;29:87–94. doi: 10.1097/MOL.0000000000000493. [DOI] [PubMed] [Google Scholar]
- 72.Bonthu S, Heistad DD, Chappell DA, Lamping KG, Faraci FM. Atherosclerosis, vascular remodelling, and impairment of endothelium-dependent relaxation in genetically altered hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 1997;17:2333–2340. doi: 10.1161/01.atv.17.11.2333. [DOI] [PubMed] [Google Scholar]
- 73.Witting PK, Pettersson K, Ostlund-Lindqvist AM, Westerlund C, Eriksson AW, Stocker R. Inhibition by a co-antioxidant of aortic lipoprotein lipid peroxidation and atherosclerosis in apolipoprotein E and low-density lipoprotein receptor gene double knockout mice. FASEB J. 1999;13:667–675. doi: 10.1096/fasebj.13.6.667. [DOI] [PubMed] [Google Scholar]
- 74.Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low-density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci U S A. 1994;91:4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hersberger M, Patarroyo-White S, Qian X, Arnold KS, Rohrer L, Balestra ME, Innerarity TL. Regulatable liver expression of the rabbit apolipoprotein B mRNA-editing enzyme catalytic polypeptide 1 (APOBEC-1) in mice lacking endogenous APOBEC-1 leads to aberrant hyperediting. Biochem J. 2003;369:255–262. doi: 10.1042/BJ20020694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fazio S, Linton MF. Mouse models of hyperlipidemia and atherosclerosis. Front Biosci. 2001;6:D515–25. doi: 10.2741/fazio. [DOI] [PubMed] [Google Scholar]
- 77.Hirano K, Young SG, Farese RV Jr, Ng J, Sande E, Warburton C, Powell-Braxton LM, Davidson NO. Targeted disruption of the mouse apobec-1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J Biol Chem. 1996;271:9887–9890. doi: 10.1074/jbc.271.17.9887. [DOI] [PubMed] [Google Scholar]
- 78.Sanan DA, Newland DL, Tao R, Marcovina S, Wang J, Mooser V, Hammer RE, Hobbs HH. Low-density lipoprotein receptor-negative mice expressing human apolipoprotein B-100 develop complex atherosclerotic lesions on a chow diet: no accentuation by apolipoprotein(a) Proc Natl Acad Sci U S A. 1998;95:4544–9. doi: 10.1073/pnas.95.8.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hellberg S, Sippola S, Liljenbäck H, Virta J, Silvola JMU, Ståhle M, Savisto N, Metso J, Jauhiainen M, Saukko P, Ylä-Herttuala S, Nuutila P, Knuuti J, Roivainen A, Saraste A. Effects of atorvastatin and diet interventions on atherosclerotic plaque inflammation and [18F]FDG uptake in Ldlr-/-Apob100/100 mice. Atherosclerosis. 2017;263:369–376. doi: 10.1016/j.atherosclerosis.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 80.Chevrier G, Mitchell PL, Rioux LE, Hasan F, Jin T, Roblet CR, Doyen A, Pilon G, St-Pierre P, Lavigne C, Bazinet L, Jacques H, Gill T, McLeod RS, Marette A. Low-molecular-weight peptides from salmon protein prevent obesity-linked glucose intolerance, inflammation, and dyslipidemia in LDLR-/-/ApoB100/100 mice. J Nutr. 2015;145:1415–22. doi: 10.3945/jn.114.208215. [DOI] [PubMed] [Google Scholar]
- 81.Ramirez C, Sierra S, Tercero I, Vazquez JA, Pineda A, Manrique T, Burgos JS. ApoB100/LDLR-/- hypercholesterolaemic mice as a model for mild cognitive impairment and neuronal damage. PLoS One. 2011;6:e22712. doi: 10.1371/journal.pone.0022712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, Davidson NO. A mouse model for familial hypercholesterolemia: markedly elevated low-density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998;4:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- 83.Miyajima C, Iwaki T, Umemura K, Ploplis VA, Castellino FJ. Characterization of atherosclerosis formation in a murine model of type IIa human familial hypercholesterolemia. Biomed Res Int. 2018;2018:1878964. doi: 10.1155/2018/1878964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schwartz SM, Bennett MR. Death by any other name. Am J Pathol. 1995;147:229–234. [PMC free article] [PubMed] [Google Scholar]
- 85.van den Maagdenberg AM, Hofker MH, Krimpenfort PJ, de Bruijn I, van Vlijmen B, van der Boom H, Havekes LM, Frants RR. Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia. J Biol Chem. 1993;268:10540–10545. [PubMed] [Google Scholar]
- 86.Lardenoye JH, Delsing DJ, de Vries MR, Deckers MM, Princen HM, Havekes LM, van Hinsbergh VW, van Bockel JH, Quax PH. Accelerated atherosclerosis by placement of a perivascular cuff and a cholesterol-rich diet in ApoE*3Leiden transgenic mice. Circ Res. 2000;87:248–253. doi: 10.1161/01.res.87.3.248. [DOI] [PubMed] [Google Scholar]
- 87.van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, van der Boom H, HogenEsch H, Frants RR, Hofker MH, Havekes LM. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1994;93:1403–1410. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Paalvast Y, Gerding A, Wang Y, Bloks VW, van Dijk TH, Havinga R, Groen AK. Male apoE*3-Leiden. CETP mice on high-fat high-cholesterol diet exhibit a biphasic dyslipidemic response, mimicking the changes in plasma lipids observed through life in men. Physiol Rep. 2017;5:e13376. doi: 10.14814/phy2.13376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van der Hoogt CC, de Haan W, Westerterp M, Hoekstra M, Dallinga-Thie GM, Romijn JA, Rensen PC. Fenofibrate increases HDL-cholesterol by reducing cholesteryl ester transfer protein expression. J Lipid Res. 2007;48:1763–71. doi: 10.1194/jlr.M700108-JLR200. [DOI] [PubMed] [Google Scholar]
- 90.de Haan W, van der Hoogt CC, Westerterp M, Hoekstra M, Dallinga-Thie GM, Princen HM, Rensen PC. Atorvastatin increases HDL cholesterol by reducing CETP expression in cholesterol-fed APOE*3-Leiden. CETP mice. Atheroscler. 2008;197:57–63. doi: 10.1016/j.atherosclerosis.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 91.van der Hoorn JW, de Haan W, Berbée JF, Havekes LM, Jukema JW, Rensen PC, Princen HM. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden. CETP mice. Arterioscler Thromb Vasc Biol. 2008;28:2016–22. doi: 10.1161/ATVBAHA.108.171363. [DOI] [PubMed] [Google Scholar]
- 92.Lutgens E, Daemen M, Kockx M, Doevendans P, Hofker M, Havekes L, de Muinck ED. Atherosclerosis in APOE*3-Leiden transgenic mice: from proliferative to atheromatous stage. Circulation. 1999;99:276–283. doi: 10.1161/01.cir.99.2.276. [DOI] [PubMed] [Google Scholar]
- 93.Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, Bernal JA. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–59. doi: 10.1161/ATVBAHA.114.303617. [DOI] [PubMed] [Google Scholar]
- 94.Akram ON, Bernier A, Petrides F, Wong G, Lambert G. Beyond LDL cholesterol, a new role for PCSK9. Arterioscler Thromb Vasc Biol. 2010;30:1279–81. doi: 10.1161/ATVBAHA.110.209007. [DOI] [PubMed] [Google Scholar]
- 95.Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, Parker R, Prat A, Seidah NG. Dissection of the endogenous cellular pathways of PCSK9-induced LDL receptor degradation: evidence for an intracellular route. J Biol Chem. 2009;284:28856–28864. doi: 10.1074/jbc.M109.037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J, Tremblay M, Jacques H, Jin W, Davignon J, Seidah NG, Prat A. PCSK9: hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. J Hepatol. 2008;48:646–654. doi: 10.1002/hep.22354. [DOI] [PubMed] [Google Scholar]
- 98.Naoumova RP, Tosi I, Patel D, Neuwirth C, Horswell SD, Marais AD, Soutar AK. Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: long-term follow-up and treatment response. Arterioscler Thromb Vasc Biol. 2005;25:2654–2660. doi: 10.1161/01.ATV.0000190668.94752.ab. [DOI] [PubMed] [Google Scholar]
- 99.Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, Muriana FJ, Fuster V, Ibáñez B, Bernal JA. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–59. doi: 10.1161/ATVBAHA.114.303617. [DOI] [PubMed] [Google Scholar]
- 100.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnas-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 101.Goettsch C, Hutcheson JD, Hagita S, Rogers MA, Creager MD, Pham T, Choi J, Mlynarchik AK, Pieper B, Kjolby M, Aikawa M, Aikawa E. A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atheroscler. 2016;251:109–118. doi: 10.1016/j.atherosclerosis.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci. 2002;115:2817–2828. doi: 10.1242/jcs.115.14.2817. [DOI] [PubMed] [Google Scholar]
- 103.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van Herck JL, De Meyer GR, Martinet W, Van Hove CE, Foubert K, Theunis MH, Apers S, Bult H, Vrints CJ, Herman AG. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E-deficient mice. Circulation. 2009;120:2478–2487. doi: 10.1161/CIRCULATIONAHA.109.872663. [DOI] [PubMed] [Google Scholar]
- 105.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gargiulo S, Gramanzini M, Mancini M. Molecular imaging of vulnerable atherosclerotic plaques in animal models. Int J Mol Sci. 2016;17:1511. doi: 10.3390/ijms17091511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Van der Donckt C, Van Herck JL, Schrijvers DM, Vanhoutte G, Verhoye M, Blockx I, Van Der Linden A, Bauters D, Lijnen HR, Sluimer JC, Roth L, Van Hove CE, Fransen P, Knaapen MW, Hervent AS, De Keulenaer GW, Bult H, Martinet W, Herman AG, De Meyer GR. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. 2014;36:1049–58. doi: 10.1093/eurheartj/ehu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque haemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 109.Daeichin V, Sluimer JC, van der Heiden K, Skachkov I, Kooiman K, Janssen A, Janssen B, Bosch JG, de Jong N, Daemen MJ, van der Steen AF. Live observation of atherosclerotic plaque disruption in apolipoprotein E-Deficient mouse. Ultrasound Int Open. 2015;1:E67–E71. doi: 10.1055/s-0035-1565092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gargiulo S, Gramanzini M, Mancini M. Molecular imaging of vulnerable atherosclerotic plaques in animal models. Int J Mol Sci. 2016;17:E1511. doi: 10.3390/ijms17091511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fan J, Kitajima S, Watanabe T, Xu J, Zhang J, Liu E, Chen YE. Rabbit models for the study of human atherosclerosis: from pathophysiological mechanisms to translational medicine. Pharmacol Ther. 2014;146:104–19. doi: 10.1016/j.pharmthera.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hoeg JM, Santamarina-Fojo S, Berard AM, Cornhill JF, Herderick EE, Feldman SH, Haudenschild CC, Vaisman BL, Hoyt RF, Demosky SJ, Kauffman RD. Overexpression of lecithin: cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci U S A. 1996;93:11448–11453. doi: 10.1073/pnas.93.21.11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szeto A, Rossetti MA, Mendez AJ, Noller CM, Herderick EE, Gonzales JA, Schneiderman N, McCabe PM. Oxytocin administration attenuates atherosclerosis and inflammation in Watanabe heritable hyperlipidemic rabbits. Psychoneuroendocrinology. 2013;38:685–693. doi: 10.1016/j.psyneuen.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nordestgaard BG, Lewis B. Intermediate density lipoprotein levels are strong predictors of the extent of aortic atherosclerosis in the St. Thomas’s Hospital rabbit strain. Atheroscler. 1991;87:39–46. doi: 10.1016/0021-9150(91)90230-z. [DOI] [PubMed] [Google Scholar]
- 115.Shiomi M, Ito T. The Watanabe heritable hyperlipidaemic (WHHL) rabbit, its characteristics and history of development: a tribute to the late Dr Yoshio Watanabe. Atheroscler. 2009;207:1–7. doi: 10.1016/j.atherosclerosis.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 116.Fan J, McCormick SP, Krauss RM, Taylor S, Quan R, Taylor JM, Young SG. Overexpression of human apolipoprotein B-100 in transgenic rabbits results in increased levels of LDL and decreased levels of HDL. Arterioscler Thromb Vasc Biol. 1995;15:1889–1899. doi: 10.1161/01.atv.15.11.1889. [DOI] [PubMed] [Google Scholar]
- 117.Brousseau ME, Hoeg JM. Transgenic rabbits as models for atherosclerosis research. J Lipid Res. 1999;40:365–375. [PubMed] [Google Scholar]
- 118.Prather RS, Lorson M, Ross JW, Whyte JJ, Walters E. Genetically engineered pig models for human diseases. Annu Rev Anim Biosci. 2013;1:203–19. doi: 10.1146/annurev-animal-031412-103715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fan N, Lai L. Genetically modified pig models for human diseases. J Genet Genomics. 2013;40:67–73. doi: 10.1016/j.jgg.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 120.Lai L, Kolber-Simonds D, Park KW, Cheong HT, Greenstein JL, Im GS, Samuel M, Bonk A, Rieke A, Day BN, Murphy CN, Carter DB, Hawley RJ, Prather RS. Production of α-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science. 2002;295:1089–1092. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- 121.Dai Y, Vaught TD, Boone J, Chen SH, Phelps CJ, Ball S, Cowell-Lucero JL. Targeted disruption of the α1,3-galactosyltransferase gene in cloned pigs. Nat Biotechnol. 2002;20:251–255. doi: 10.1038/nbt0302-251. [DOI] [PubMed] [Google Scholar]
- 122.Al-Mashhadi RH, Sorensen CB, Kragh PM, Christoffersen C, Mortensen MB, Tolbod LP, Moldt B. Familial hypercholesterolemia and atherosclerosis in cloned minipigs created by DNA transposition of a human PCSK9 gain-of-function mutant. Sci Transl Med. 2013;5:166ra1. doi: 10.1126/scitranslmed.3004853. [DOI] [PubMed] [Google Scholar]
- 123.Dixon JL, Stoops JD, Parker JL, Laughlin MH, Weisman GA, Sturek M. Dyslipidemia and vascular dysfunction in diabetic pigs fed an atherogenic diet. Arterioscler Thromb Vasc Biol. 1999;19:2981–2992. doi: 10.1161/01.atv.19.12.2981. [DOI] [PubMed] [Google Scholar]
- 124.Wernersson R, Schierup MH, Jorgensen FG, Gorodkin J, Panitz F, Staerfeldt HH, Wang J. Pigs in sequence space: a 0.66X coverage pig genome survey based on shotgun sequencing. BMC Genomics. 2005;6:70. doi: 10.1186/1471-2164-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen F, Wang Y, Yuan Y, Zhang W, Ren Z, Jin Y, Liu X, Xiong Q, Chen Q, Zhang M, Li X, Zhao L, Li Z. Generation of B cell-deficient pigs by highly efficient CRISPR/Cas9-mediated gene targeting. J Genet Genomics. 2015;42:437–44. doi: 10.1016/j.jgg.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 126.Mohammadpour AH, Akhlaghi F. Future of cholesteryl ester transfer protein (CETP) inhibitors: a pharmacological perspective. Clin Pharmacokinet. 2013;52:615–626. doi: 10.1007/s40262-013-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vilahur G, Padro T, Badimon L. Atherosclerosis and thrombosis: insights from large animal models. J Biomed Biotechnol. 2011;2011:907575. doi: 10.1155/2011/907575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Patel D, Hamamdzic D, Llano R, Patel D, Cheng L, Fenning RS, Wilensky RL. Subsequent development of fibroatheromas with inflamed fibrous caps can be predicted by intracoronary near-infrared spectroscopy. Arterioscler Thromb Vasc Biol. 2013;33:347–353. doi: 10.1161/ATVBAHA.112.300710. [DOI] [PubMed] [Google Scholar]
- 129.Thim T, Hagensen MK, Wallace-Bradley D, Granada JF, Kaluza GL, Drouet L, Falk E. Unreliable assessment of necrotic core by virtual histology intravascular ultrasound in porcine coronary artery disease. Circ Cardiovasc Imaging. 2010;3:384–391. doi: 10.1161/CIRCIMAGING.109.919357. [DOI] [PubMed] [Google Scholar]
- 130.Davis BT, Wang XJ, Rohret JA, Struzynski JT, Merricks EP, Bellinger DA, Rogers CS. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS One. 2014;9:e93457. doi: 10.1371/journal.pone.0093457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pedrigi RM, Poulsen CB, Mehta VV, Ramsing Holm N, Pareek N, Post AL, Kilic ID, Banya WA, Dall’Ara G, Mattesini A, Bjørklund MM, Andersen NP, Grøndal AK, Petretto E, Foin N, Davies JE, Di Mario C, Fog Bentzon J, Erik Botker H, Falk E, Krams R, de Silva R. Inducing persistent flow disturbances accelerates atherogenesis and promotes thin cap fibroatheroma development in D374Y-PCSK9 hypercholesterolemic minipigs. Circulation. 2015;132:1003–12. doi: 10.1161/CIRCULATIONAHA.115.016270. [DOI] [PubMed] [Google Scholar]
- 132.Poulsen CB, Pedrigi RM, Pareek N, Kilic ID, Holm NR, Bentzon JF, Botker HE, Falk E, Krams R, de Silva R. Plaque burden influences accurate classification of fibrous cap atheroma by in vivo optical coherence tomography in a porcine model of advanced coronary atherosclerosis. EuroIntervention. 2018;14:1129–1135. doi: 10.4244/EIJ-D-17-01028. [DOI] [PubMed] [Google Scholar]
- 133.Li Y, Fuchimoto D, Sudo M, Haruta H, Lin QF, Takayama T, Morita S, Nochi T, Suzuki S, Sembon S, Nakai M, Kojima M, Iwamoto M, Hashimoto M, Yoda S, Kunimoto S, Hiro T, Matsumoto T, Mitsumata M, Sugitani M, Saito S, Hirayama A, Onishi A. Development of human-like advanced coronary plaques in low-density lipoprotein receptor knockout pigs and justification for statin treatment before formation of atherosclerotic plaques. J Am Heart Assoc. 2016;5:e002779. doi: 10.1161/JAHA.115.002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ogita M, Miyauchi K, Onishi A, Tsuboi S, Wada H, Konishi H, Naito R, Dohi T, Kasai T, Kojima Y, Schwartz RS. Development of accelerated coronary atherosclerosis model using low density lipoprotein receptor knock-out swine with balloon injury. PLoS One. 2016;11:e0163055. doi: 10.1371/journal.pone.0163055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maeng M, Olesen PG, Emmertsen NC, Thorewest M, Nielsen TT, Falk E, Kristensen BO, Falk E, Andersen HR. Time course of vascular remodelling, formation of neointima, and formation of neoadventitia after angioplasty in a porcine model. Coron Artery Dis. 2001;12:285–293. doi: 10.1097/00019501-200106000-00004. [DOI] [PubMed] [Google Scholar]
- 136.Fang B, Ren X, Wang Y, Li Z, Zhao L, Zhang M, Li C, Zhang Z, Chen L, Li X, Liu J, Xiong Q, Zhang L, Jin Y, Liu X, Li L, Wei H, Yang H, Li R, Dai Y. Apolipoprotein E deficiency accelerates atherosclerosis development in miniature pigs. Dis Model Mech. 2018;11 doi: 10.1242/dmm.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shim J, Poulsen CB, Hagensen MK, Larsen T, Heegaard PM, Christoffersen C, Li R. Apolipoprotein E deficiency increases remnant lipoproteins and accelerates progressive atherosclerosis, but not xanthoma formation, in gene-modified minipigs. JACC Basic Transl Sci. 2017;2:591–600. doi: 10.1016/j.jacbts.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang L, Hua Z, Xiao H, Cheng Y, Xu K, Gao Q, Xia Y, Liu Y, Zhang X, Zheng X, Mu Y, Li K. CRISPR/Cas9-mediated ApoE-/- and LDLR-/- double gene knockout in pigs elevates serum LDL-C and TC levels. Oncotarget. 2017;8:37751–37760. doi: 10.18632/oncotarget.17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lo Sasso G, Schlage WK, Boué S, Veljkovic E, Peitsch MC, Hoeng J. The Apoe(-/-) mouse model: a suitable model to study cardiovascular and respiratory diseases in the context of cigarette smoke exposure and harm reduction. J Transl Med. 2016;14:146. doi: 10.1186/s12967-016-0901-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Van der Heiden K, Hoogendoorn A, Daemen MJ, Gijsen FJ. Animal models for plaque rupture: a biomechanical assessment. Thromb Hemost. 2016;115:501–508. doi: 10.1160/TH15-07-0614. [DOI] [PubMed] [Google Scholar]
- 141.Bond AR, Jackson CL. The fat-fed apolipoprotein E knockout mouse brachiocephalic artery in the study of atherosclerotic plaque rupture. BioMed Res Int. 2011;2011:379069. doi: 10.1155/2011/379069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, Wang Y. Hyperlipidemia and atherosclerotic lesion development in Ldlr-deficient mice on a long-term high-fat diet. PLoS One. 2012;7:e35835. doi: 10.1371/journal.pone.0035835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kostogrys RB, Johann C, Czyżyńska I, Franczyk-Żarów M, Drahun A, Maślak E, Wybrańska I. Characterisation of atherogenic effects of low carbohydrate, high protein diet (LCHP) in apoE/LDLR-/- mice. J Nutr Health Aging. 2015;19:710–718. doi: 10.1007/s12603-015-0543-7. [DOI] [PubMed] [Google Scholar]
- 144.Jawien J. Mouse models of experimental atherosclerosis as a tool for checking a putative anti-atherogenic action of drugs. InAtherogenesis. 2012 IntechOpen. [Google Scholar]
- 145.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 146.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnæs-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 147.Daugherty A, Tabas I, Rader DJ. Accelerating the pace of atherosclerosis research. Arterioscler Thromb Vasc Biol. 2015;35:11–12. doi: 10.1161/ATVBAHA.114.304833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Davis BT, Wang XJ, Rohret JA, Struzynski JT, Merricks EP, Bellinger DA, Rohret FA, Nichols TC, Rogers CS. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS One. 2014;9:e93457. doi: 10.1371/journal.pone.0093457. [DOI] [PMC free article] [PubMed] [Google Scholar]