

Abstract
Recently, heat shock transcription factor 1 (HSF1) is observed to be involved in the process of cellular metabolism in cancer. However, the roles of HSF1 in the metabolic alteration of hepatocellular carcinoma (HCC) in tumor microenvironment remain elusive. Here, HCC cells were co-cultured with tumor-associated macrophages (TAM). The levels of glucose uptake, the lactate release, reactive oxygen species (ROS) and mtDNA content were measured by the associated Kits; all detected protocols were correspondingly according to the manufacturers’ instructions. Recombinant lentiviruses with shRNA against HSF1 and MCT4 were transfected into HCC cells or TAMs. Western blot analysis was conducted to detect the relative levels of HSF1, MCT1 and MCT4 proteins. CCK-8 assay was utilized to assess cell proliferation. Based on the co-culture system with HCC cells and TAMs, metabolic alteration of HCC cells after co-culture with TAMs was observed. Furthermore, glucose consumption rate, lactate production rate and intercellular ROS level were decreased, while the copy number of mtDNA was increased in HSF1-knockdown HCC cells. Besides, metabolic crosstalk between HCC cells and TAMs was induced by HSF1 not only in HCC cells but also in TAMs through regulating individually MCT1 and MCT4 expressions. To the best of our knowledge, this is an important study to demonstrate the roles of HSF1 in regulating metabolic alteration of HCC cells induced by TAMs, which implies the potential use of HSF1 as a target modulating malignant behaviors of HCC cells.
Keywords: Hepatocellular carcinoma, heat shock transcription factor 1, tumor-associated macrophages, metabolic phenotype
Introduction
The metabolic reprogramming has been recognized as the new hallmark of tumor [1], which results in a series of intracellular changes, contributing not only to providing energy source for rapid cell growth, but also establishing a foundation for effective adaptation to the altered microenvironment [2]. Previous researchers assume that the altered cellular metabolism is merely the trivial phenomena of tumor cytogenetic alterations [3]. However, mounting recent evidences suggest that energetic metabolism acts as the downstream components of oncogene activation pathways, and in contrast, can also function as the upstream components, affecting signaling pathways of cell growth. In other words, metabolic reprogramming is not an accompanying phenomenon with oncogenesis, but a central node of cellular oncogenesis [4].
Metabolic reprogramming of tumor cells is triggered largely by activation of oncogenes or inactivation of tumor suppressor genes [5], which also means tumor cells doesn’t fabricate new “metabolic reactions” or “metabolic pathway” and just selectively turns on/off the specific metabolic pathways originally existed in cells [6]. Current studies in tumor metabolism field often paid more attention on oncogene activation, anti-oncogene inactivation and metabolic enzyme expression abnormalities [7]; in contrast, few studies have focused on roles and functions of non-oncogenes, which may not directly mediate alteration of metabolism in oncogenesis, but can regulate the activation of oncogene-driven metabolic pathway [8]. Hence, researches on the involvement of non-oncogenes in metabolic reprogramming of tumor cells will provide a new perspective for elucidating the mechanisms of tumor metabolic regulation and meanwhile offer new potential targets for the treatment of malignant tumor.
It was previously deemed that “Warburg effect” is only confined to tumor cells; however, tumor cells constantly interact with the surrounding microenvironment in vivo, which could induce the neighboring cells’ metabolic alteration through oxidative stress and mitochondrial autophagy [9]. Meanwhile, peritumoral cells with high-expressed monocarboxylate transporter 4 (MCT4) secret large amounts of lactate and pyruvate, which can be utilized by tumor cells through monocarboxylate transporter 1 (MCT1) to maintain rapid proliferation. The novel metabolic pattern was termed as the “Reverse Warburg Effect” [10,11]. So far, this pattern has been verified by many studies in vivo and in vitro [12-14]. Among peritumoral cells, tumor-associated macrophage (TAM) was characterized as an important component of the tumor microenvironment, promoting tumor progression and poor prognosis [15,16]. Our previous study has identified that peritumoral high MCT4 and high heat shock transcription factor 1 (HSF1) expressions led to the worst prognosis of HCC patients and the expression of peritumoral HSF1 was significantly correlated with peritumoral MCT4 [17]. Furthermore, roles of HSF1 in a variety of tumor cells has been reported as a typical model for “non-oncogene addiction” phenomenon [18]. Recently, HSF1 has been identified as a key mediator in glucose metabolism and involved in the acidic tumor microenvironment [19,20]. Based on the studies mentioned above, it can be proposed that HSF1 may be involved in the interaction between TAMs and HCC cells through affecting metabolic reprograming. To address this issue, we co-cultured HCC cells with TAMs, and demonstrated that the metabolic conversation between HCC cells and TAMs was mediated by HSF1, indicating the potential use of HSF1 as a target modulating malignant behaviors of HCC cells.
Materials and methods
Cell co-culture and treatment
MHCC97H is hepatitis B virus (HBV)-positive HCC cell line with high metastatic potential, was established at our institute [21]. The human monocyte leukemia cell line THP-1 and HCC cell line-SMMC7721, which is HBV-negative cell line with low metastatic potential, were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China. MHCC97H, SMMC7721 and THP-1 cells, obtained from the Typical Culture Preservation Commission Cell Bank, Chinese Academy of Sciences (Shanghai, China), were cultured in Dulbecco’s modified Eagle’s (DMEM) medium and in RPMI medium (Gibco, USA); media were supplemented with 10% fetal bovine serum (FBS, Gibco, USA). Cells were cultured at 37°C in a humidified incubator of 5% CO2. For M2 polarization to producing TAMs [22], THP-1 cells were incubated in complete medium containing 200 ng/ml PMA (Sigma) for 24 h and cultured in fresh complete medium for another 24 h to produce THP-1-derived macrophages; these macrophages were cultured fresh complete medium with 20 ng/ml IL-4 (Peprotech) and 20 ng/ml IL-13 (Peprotech) for 72 h and then refreshed the culture medium with serum-free DMEM for another 24 hours. These adherent TAMs were collected for the following experiments. As for 2-DG treatment, medium was changed for low glucose medium containing 1 g/L glucose with L-lactate (5 mM). The separation of TAMs and co-culture with MHCC97H cells system were established in our institute with reference to previous studies [23].
Reagents and antibodies
Sodium L-lactate and glycolysis inhibitor 2-Deoxy-D-glucose (2-DG) were purchased from Sigma-Aldrich. Rabbit anti-human HSF1 (#4356) and β-tubulin (#2128) monoclonal antibody were obtained from Cell Signaling Technology (Danvers, MA, USA). Mouse anti-human MCT1 (sc-365501) and MCT4 (sc-376140) monoclonal antibodies were obtained from Santa Cruz Biotech. Anti-rabbit Anti-mouse HRP-linked secondary antibody IgG (#7074 and #7076) were purchased from Cell Signaling Technology.
Western blotting
Harvested cells were homogenized with lysis buffer and protease inhibitor cocktail (Roche, Mannheim, Germany), then cell lysates were centrifuged at 4°C for 30 minutes at 12,000 rpm. The supernatant was placed in fresh tubes and quantified using the protein assay Bradford reagent (Bio-Rad, Hercules, CA). Equal amounts of protein (40 μg) were firstly separated by 10% SDS-PAGE and then transferred onto PVDF membrane (Millipore, Billerica, USA). After blocking for nonspecific binding in 1 × TBS with 5% BSA or 5% non-fat milk containing 0.1% Tween-20 (TBS-T) for 1 h at room temperature, the membranes were incubated with a primary antibody (1:1000) overnight at 4°C. A β-tubulin antibody (1:1,000) was used as an internal control. Followed an extensively washed with TBS-T, the membranes were then incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. After washing in TBS-T for three times, the membranes were finally visualized using the ECL western blot system (Super Signal West Pico Chemiluminescent Substrate, Pierce, Rockford, IL, USA) according to the manufacturer’s instruction and quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Cell proliferative assay
MCT4-sh TAMs and MHCC97H cells were co-cultured at indicated times, and then separated each other. Both MCT4-sh TAMs and MHCC97H cells were seeded at 5 × 103 cells/well (100 μl) in a 96-well plate respectively. The Cell Counting Kit-8 (CCK8, Dojindo, Kumamoto, Japan) was used to determine the relative growth of cell groups. As for sensibility to glycolysis inhibitor 2-DG, MHCC97H and SMMC7721 cells (5 × 103 cells/well, 100 μl) from monoculture and co-culture condition were seeded at 96-well plate and incubated with the concentration gradient of 2-DG (0, 5, and 10 mmol/L) at indicated times. After treatments, all cells were stained with 10 μl CCK8 for 1 h at 37°C. Absorbances were measured at 450 nm using the microplate reader, Infinite 200 PRO NanoQuant (TECAN, Männedorf, Switzerland). Cells incubated with only complete medium were used as the negative control and wells with complete medium but without cells as the blank control. Each treatment was repeated 4 wells and cell proliferation activity was measured according to the equation: Cell proliferation activity = [(experimental absorbance - blank control absorbance)/(negative control absorbance - blank control absorbance)] × 100%.
Glucose uptake assays
The ability of glucose uptake in different groups was measured using the Glucose Uptake Cell-Based Assay Kit (Cayman, Ann Arbor, Michigan, USA) according to the manufacturer’s instructions. Briefly, the cells were seeded at a density of 5 × 103 cells/well in 96-well flat-bottomed plates and incubated overnight. Then, cells were treated with 100 μL glucose-free media containing 150 μg/mL of the fluorescent glucose analog 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose (2-NBDG) for 48 h. After treatment, the plates were centrifuged for 5 min at 400 × g at room temperature, and the supernatant was aspirated. Cell-Based Assay Buffer was added to each well (100 μL/well). The reading of each well was immediately measured using a VICTOR2 1420 multilabel counter (PerkinElmer Life Sciences, Downers Grove, IL, USA) at 485 nm excitation and 535 nm emission.
Lactate release assays
The cell culture media of different groups were collected for lactate measurement at indicated times. Samples were precipitated with 200 μl of 10% perchloric acid (Fisher Scientific, Cat No A-229) for 15 min and neutralized with 150 μl of 9.13% KOH (BDH Inc, Canada, Cat No 0260) for another 15 min at room temperature, then centrifuged at 13,000 rpm for 3 min. The lactate assay was performed using an EnzyChrome lactate assay kit (ECLC-100) according to the manufacturer’s instructions (BioAssay Systems), and the absorbance at 565 nm was measured using a microplate reader (Infinite 200 PRO NanoQuant, TECAN, Männedorf, Switzerland).
Reactive oxygen species assessment
The reactive oxygen species (ROS) in live cells was measured by Cell ROX Oxidative Stress Reagents fluorogenic probe (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s specifications. This cell-permeable dye is weakly fluorescent in reduced state while in the presence of ROS, it is converted to the oxidized form which emits a fluorescence signal directly proportional to oxidation and thus to the amount of ROS.
Measurement of mtDNA
mtDNA content was measured by Human Mitochondrial DNA Fluorescence quantitative Polymerase Chain Reaction (PCR) Diagnostic Kit (Suoao Biotech., Beijing, China) according to manufacturer’s specifications. The precise mtDNA content per cell was reflected by the ratio of mtDNA to nuclear DNA. PCR was performed in Stepone Plus system (Applied Biosystems, Foster City, CA).
Plasmids and viral transductions
We used the Lentiviral vectors pLKO.1 TRC and pWPI.1 to construct recombinant lentiviruses. Oligonucleotides encoding hairpin precursors for shMCT4 (target sequence: 5’-CGTCTACATGTACGTGTTCAT-3’) were used for generating short interference RNA (shRNA) constructs. A scrambled non-targeting sequence was used as a control (Mock). The HSF1 knockdown by shRNA transfection in HCC cells and TAMs was carried out as previously described [17]. Recombinant lentivirus was amplified in HEK293T cells. The site-directed mutagenic plasmid of HSF1 at Ser303/307 was generated using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA) according to manufacturer’s protocol.
Statistical analysis
Statistical analysis was performed with SPSS 16.0 for Windows (SPSS, Chicago, IL). The Fisher’s exact test was used to compare qualitative variables, and quantitative variables were expressed as the means ± SEM. Nonparametric methods including Wilcoxon rank test were applied to perform a comparison between monoculture and coculture groups and time was set as a covariance. A value of P<0.05 or P<0.01 was considered as significant difference or extremely significant difference, respectively.
Results
Metabolic phenotype of HCC cells altered from glycolysis phenotype to OXPHOS phenotype after co-culture with TAMs
After the differentiation of TPH-1 into TAMs, the associated markers CD68 and CD204 levels were increased, suggesting the phenotype of TAMs (Figure S1). HCC cells-MHCC97H or SMMC7721 were co-cultured with TAMs for 4 days, and then were separated; glucose consumption (G) and lactate release (L) were detected in both monoculture and co-culture HCC cells; afterwards, the L/G ratio was calculated. The results showed that, in monoculture condition, the L/G ratio of MHCC97H or SMMC7721 cells was close to 2 (L/G≈2 means the glycolysis phenotype) and these HCC cells were sensitive to the glycolysis inhibitor 2-DG, the inhibition of cell proliferation was dose-dependent (Figure 1A, 1B and 1D, 1E). In contrast, in co-culture condition, MHCC97H or SMMC7721 cells weren’t sensitive to 2-DG and the values of L and G were lower than monoculture, and L/G ratio in MHCC97H and SMMC7721 cells were respectively 2.75 and 2.93, deviated far from 2.0 (Figure 1C, 1F); while TAMs did not presented obviously deviation, but L/G significantly decreased in the co-culture condition and proliferation ability was not affected by the existence of HCC cells (Figure S2). These results suggested that metabolic phenotype of HCC cells altered from glycolysis phenotype to OXPHOS phenotype after co-culture with TAMs.
Figure 1.
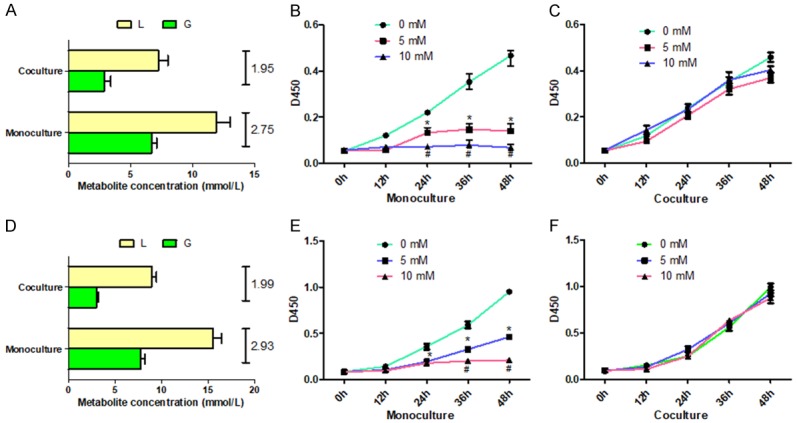
Metabolic alteration of HCC cells after co-culture with TAMs. Co-cultured TAMs with HCC cells including MHCC97H (A) and SMMC7721 (D) for 4 days, and then separated the HCC cells, glucose consumption (G) and lactate production (L) were detected in both monoculture and co-culture MHCC97H cells, afterwards, calculate the L/G ratio. Proliferation of MHCC97H (B, C) and SMMC7721 (E, F) cells under monoculture or co-culture condition after adding pre-set concentration of 2-DG was measured by using CCK8 assay. *P<0.05.
HSF1 functions in metabolism reprogramming of HCC cells co-cultured with TAMs
Accumulating evidences have showed that HSF1 regulates cellular metabolism in tumor development [19,24]. To further validate the relationship between HSF1 and metabolism alteration in HCC cells, we knock-downed significantly HSF1 expression in HCC cells (Figure 2A, 2B) and found that glucose consumption rate, lactate production rate and intercellular ROS level (by-product of OXPHOS) were decreased in HSF1-knockdown HCC cells, while the copy number of mtDNA was increased, compared with control and mock cells (Figure 2C-F); meanwhile the expressions of some critical glycolysis-related enzymes including HK1, LDHA and PKM2 were downregulated after HSF1 knockdown in HCC cells (Figure 2G, 2H). In addition, these metabolic indicators were detected again in HCC cells with reintroducing HSF1-cDNA after knockdown (shRES). The results showed that reintroduction of HSF1 partly recovered the glucose consumption and lactate production rate (Figure 2I, 2J), suggesting further the roles of HSF1 in regulating metabolism alteration of HCC cells.
Figure 2.
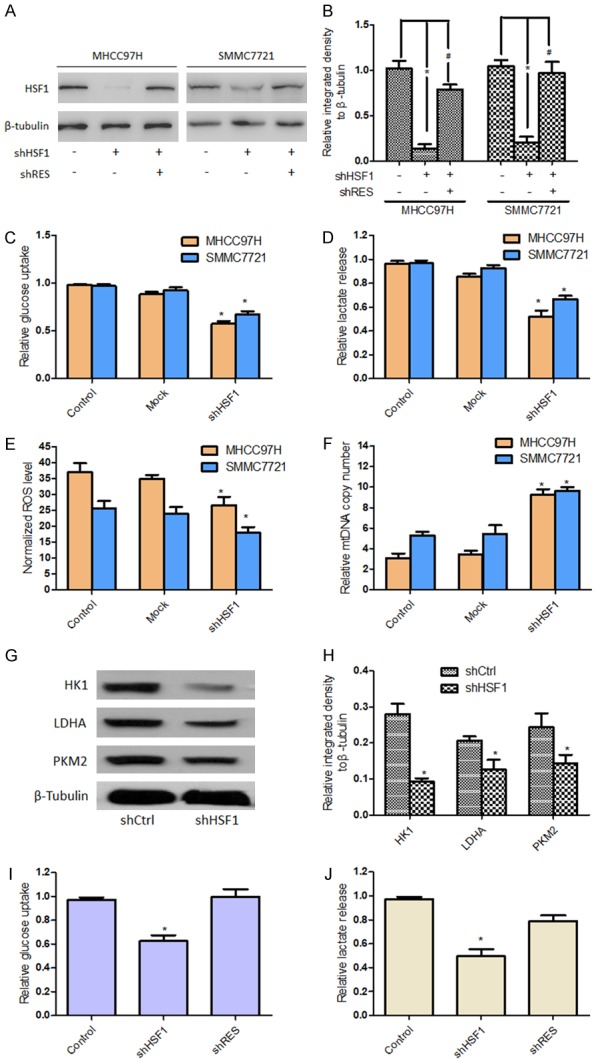
HSF1 played an important role in regulating energy metabolism of HCC cells. HSF1 expression in MHCC97H and SMMC7721 cells were significantly knock-downed, in indicated conditions reintroduced HSF1-cDNA (shRES) after knockdown for subsequent assays (A, B). After these HCC cells were transinfected with sh-HSF1 or Mock lentivirus or control cells without treatment, the glucose consumption rate (C), lactate production rate (D), the expression of ROS (E) and the copy number of mtDNA (F) were measured by relevant Kit according to the manufacturer’s instructions. Expressions of some metabolism-related enzymes including HK1, LDHA and PKM2 were examined by western blotting (G, H). Moreover, HSF1 wild-type (HSF1-WT), HSF1 knockdown (HSF1-sh), reintroducing HSF1-cDNA (HSF1-R) MHCC97H were established to detect glucose consumption (I) and lactate production rate (J). *P<0.05 vs Control, #P<0.05 vs shRES.
HSF1 was involved in the conversation of energy metabolism between TAMs and HCC cells
As mentioned above, MCT4 in peritumoral cells has a responsibility for lactate and pyruvate release, which can be transported into cells by MCT1 in tumor cells [10]. Here, TAMs were co-cultured with HCC cells for 4 days and it was found the increased expression of MCT4 in TAMs and MCT1 in HCC cells (Figure 3A, 3B); moreover, to mimic the effect of co-culture condition on the expression of MCT1 in HCC cells, L-lactate (5 mM) was added in glucose-restriction (1 g/L) condition media for 72 h and it was observed the expression of MCT1 in HCC cells was significantly up-regulated (Figure 3C). Then, to further demonstrate the effect of TAMs on the altered metabolism of HCC cells, TAMs were treated with shRNA against MCT4 (shMCT4-TAMs). The results showed that knockdown of MCT4 in TAMs did not down-regulate HSF1 expression but inhibit cell proliferation (Figure 3D-F); meanwhile, led to the decrease of relative lactate release rate (based on same number of detected cells) (Figure 3G). When HCC cells were co-cultured with shMCT4-TAMs, ROS level was decreased and MCT1 expression was decreased but MCT4 and HSF1 expression was not significantly changed in HCC cells (Figure 3H-J).
Figure 3.
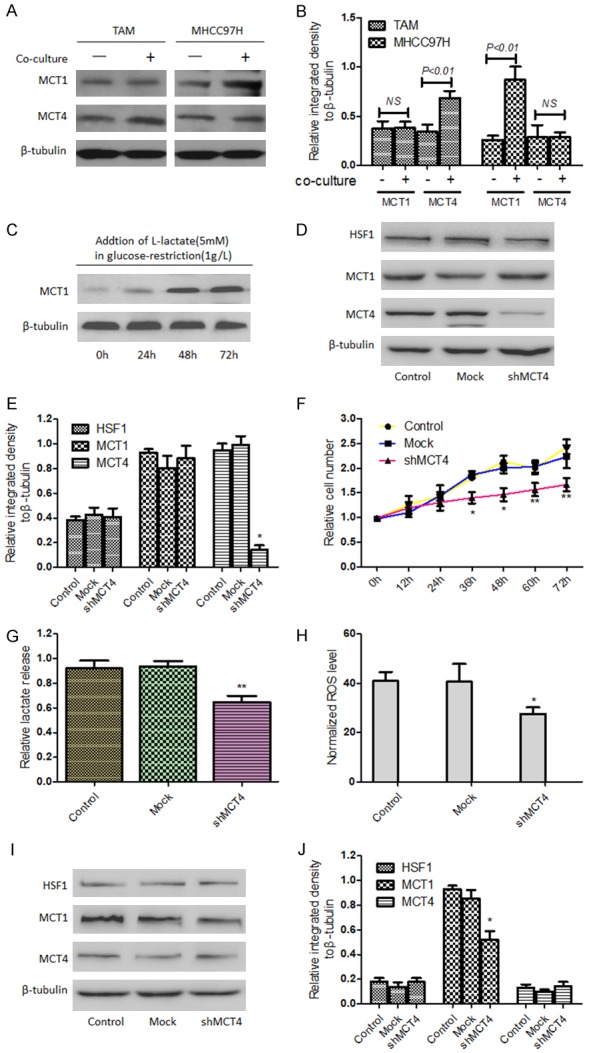
HSF1 was involved in the conversation of energy metabolism between TAMs and HCC cells. When MHCC97H cells were cocultured with TAMs, the expressions of MCT1 and MCT4 were separately detected and relatively quantitative analysis was performed (A, B). The protein levels of MCT1 in HCC cells were analyzed by Western blotting after adding L-lactate (5 mM) in glucose-restriction (1 g/L) condition at indicated times (C). After TAMs were treated with shRNA against MCT4 (shMCT4-TAMs), expressions of HSF1, MCT1 and MCT4 in TAMs were analyzed by Western blotting (D, E) and proliferation of TAMs was determined by CCK-8 assay (F). In addition, lactate release rate of TAMs was determined after transfection (G). When MHCC97H cells were co-cultured with shMCT4-TAMs, ROS level was detected by relevant Kit according to the manufacturer’s instructions (H) and MCT1, MCT4 and HSF1 expression in MHCC97H cells was detected by Western blotting (I, J). *P<0.05 or **P<0.01 vs Mock.
Subsequently, to investigate the effects of HSF1 on regulating the expressions of MCT1 and MCT4 when HCC cells were co-cultured with TAMs, HCC cells were firstly co-cultured with HSF1-knockdown TAMs (shHSF1-TAMs) and it was found that HSF1-knockdown downregulated MCT1, but had no influence on the expression of MCT4 in TAMs; meanwhile such co-cultured condition didn’t lead to the upregulation of MCT1 and MCT4 expression in HCC cells (Figure 4A, 4B). In contrast, the expression of MCT1 in TAMs was not significantly altered, although MCT4 expression presented non-significant decrease, when co-cultured with shHSF1-HCC cells, in which HSF1-knockdown downregulated both MCT1 and MCT4 (Figure 4C, 4D). Notably, both of the co-culture conditions from shHSF1-TAMs with HCC cells and shHSF1-HCC cells with TAMs led to decreased glucose uptake and lactate release rate of HCC cells, compared to the co-culture of HCC cells with TAMs (Figure 4E, 4F). Altogether, these results above suggested that HSF1 participated in the conversation of energy metabolism between TAMs and HCC cells through regulating individually MCT1 and MCT4 expression.
Figure 4.
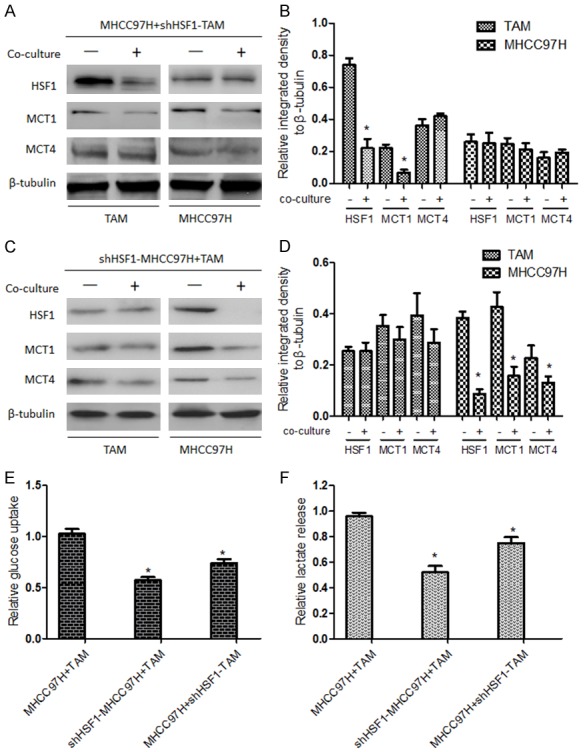
Effects of HSF1 on regulating the expressions of MCT1 and MCT4 when HCC cells were co-cultured with TAMs. When MHCC97H cells were co-cultured with shHSF1-TAMs (A, B) or shHSF1-MHCC97H cells were co-cultured with TAMs (C, D), MCT1, MCT4 and HSF1 expression in MHCC97H cells was detected by Western blotting. *P<0.05 vs monoculture. Then MHCC97H cells were separated and glucose uptake (E) and lactate release rate (F) of HCC cells, compared to those of HCC cells from the co-culture of HCC cells with TAMs, were detected by relevant Kits according to the manufacturer’s instructions. *P<0.05 vs MHCC97H cells from the co-culture of HCC cells with TAMs.
Discussion
In this study, based on the co-culture system with HCC cells and TAMs, the effects of HSF1 on metabolic crosstalk between HCC cells and TAMs was investigated and several novel findings were obtained. Firstly, HCC cells achieved metabolism alteration when co-cultured with TAMs. Secondly, HSF1 plays an important role in regulating metabolism of HCC cells. Thirdly, metabolic crosstalk between HCC cells and TAMs was induced by HSF1 expression not only in HCC cells but also in TAMs, possibly associated with regulating MCT1 and MCT4 expression. To the best of our knowledge, this is an important study to demonstrate the roles of HSF1 in metabolic reprograming of HCC cells induced by TAMs.
Over the past decade, researches in tumor metabolism have gained more and more attention. The metabolic reprogramming of tumor cells has previously been assumed as the trivial phenomena of tumor cytogenetic alterations. Some metabolites and mutated metabolic enzymes including isocitrate dehydrogenase (IDH) and pyruvate kinase (PKM) have been recognized as oncogenic factors closely related to tumor development [25-28]. However, recent evidences suggested that HSF1 supports malignant phenotypes of tumor cells by orchestrating a network of core cellular functions [29], linked to an altered metabolism in a variety of tumors by influencing cellular energy homeostasis [30]. For example, HSF1 has been identified as a critical transcriptional regulator of NAD+ metabolism and can uncouple metabolic control from proteostatic regulation [31]; moreover, the malfunctions of HSF1 can enhance insulin sensitivity and promote activation of AMP-activated protein kinase (AMPK), an important regulator of energy homeostasis and inhibitor of lipid synthesis, finally involving in the regulation of hepatic metabolism [32]. In the study, we found that HSF1 knockdown was associated with the down-regulation of glycolysis, whereas OXPHOS level was elevate to some extent in HCC cells. These results suggested that HSF1 functions as a mediator in regulating energy metabolism of HCC cells.
In 2009, a new metabolic pattern “Warburg effect takes place in stroma cells rather than tumor cells” was firstly described [33]; hereafter, it was termed as “Reverse Warburg Effect”, which stresses the aerobic glycolysis of tumor stroma, the symbiosis relationship of metabolism between tumor cells and stroma [9]. In the model, some noncancerous cells surrounding tumor tissue experience genetic and epigenetic alteration, playing a potential role in regulating the metabolic reprogramming of tumor cells [34]. Herein, as an important component and effector of tumor microenvironment, TAMs was introduced through co-culture system to investigate the effects of noncancerous cells on metabolic reprogramming of HCC cells and the results showed metabolic phenotype switch of HCC cells; meanwhile upregulated expression MCT4 in TAMs and MCT1 in HCC cells after co-culture, in accordance with the “Reverse Warburg Effect”, in which stromal cells and tumor cells achieved energy resource shuttle through MCT4 and MCT1 [35]. Furthermore, it was found that knockdown of MCT4 in TAMs led to reduced lactate release rate; furthermore, after adding L-lactate in glucose-restriction condition, the proliferation of MCT4-sh TAMs was inhibited, suggesting a positive feedback mechanism in macrophages to maintain a high glycolytic rate [36]. Meanwhile, MCT1 expression in HCC cells was significantly up-regulated in our study, which may be due to effective adaption of HCC cells to the microenvironments that include such stressors as acidosis and nutrient deprivation [22,37].
In addition, our previous study demonstrated that HSF1 can regulate MCT4 expression in HCC cells in stress condition [17]. In present study, it was observed that TAMs with HSF1 knockdown unable to up-regulate MCT1 expression in HCC cells in co-cultured condition; meanwhile MCT4 expression in TAMs was not significantly altered when co-cultured with HSF1-knockdown HCC cells. These results suggested that HSF1 participates in the conversation of energy metabolism between TAMs and HCC cells through regulating conditionally and correspondingly MCT1 and MCT4 expression.
Nevertheless, there are some limitations in our study which will be improved in the future. For example, the cytokines and/or chemokines which possible induce the association between TAM and HCC cells will be profiled. Moreover, the molecular mechanism of HSF1 regulating MCT1 or MCT4 will be elucidated in subsequent experiments.
In conclusion, our results suggested that interaction between TAMs and HCC cells altered energy metabolism of HCC cells, providing necessary energy-rich microenvironment for facilitating tumor growth. The complicated effects of HSF1 on metabolic reprograming of HCC cells induced by TAMs and the crosstalk between HCC cells and TAMs implied the potential use of HSF1 as a target modulating malignant behaviors of HCC cells. At present, the new mechanism of tumorigenesis is being elucidated from the perspective of metabolic abnormality. The correction of cellular metabolic abnormalities is becoming new ideas for tumor prevention and treatment. Fortunately, some targeted drugs against cellular metabolism have entered the clinical stage, such as TLN-232/CAP-232 targeting PKM2, AZD3965 targeting MCT1, and PU-H71 targeting HSP90, a downstream of HSF1, etc [38,39].
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81872492) and Shanghai Pujiang Program (No. 15PJD007).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux SP, Wilson GL, Tan M. Emerging metabolic targets in cancer therapy. Front Biosci (Landmark Ed) 2011;16:1844–1860. doi: 10.2741/3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otto AM. Warburg effect(s)-a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016;4:5. doi: 10.1186/s40170-016-0145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy P, Bartosch B. Metabolic reprogramming: a hallmark of viral oncogenesis. Oncogene. 2016;35:4155–4164. doi: 10.1038/onc.2015.479. [DOI] [PubMed] [Google Scholar]
- 5.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, Jones LW, Ko YH, Le A, Lea MA, Locasale JW, Longo VD, Lyssiotis CA, McDonnell E, Mehrmohamadi M, Michelotti G, Muralidhar V, Murphy MP, Pedersen PL, Poore B, Raffaghello L, Rathmell JC, Sivanand S, Vander Heiden MG, Wellen KE Target Validation Team. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. 2015;35(Suppl):S129–S150. doi: 10.1016/j.semcancer.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagel R, Semenova EA, Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016;17:1516–1531. doi: 10.15252/embr.201643030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol. 2014;41:195–216. doi: 10.1053/j.seminoncol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, Martinez-Outschoorn U. Metabolic coupling and the Reverse Warburg Effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44:198–203. doi: 10.1053/j.seminoncol.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez CD, Alvarez S, Ropolo A, Rosenzvit C, Bagnes MF, Vaccaro MI. Autophagy, Warburg, and Warburg reverse effects in human cancer. Biomed Res Int. 2014;2014:926729. doi: 10.1155/2014/926729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Philp NJ, Pestell RG, Lisanti MP. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–1454. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–1117. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez-Outschoorn UE, Whitaker-Menezes D, Valsecchi M, Martinez-Cantarin MP, Dulau-Florea A, Gong J, Howell A, Flomenberg N, Pestell RG, Wagner J, Arana-Yi C, Sharma M, Sotgia F, Lisanti MP. Reverse Warburg effect in a patient with aggressive B-cell lymphoma: is lactic acidosis a paraneoplastic syndrome? Semin Oncol. 2013;40:403–418. doi: 10.1053/j.seminoncol.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 15.El Kasmi KC, Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol. 2015;27:267–275. doi: 10.1016/j.smim.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43:435–449. doi: 10.1016/j.immuni.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Zhang JB, Guo K, Sun HC, Zhu XD, Zhang B, Lin ZH, Zhang BH, Liu YK, Ren ZG, Fan J. Prognostic value of peritumoral heat-shock factor-1 in patients receiving resection of hepatocellular carcinoma. Br J Cancer. 2013;109:1648–1656. doi: 10.1038/bjc.2013.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets. 2009;13:469–478. doi: 10.1517/14728220902832697. [DOI] [PubMed] [Google Scholar]
- 19.Dai C, Sampson SB. HSF1: guardian of proteostasis in cancer. Trends Cell Biol. 2016;26:17–28. doi: 10.1016/j.tcb.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang ZY, Ye SL, Liu YK, Qin LX, Sun HC, Ye QH, Wang L, Zhou J, Qiu SJ, Li Y, Ji XN, Liu H, Xia JL, Wu ZQ, Fan J, Ma ZC, Zhou XD, Lin ZY, Liu KD. A decade’s studies on metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2004;130:187–196. doi: 10.1007/s00432-003-0511-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antonio MJ, Le A. Different tumor microenvironments lead to different metabolic phenotypes. Adv Exp Med Biol. 2018;1063:119–129. doi: 10.1007/978-3-319-77736-8_9. [DOI] [PubMed] [Google Scholar]
- 23.Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL, Zhao YM, Xiao YS, Sun QM, Ding ZB, Fan J. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol. 2015;46:587–596. doi: 10.3892/ijo.2014.2761. [DOI] [PubMed] [Google Scholar]
- 24.Carpenter RL, Paw I, Dewhirst MW, Lo HW. Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene. 2015;34:546–557. doi: 10.1038/onc.2013.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, Goodell MA, Taegtmeyer H. Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci U S A. 2016;113:10436–10441. doi: 10.1073/pnas.1601650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 27.Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, Feigenbaum AS, Grange DK, Hofstede FC, Holme E, Kirk EP, Korman SH, Morava E, Morris A, Smeitink J, Sukhai RN, Vallance H, Jakobs C, Salomons GS. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330:336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- 28.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sourbier C, Scroggins BT, Ratnayake R, Prince TL, Lee S, Lee MJ, Nagy PL, Lee YH, Trepel JB, Beutler JA, Linehan WM, Neckers L. Englerin A stimulates PKCtheta to inhibit insulin signaling and to simultaneously activate HSF1: pharmacologically induced synthetic lethality. Cancer Cell. 2013;23:228–237. doi: 10.1016/j.ccr.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantó C. The heat shock factor HSF1 juggles protein quality control and metabolic regulation. J Cell Biol. 2017;216:551–553. doi: 10.1083/jcb.201701093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011;14:91–103. doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 34.Suh DH, Kim HS, Kim B, Song YS. Metabolic orchestration between cancer cells and tumor microenvironment as a co-evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. Biochem Pharmacol. 2014;92:43–54. doi: 10.1016/j.bcp.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan Z, Xie N, Banerjee S, Cui H, Fu M, Thannickal VJ, Liu G. The monocarboxylate transporter 4 is required for glycolytic reprogramming and inflammatory response in macrophages. J Biol Chem. 2015;290:46–55. doi: 10.1074/jbc.M114.603589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78:5492–5503. doi: 10.1158/0008-5472.CAN-18-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16:425–441. doi: 10.1038/s41571-019-0203-7. [DOI] [PubMed] [Google Scholar]
- 39.Kourtis N, Lazaris C, Hockemeyer K, Balandrán JC, Jimenez AR, Mullenders J, Gong Y, Trimarchi T, Bhatt K, Hu H, Shrestha L, Ambesi-Impiombato A, Kelliher M, Paietta E, Chiosis G, Guzman ML, Ferrando AA, Tsirigos A, Aifantis I. Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat Med. 2018;24:1157–1166. doi: 10.1038/s41591-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.