

Abstract
Apolipoprotein E (apoE) and apoE-mimetic peptides exert prominent anti-inflammatory effects. We determined the anti-inflammatory effects of novel apoE receptor mimetics, composed of the LDL receptor-binding domain of apoE (aa 133-152, ApoEp) or ApoEp with 6 lysines (6KApoEp) or 6 aspartates added at the N-terminus (6DApoEp). BV2 microglia and human THP-1 monocytes were treated with lipopolysaccharide (LPS) in the absence or presence of ApoEp, 6KApoEp or 6DApoEp, followed by determination of pro-inflammatory tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) release by ELISA. As signaling intermediates of inflammation, Signal Transducer and Activator of Transcription 3 (STAT3), Janus-Activated Kinase2 (JAK2) and p38 and p44/42 MAPK phosphorylation levels were determined by Western blot analysis. In addition, we isolated splenocytes from female htau mice treated with 6KApoEp or 6K for 28 weeks, followed by determination of concanavalinA (conA)-mediated interferon gamma (IFNγ) release. 6KApoEp starting at 2.5 µM significantly reduced LPS-mediated TNFα and IL-6 secretion in BV2 and THP-1 cells in a dose-dependent manner. In BV2 cells, 6KApoEp reduced TNFα secretion more effectively than 6DApoEp and ApoEp, which was blocked by PCSK9 treatment, suggesting a role for LDL receptors. 6KApoEp also inhibited LPS-induced p44/42 MAPK, JAK2 and STAT3 phosphorylation, while enhancing p38 MAPK phosphorylation. In addition, conA induced significantly less IFNγ release in splenocytes derived from htau mice treated with 6KApoEp compared with those treated with 6K. Thus, 6KApoEp most effectively reduces LPS-mediated neuroinflammation by interacting with LDL receptors, thus representing a novel anti-inflammatory agent for treatment of neurodegenerative disease.
Keywords: Apolipoprotein E, tumor necrosis factor α (TNFα), interleukin-6 (IL6), janus-activated kinase 2 (JAK2), signal transducer and activator of transcription 3 (STAT3), mitogen-activated protein kinase (p38 and p44/42)
Introduction
Several lines of evidence show that apolipoprotein E (apoE) is involved in various functions in the periphery and central nervous system (CNS) including cholesterol metabolism and protection against oxidation and atherosclerosis [1-3]. ApoE is a 299 amino acid glycosylated protein of 34-kDa that serves as a ligand for low density lipoprotein family of receptors (LDLR) [4]. The LDLR-binding domain of apoE is between residues 133-152 of the protein, where multiple basic amino acids are present [5]. LDLR is a trans-membrane receptor that facilitates the endocytosis of plasma lipoprotein particles containing apoB100 and apoE in the periphery and brain, respectively [6,7].
ApoE in the CNS is synthesized and secreted by astrocytes and microglia [8]. Microglia are the primary immunocompetent cells within the brain and are believed to play a significant role in mediating neuronal injury in neurodegenerative disease and inflammation [9]. It is reported that microglia can be activated by LPS, promoting the production of inflammatory cytokines such as IL-6 and TNFα and reactive oxygen species [10]. One mechanism by which apoE might affect acute and chronic neurological disease is by modulating microglia and astrocytes [11-14]. Indeed, apoE knockout enhances microglial and astrocyte-mediated inflammatory responses in the brains of neonatal mice [15]. Thus, therapeutic agents targeting apoE and its receptors hold great promise as treatments to prevent or reduce neuroinflammation associated with neurodegenerative diseases such as Alzheimer’s disease.
TNFα, IL-6 and IFNγ are among the major pro-inflammatory cytokines which when administered to humans produce fever, inflammation, tissue destruction and, in some cases, shock and death [16,17]. p38 and p44/42 MAPK, Signal Transducer and Activator of Transcription 3 (STAT3) and Janus-Activated Kinases (JAK) play a role in LPS-induced IL-6 and TNFα expression [18-20], while IL-6 preferentially activates STAT3 phosphorylation through the activation of JAK [21]. IFNγ also induces two parallel but coordinated pathways which regulate the expression of proinflammatory cytokines, transcript synthesis by the JAK-STAT pathway and transcript stabilization by the p38 MAPK pathway [22]. Reducing the biological levels and activities of proinflammatory cytokines and its signaling intermediates should be considered in any potential anti-inflammatory strategy.
In the present study, we determined and compared the anti-inflammatory effects of small bioactive peptides formed from the LDLR binding domain of ApoE (ApoEp) alone or after modification by addition of 6 lysine or 6 aspartate residues at its N-terminus (6KApoEp and 6DApoEp). We compared the effectiveness of ApoEp, 6DApoEp and 6KApoEp in reducing LPS-induced proinflammatory TNF-α and IL-6 secretion in BV2 microglia and THP-1 monocytes. In addition, we determined the effectiveness of 6KApoEp in suppressing LPS-induced MAPK, JAK2 and STAT3 phosphorylation. Finally, we determined if treatment of htau mice with 6KApoEp reduces concanavalin A-mediated release of IFNγ from isolated splenocytes.
Materials and methods
Materials and reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin, trypsin-ethylenediaminetetraacetic acid (EDTA) and lipopolysaccharide (LPS) were purchased from Millipore-Sigma (Burlington, MA, USA). RPMI media was purchased from ATCC (Manassas, VA, USA). Antibodies for Western blot analysis were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). The mouse IL-6, TNFα and IFNγ enzyme-linked immunosorbent assays (ELISA) and RBC lysis kits were obtained from eBioscience Inc. Pharmingen (San Diego, CA, USA) and the human TNFα ELISA kit was obtained from Proteintech Group Inc. (Rosemont, IL USA). BV2 mouse microglia and THP1 human monocytes were obtained from ATCC.
BV2 microglia and THP-1 monocyte culture
BV2 microglia and THP-1 monocytes were maintained in DMEM or RPMI media, respectively, containing 10% FBS, 100 U/ml penicillin-streptomycin and 5% CO2 at 37°C. For treatment, cells were cultured in 24 well plates at 1 × 105 cells/well containing DMEM and 5% FBS overnight and then treated with LPS at 100 ng/ml in the absence or presence of 6KApoEp at 2.5, 5 or 10 µM for 3 h. In addition, BV2 microglia were treated with 6KApoEp, 6DApoEp or ApoEp at 10 µM alone or in combination with LPS (100 ng/ml). In order to determine the role of LDL receptors in the anti-inflammatory effect of 6KApoEp, BV2 microglia were pretreated with proprotein convertase subtilisin/kexin type 9 (PCSK9 [23,24]) at 10 µg/ml for 2 h followed by treatment with LPS (100 ng/ml) in the absence or presence of 6KApoEp (10 µM) for 1 h. Following treatment, the cultured media was collected for TNFα and IL-6 ELISA.
In order to determine the effect of 6KApoEp on STAT3, JAK2 and p38 and p44/42 MAPK phosphorylation, BV2 cells were treated with LPS (100 ng/ml) in the absence or presence of 6KApoEp (10 µM) for 15, 30 or 60 min, followed by Western blot analysis of cell lysates. BV2 cells were also treated with 6KApoEp at 10 µM, the p38 MAPK inhibitor SB203580 at 20 µM or the p44/42 MAPK inhibitor PD98059 at 50 µM for 1 h followed by treatment with LPS (100 ng/ml) for 1 h.
Primary splenocyte culture
All animal experiments were performed in accordance with the USF Institutional Animal Care and Use Committee and approved protocols and guidance of the National Institutes of Health. Transgenic female htau mice harboring all six isoforms of human MAPT (B6.Cg-Mapttm1(EGFP)Klt Tg(MAPT)8cPdav/J, Stock #005491, The Jackson laboratory, Bar Harbor, ME, USA) at 10 wk of age were treated with 6KApoEp (n = 6) or 6K (n = 6) daily at 10 µg/mouse/day for 28 wk via intra-nasal administration. Briefly, with a good grip on the loose skin over the scruff, the mouse was turned on its back with the neck parallel to the ground and 2 µl of 6KApoEp or 6K dissolved in PBS at 2.5 µg/µl was administered to each nostril via a micropipette held at a 45 degree angle and with an interval of 10-15 sec between nostrils.
After treatment, mice were euthanized and their spleens were harvested, homogenized in PBS and filtered through a strainer to obtain a single cell suspension. The splenocytes were centrifuged at 300-400×g at 4°C, resuspended in 5 ml RBC lysis buffer and allowed to stand at RT for 4-5 min. Twenty milliliters of PBS were then added to stop the reaction, the suspension was centrifuged again and the cells were resuspended in culture medium on 24-well plates at 3×106/well. These cells were treated with concanavalin A (conA) at 5 µg/ml between 24 and 48 h, followed by collection of conditioned media for IFNγ ELISA.
ELISA
The levels of IL-6, TNFα and IFNγ in conditioned media were measured using a mouse IL-6, mouse and human TNFα and mouse IFNγ ELISA kits according to the manufacturer’s instructions.
Western blot
For determination of the effects of 6KApoEp on STAT3, JAK2 and p38 and p44/42 MAPK phosphorylation, treated BV2 cells were washed in ice-cold PBS, lysed in cell lysis buffer and centrifuged at 4°C for 20 min. Supernatants were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and proteins were transferred onto PVDF membranes, blocked with blocking buffer, hybridized with anti-pSTAT3, -pJAK2, -p38 or -p42/44 MAPK antibody (1:1,000) overnight at 4°C, and then incubated with secondary antibody (1:500). Bands were visualized using chemiluminescence and band density was analyzed by Image J for Windows. Total protein concentration was determined by BCA assay.
Statistical analysis
All experiments were repeated a minimum of three times. For data analyses, SPSS (version 16.0; SPSS, Inc., Chicago, IL, USA) was used to test for data normality and homogeneity of variance. All data are present as mean ± S.E.M. Comparisons between two sample means were made using a t-test, while comparisons between multiple means were made by one-way analysis of variance (ANOVA). A value of P < 0.05 was considered to indicate a statistically significant difference.
Results
ApoE receptor mimetic 6KApoEp reduces LPS-stimulated TNFα and IL-6 production via activating LDL receptors
Previous studies have found that apoE reduces neuroinflammation in cell culture and mouse models [11-15]. In the present study, we developed a novel apoE mimetic (6KApoEp), derived from the apoE LDLR binding domain with 6 lysine residues attached at the N-terminus. We then treated BV2 microglia and THP-1 monocytes with 6KApoEp at 10 µM alone or LPS at 100 ng/ml in the absence or presence of 6KApoEp at 2.5, 5 or 10 µM for 3 h followed by determination of TNFα and IL-6 concentrations in conditioned media by ELISA (Figure 1A-C). In BV2 microglia, LPS increased TNFα from 85.7 ± 16.0 to 778 ± 27 pg/ml and IL-6 from 55.5 ± 7.0 to 446 ± 18.8 pg/ml. LPS also increased TNFα from 21.8 ± 7.5 to 925.0 ± 27.8 pg/ml in THP-1 monocytes. 6KApoEp reduced LPS-induced TNFα and IL-6 in a dose dependent fashion, starting at 5 µM for BV2 microglia and 2.5 µM for THP-1 monocytes. 6KApoEp at 10 µM maximally reduced LPS-induced TNFα and IL-6 by approximately 50% in BV2 microglia and at 5 µM maximally reduced LPS-induced TNFα by 90% in THP-1 monocytes.
Figure 1.
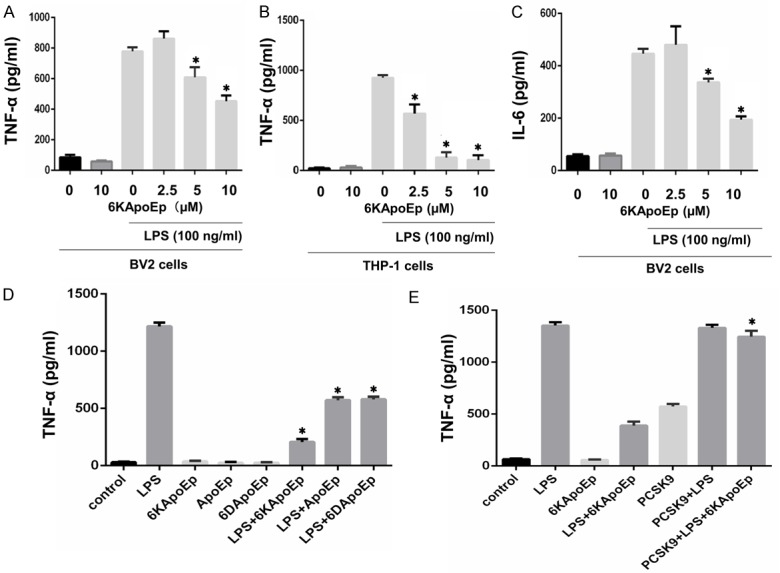
6KApoEp reduces LPS-induced TNFα and IL-6 secretion. Mouse microglia (BV2) and human monocytes (THP-1) were treated with 6KApoEp alone at 10 µM or LPS at 100 ng/ml in the absence or presence of 6KApoEp at 2.5, 5 or 10 µM for 3 hours followed by determination of TNFα and IL-6 release by ELISA (A-C). In addition, BV2 cells were treated with 6KApoEp, ApoEp or 6DApoEp at 10 µM in the absence or presence of LPS at 100 ng/ml for 3 h (D). In order to determine the role of LDLR in the anti-inflammatory effect of 6KApoEp, BV2 cells were treated with LPS at 100 ng/ml, 6KApoEp at 10 µM or LPS and 6KApoEp for 1 h alone or after pretreatment with PCSK9 at 10 µg/ml for 2 h (E). 6KApoEp reduced LPS-stimulated release of TNFα and IL-6 in a dose-dependent fashion more effectively than ApoEp and 6DApoEp, which was diminished by PCSK9. Results expressed as means ± S.E.M. of 3-5 independent experiments. Asterisk indicates P < 0.05 compared with LPS treatment alone (A-D) or LPS + 6KApoEp treatment (E).
In additional studies, we compared the effectiveness of ApoEp, 6KApoEp and 6D (6 aspartate residues) ApoEp in reducing LPS-induced TNFα production in BV2 cells (Figure 1D). While 6KApoEp, ApoEp and 6DApoEp alone had no effect on TNFα production, these peptides reduced LPS-induced TNFα production by approximately 80%, 50% and 50%, respectively, underscoring the superior effectiveness of 6KApoEp in reducing LPS-mediated neuroinflammation. We also pretreated BV2 cells with PCSK9 at 10 µg/ml for 2 h to downregulate LDL receptors before treatment with LPS in the absence or presence of 6KApoEp (Figure 1E). While PCSK9 alone had no effect on LPS-induced TNFα production, 6KApoEp failed to decrease TNFα production in the presence of PCSK9. Thus, 6KApoEp appears to reduce LPS-induced TNFα production by activating LDL receptors.
6KApoEp reduces LPS-induced p44/42 MAPK and JAK2/STAT3 phosphorylation
Previous studies found that LPS-induced TNFα release required phosphorylation of both p38 and p44/42 MAPK [25,26]. Thus, we determined the effect of 6KApoEp on LPS-induced p38 and p44/42 MAPK phosphorylation in BV2 microglia. BV2 microglia were treated with LPS at 100 ng/ml for 0, 15, 30 and 60 min, in the absence or presence of 6KApoEp at 10 µM, followed by determination of p38 and p44/42 MAPK phosphorylation by Western blot (Figure 2A-D). LPS alone increased both p38 and p44/42 MAPK phosphorylation in a time-dependent manner, reaching maximal phosphorylation at 60 min of treatment. 6KApoEp enhanced LPS-induced p38 MAPK phosphorylation between 15 and 60 min of treatment and p44/42 MAPK phosphorylation at 15 min of treatment, but reduced p44/42 MAPK phosphorylation at 60 min of treatment.
Figure 2.

6KApoEp attenuates LPS-induced p44/42 MAPK phosphorylation. BV2 microglia were treated with LPS at 100 ng/ml in the absence or presence of 6KApoEp at 10 µM for 0, 15, 30 or 60 min. Total (p38 and p44/42) and phosphorylated p38 and p44/42 MAPK (P-p38 and P-p44/42) in cell lysates was determined by WB, utilizing anti-total p38 (Cell Signaling, #9212), anti-total p44/42 (#4695), anti-phospho p38 (#4511) and anti-phospho p44/42 antibodies (#4370) (A-D). In addition, BV2 cells were pretreated with the p38 MAPK inhibitor SB203580 at 20 µM, the p44/42 MAPK inhibitor PD98059 at 50µM or 6KApoEp at 10µM for 60 min followed by treatment with LPS at 100 ng/ml for 60 min (E-H). Representative blots are shown for each condition and band density ratio of phospho-p38 to total p38 and phospho-p44/42 to total p44/42 MAPK determined by densitometry analysis are shown below each blot. Full non-adjusted images of WB shown in Figure S1. PD98059 and 6KApoEp reduced LPS-induced p44/42 MAPK phosphorylation while enhancing p38 MAPK phosphorylation. In contrast, SB203580 reduced LPS-induced p38 MAPK phosphorylation while enhancing p44/42 MAPK phosphorylation. Total p38 and p44/42 MAPK were similar across conditions. Results expressed as means ± S.E.M of three experiments. Asterisk indicates P < 0.05 compared with LPS alone.
Further studies suggest that p38 and p44/42 MAPK reciprocally regulate each other by a cross talk mechanism [27,28]. In order to determine if 6KApoEp reciprocally regulate p38 and p44/42 MAPK phosphorylation, we treated BV2 cells with LPS in the absence or presence of the p38 MAPK inhibitor SB203580, the p44/42 MAPK inhibitor PD98059 or 6KApoEp followed by determination of p38 and p42/44 MAPK phosphorylation by WB (Figure 2E-H). SB203580 decreased LPS-induced p38 MAPK phosphorylation, while reciprocally enhancing p44/42 MAPK phosphorylation. In contrast, PD98059 and 6KApoEp decreased LPS-induced p44/42 MAPK phosphorylation, while reciprocally enhancing p38 MAPK phosphorylation. Thus, 6KApoEp appears to reciprocally enhance p38 MAPK phosphorylation by inhibiting p44/42 MAPK.
In addition to activation of MAPK, the proinflammatory effect of LPS may be mediated by activation of the JAK2/STAT3 pathway [19-21]. In order to determine if 6KApoEp inhibits this pathway, we treated BV2 cells with LPS in the absence or presence of 6KApoEp, followed by determination of JAK2 and STAT3 phosphorylation by WB (Figure 3A-D). LPS increased JAK2 and STAT3 phosphorylation in a time-dependent manner, reaching maximal phosphorylation at 30-60 min of treatment. 6KApoEp treatment blocked LPS-induced JAK2 phosphorylation, while reducing STAT3 phosphorylation, between 15 and 60 min of treatment. Thus, the anti-inflammatory effect of 6KApoEp may be mediated by inhibition of the LPS-induced JAK2/STAT3 pathway.
Figure 3.
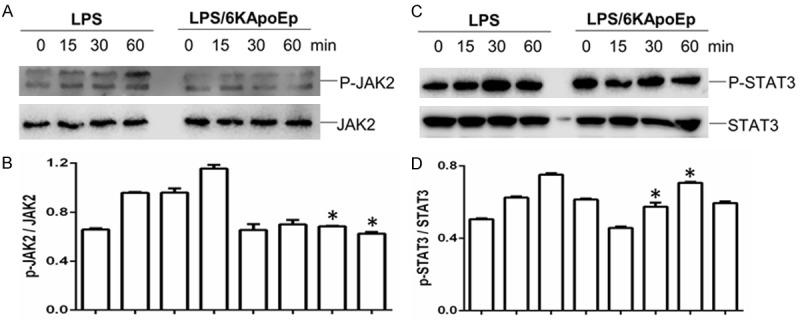
6KApoEp attenuates LPS-induced JAK2 and STAT3 phosphorylation. BV2 cells were treated with LPS at 100 ng/ml in the absence or presence of 6KApoEp at 10 µM for 0, 15, 30 or 60 min. Total (JAK2 and STAT3) and phosphorylated JAK2 and STAT3 (p-JAK2 and p-STAT3) in cell lysates was then determined by WB, utilizing anti-total JAK2 (Cell Signaling, #3230), anti-total STAT3 (#9132), anti-phospho-JAK2 (#3774) and anti-phospho STAT3 antibodies (#9131). Representative blots are shown for each condition and band density ratios of p-JAK2 to JAK2 and p-STAT3 to STAT3 determined by densitometry analysis are shown below each blot. Full non-adjusted images of WB shown in Figure S2. LPS-induced JAK2 and STAT3 phosphorylation was reduced by 6KApoEp between 15 and 60 min of treatment. Total JAK2 and STAT3 levels were similar across treatment conditions. Results are expressed as mean ± S.E.M. Asterisk indicate P < 0.05 compared with LPS alone.
Splenocyte IFNγ release is reduced by 6KApoEp treatment in vivo
In order to determine if 6KApoEp has an anti-inflammatory effect in vivo, we treated female htau mice with 6KApoEp or 6K daily at 10 μg/mouse/day for 28 wk by intranasal administration. After treatment, mice were euthanized and splenocytes were isolated and treated with concanavalin A (conA) at 5 µg/ml overnight. IFNγ release from splenocytes isolated from 6KApoEp-treated mice was significantly reduced compared with those isolated from 6K-treated mice (Figure 4). Thus, 6KApoEp treatment in vivo reduces splenocyte release of IFNγ.
Figure 4.
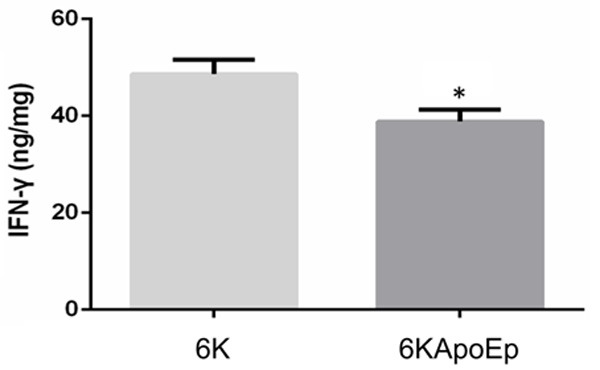
6KApoEp treatment in vivo reduces splenocyte IFN-γ release. Htau female mice at 10 weeks of age were treated with 6KApoEp (n = 6) or 6K (n = 6) daily at 10 µg/mouse/day for 28 wk by intranasal administration, followed by isolation of splenocytes, overnight treatment with concanavalin A (ConA) and determination of IFNγ levels in conditioned media by ELISA Splenocyte IFNγ release was significantly reduced by 6KApoEp compared with 6K treatment. ELISA results presented as mean ± S.E.M. of IFNγ (ng/mg total intracellular protein). Asterisk indicates P < 0.05 compared with 6K treatment.
Discussion
In the present study, we tested the anti-inflammatory properties of a novel apoE mimetic peptide (6KApoEp), consisting of the apoE LDLR binding domain (aa 133-152, ApoEp) linked with 6 lysine residues at its N-terminus. 6KApoEp reduced LPS-induced TNFα and IL6 production in a dose-dependent fashion starting at 5 µM in BV2 microglia and 2.5 µM in THP-1 monocytes (Figure 1A-C), together with reduced p44/42 MAPK, JAK2 and STAT3 phosphorylation (Figures 2 and 3). This reduction of LPS-induced TNFα elicited by 6KApoEp was greater than that elicited by ApoEp alone or by 6DApoEp, consisting of ApoEp with 6 aspartates linked at its N-terminus (Figure 1D). In addition, this anti-inflammatory effect of 6KApoEp was blocked by treatment with PCSK9, which downregulates LDLR [23,24], suggesting a role for LDLR activation (Figure 1E). 6KApoEp treatment of htau mice by intra-nasal administration also reduced ConA-mediated release of IFNγ by isolated splenocytes (Figure 4). Together these results indicate that 6KApoEp may be a novel anti-inflammatory peptide for treatment of neurodegenerative diseases.
Inflammation in the central nervous system is a common feature of many age-related neurodegenerative diseases. Activation of microglia and p38 and p44/42 MAPK pathways occur in most chronic neurodegenerative diseases, such as AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), as well as in their animal models [29]. LPS directly activates p38 and p44/42 MAPK (ERK1/2) in microglial cell culture, increasing the production of proinflammatory cytokines such as TNFα that are believed to be involved in neuronal damage [30]. Since excessive proinflammatory cytokine production can further activate p38 MAPK in microglia, astrocytes and neurons, a potential positive feedback loop can result that exacerbates disease. IL-6 is a member of the IL-6-type cytokine family with pro- and anti-inflammatory properties. They activate Janus tyrosine Kinase family members (JAK1 and JAK2), leading to the activation of transcription factors of the Signal Transducer and Activator of Transcription (STAT) family [31,32]. Conversely, JAK2 activation can further mediate IL-6 production, since JAK2-specific inhibition blocks IL6 production as well as STAT3 phosphorylation in the peripheral circulation and cancer cells [33,34]. The present study suggests that 6KApoEp potentially reduces neuroinflammation by reducing p44/42 MAPK, JAK2 and STAT3 phosphorylation.
Apolipoprotein E has emerged as a pivotal anti-inflammatory agent in a number of neurodegenerative diseases, including AD, PD, cerebrovascular disease (CVD), multiple sclerosis (MS), traumatic brain injury (TBI), HIV-encephalitis, cerebral palsy (CP), vascular dementia, and ischemic stroke [13,35,36]. In addition, an increasing body of evidence suggests that apoE and apoE-mimetic peptides exert prominent anti-inflammatory effects [12]. ApoE signaling via VLDL-R or apoER2 promotes macrophage conversion from the pro-inflammatory M1 to the anti-inflammatory M2 phenotype, representing a novel anti-inflammatory activity of apoE [37]. In addition, there is mounting evidence that apoE influences innate as well as acquired immunity. Effects on innate immunity have been demonstrated by the ability of apoE to inhibit stimulation of cultured neutrophils by urate crystals [38] as well as lymphocyte proliferation by interleukin-2 and -4 [39,40]. In addition, apo E-deficient mice have impaired innate immune responses to Listeria monocytogenes [41] and reduced production of anti-inflammation factors such as IL-10 mainly from microglia and TGF-beta mainly from astrocytes, resulting in severe inflammation [15]. Thus, targeting apoE may be a potential anti-inflammatory approach for the prevention and treatment of various neurodegenerative and peripheral diseases in humans [42].
It is notable that 6KApoEp enhanced p38 MAPK while reducing p44/42 MAPK phosphorylation in BV2 microglia, suggesting that 6KApoEp has a primary action to reduce p44/42 MAPK phosphorylation, which reciprocally enhances p38 MAPK phosphorylation by a crosstalk mechanism. Likewise, treatment with the p44/42 MAPK inhibitor PD 98059 increased p38 MAPK phosphorylation, while treatment with the p38 MAPK inhibitor SB203580 increased p44/42 MAPK phosphorylation. Such reciprocal regulation or crosstalk of MAPK p38 and p44/42 has become increasingly recognized as a means to allow “reciprocal equilibrium” between MAPK p38 and p44/42 phosphorylation, involving concordant activation of protein kinases and phosphatases [27,28].
Our results using cultured BV2 microglia and THP1 monocytes support a role for 6KApoEp as an anti-inflammatory agent with potential for the treatment of neurodegenerative disease. In order to confirm these findings in vivo, we treated htau mice with 6KApoEp by daily intranasal (i.n.) administration for 28 weeks and determined its effect on IFNγ release by isolated splenocytes. Treatment with 6KApoEp reduced concanavalin A (ConA)-induced splenocyte IFNγ release compared with treatment with 6K alone (Figure 4). Inflammation of the CNS during multiple sclerosis and experimental autoimmune encephalomyelitis is characterized by increased levels of IFNγ, a cytokine not normally expressed in the CNS [43]. IFNγ has been shown to regulate T cell proliferation and cytokine production, with profound implication for onset and progression of disease [44,45]. In addition, IFNγ is known to induce two parallel but coordinated pathways regulating the expression of genes encoding proinflammatory cytokines, transcript synthesis regulated by the JAK-STAT pathway and transcript stabilization regulated by p38 MAPK [22]. By reducing IFNγ release, 6KApoEp and potentially other ApoEp derived peptides hold great promise for therapeutic management of neuroinflammation.
Acknowledgements
We acknowledge Jun Tian and Anran Fan for their technical supports and Brian Giunta for his critical discussion. Supported by NIH/NIA (R01AG050253 and R21AG055116) to Dr. Jun Tan.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Go GW, Mani A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med. 2012;85:19–28. [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson LA, Olsen RH, Merkens LS, DeBarber A, Steiner RD, Sullivan PM, Maeda N, Raber J. Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol Dis. 2014;64:150–162. doi: 10.1016/j.nbd.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Handattu SP, Monroe CE, Nayyar G, Palgunachari MN, Kadish I, van Groen T, Anantharamaiah GM, Garber DW. In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-beta pathology. J Alzheimers Dis. 2013;36:335–347. doi: 10.3233/JAD-122377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci U S A. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon H, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. 2005;74:535–562. doi: 10.1146/annurev.biochem.74.082803.133354. [DOI] [PubMed] [Google Scholar]
- 7.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 8.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa Y, Chiba K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals (Basel) 2014;7:1028–1048. doi: 10.3390/ph7121028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, Bennett ER. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. 2001;167:74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- 12.Datta G, White CR, Dashti N, Chaddha M, Palgunachari MN, Gupta H, Handattu SP, Garber DW, Anantharamaiah GM. Anti-inflammatory and recycling properties of an apolipoprotein mimetic peptide, Ac-hE18A-NH(2) Atherosclerosis. 2010;208:134–141. doi: 10.1016/j.atherosclerosis.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP, Dawson HN. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma. 2010;27:1983–1995. doi: 10.1089/neu.2010.1396. [DOI] [PubMed] [Google Scholar]
- 14.Maezawa I, Maeda N, Montine TJ, Montine KS. Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation. 2006;3:10. doi: 10.1186/1742-2094-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Xu X, Dou H, Hua Y, Xu J, Hui X. Apolipoprotein E knockout induced inflammatory responses related to microglia in neonatal mice brain via astrocytes. Int J Clin Exp Med. 2015;8:737–743. [PMC free article] [PubMed] [Google Scholar]
- 16.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 17.Popko K, Gorska E, Stelmaszczyk-Emmel A, Plywaczewski R, Stoklosa A, Gorecka D, Pyrzak B, Demkow U. Proinflammatory cytokines Il-6 and TNF-alpha and the development of inflammation in obese subjects. Eur J Med Res. 2010;15(Suppl 2):120–122. doi: 10.1186/2047-783X-15-S2-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 19.Lee C, Lim HK, Sakong J, Lee YS, Kim JR, Baek SH. Janus kinase-signal transducer and activator of transcription mediates phosphatidic acid-induced interleukin (IL)-1beta and IL-6 production. Mol Pharmacol. 2006;69:1041–1047. doi: 10.1124/mol.105.018481. [DOI] [PubMed] [Google Scholar]
- 20.Kimura A, Naka T, Muta T, Takeuchi O, Akira S, Kawase I, Kishimoto T. Suppressor of cytokine signaling-1 selectively inhibits LPS-induced IL-6 production by regulating JAK-STAT. Proc Natl Acad Sci U S A. 2005;102:17089–17094. doi: 10.1073/pnas.0508517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ke X, Hu G, Fang W, Chen J, Zhang X, Yang C, Peng J, Chen Y, Sferra TJ. Qing Hua Chang Yin inhibits the LPS-induced activation of the IL-6/STAT3 signaling pathway in human intestinal Caco-2 cells. Int J Mol Med. 2015;35:1133–1137. doi: 10.3892/ijmm.2015.2083. [DOI] [PubMed] [Google Scholar]
- 22.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 23.Blanchard V, Khantalin I, Ramin-Mangata S, Chemello K, Nativel B, Lambert G. PCSK9: from biology to clinical applications. Pathology. 2019;51:177–183. doi: 10.1016/j.pathol.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 24.Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee YB, Schrader JW, Kim SU. p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine. 2000;12:874–880. doi: 10.1006/cyto.2000.0688. [DOI] [PubMed] [Google Scholar]
- 26.Pyo H, Jou I, Jung S, Hong S, Joe EH. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. Neuroreport. 1998;9:871–874. doi: 10.1097/00001756-199803300-00020. [DOI] [PubMed] [Google Scholar]
- 27.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem. 2002;277:14884–14893. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Yang H, Tachado SD, Capo-Aponte JE, Bildin VN, Koziel H, Reinach PS. Phosphatase-mediated crosstalk control of ERK and p38 MAPK signaling in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:5267–5275. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- 29.Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- 30.van der Bruggen T, Nijenhuis S, van Raaij E, Verhoef J, van Asbeck BS. Lipopolysaccharide-induced tumor necrosis factor alpha production by human monocytes involves the raf-1/MEK1-MEK2/ERK1-ERK2 pathway. Infect Immun. 1999;67:3824–3829. doi: 10.1128/iai.67.8.3824-3829.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babon JJ, Varghese LN, Nicola NA. Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol. 2014;26:13–19. doi: 10.1016/j.smim.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, Findlay J, Ackland L, Ahmed N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer. 2009;100:134–144. doi: 10.1038/sj.bjc.6604794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, Rodgers JD, Haley P, Kantarjian H, Fridman JS, Verstovsek S. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lien E, Andersen GL, Bao Y, Gordish-Dressman H, Skranes JS, Vik T, Blackman JA. Apolipoprotein E polymorphisms and severity of cerebral palsy: a cross-sectional study in 255 children in Norway. Dev Med Child Neurol. 2013;55:372–377. doi: 10.1111/dmcn.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A, Nofer JR. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1160–1168. doi: 10.1161/ATVBAHA.111.222745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terkeltaub RA, Dyer CA, Martin J, Curtiss LK. Apolipoprotein (apo) E inhibits the capacity of monosodium urate crystals to stimulate neutrophils. Characterization of intraarticular apo E and demonstration of apo E binding to urate crystals in vivo. J Clin Invest. 1991;87:20–26. doi: 10.1172/JCI114971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelly ME, Clay MA, Mistry MJ, Hsieh-Li HM, Harmony JA. Apolipoprotein E inhibition of proliferation of mitogen-activated T lymphocytes: production of interleukin 2 with reduced biological activity. Cell Immunol. 1994;159:124–139. doi: 10.1006/cimm.1994.1302. [DOI] [PubMed] [Google Scholar]
- 40.Mistry MJ, Clay MA, Kelly ME, Steiner MA, Harmony JA. Apolipoprotein E restricts interleukin-dependent T lymphocyte proliferation at the G1A/G1B boundary. Cell Immunol. 1995;160:14–23. doi: 10.1016/0008-8749(95)80004-3. [DOI] [PubMed] [Google Scholar]
- 41.Roselaar SE, Daugherty A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res. 1998;39:1740–1743. [PubMed] [Google Scholar]
- 42.Giau VV, Bagyinszky E, An SS, Kim SY. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr Dis Treat. 2015;11:1723–1737. doi: 10.2147/NDT.S84266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Millward JM, Caruso M, Campbell IL, Gauldie J, Owens T. IFN-gamma-induced chemokines synergize with pertussis toxin to promote T cell entry to the central nervous system. J Immunol. 2007;178:8175–8182. doi: 10.4049/jimmunol.178.12.8175. [DOI] [PubMed] [Google Scholar]
- 44.Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164:2759–2768. doi: 10.4049/jimmunol.164.5.2759. [DOI] [PubMed] [Google Scholar]
- 45.Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R. Opposing Roles of Interferon-Gamma on Cells of the Central Nervous System in Autoimmune Neuroinflammation. Front Immunol. 2015;6:539. doi: 10.3389/fimmu.2015.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.