

Abstract
Sex hormone metabolism is altered during mammary gland tumorigenesis, and different metabolites may have different effects on mammary epithelial cells. This study aimed to evaluate associations between urinary sexual metabolite levels and breast cancer risk among premenopausal women of Mainland China. The molecular metabolism of the cancer-related metabolites was also explored based on the clinical data. The sex hormone metabolites in the urine samples of patients with breast cancer versus normal healthy women were analyzed comprehensively. Among many alterations of sex hormone metabolisms, 4-hydroxy estrogen (4-OH-E) metabolite was found to be significantly increased in the urine samples of patients with breast cancer compared with the normal healthy controls. This was the most important risk factor for breast cancer. Several experiments were conducted in vitro and in vivo to probe this mechanism. 4-Hydroxyestradiol (4-OH-E2) was found to induce malignant transformation of breast cells and tumorigenesis in nude mice. At the molecular level, 4-OH-E2 compromised the function of spindle-assembly checkpoint and rendered resistance to the anti-microtubule drug. Further, transgenic mice with high expression of CYP1B1, a key enzyme of 4-hydroxy metabolites, were established and stimulated with estrogen. Cancerous tissue was found to appear in the mammary gland of transgenic mice.
Keywords: Breast cancer, estrogen metabolism, 4-hydroxy estrogen, SAC, spindle-assembly checkpoint
Introduction
During the last 20 years, studies have suggested several hypotheses about how estrogen metabolism might influence the risk of breast cancer. Accruing evidence, primarily laboratory based, suggests substantial differences in the genotoxic, mutagenic, and proliferative activities of various estrogen metabolites (EMs) and their contributions to mammary carcinogenesis [1-4]. Estrogenic hormones, estrone (E1), estradiol (E2), and estriol (E3), are metabolized in the C-2, C-4, or C-16 pathways by a series of oxidizing enzymes in the cytochrome P450 family, resulting in 2-hydroxyestrogen (2-OH-E), 4-hydroxyestrogen (4-OH-E) and 16-hydroxyestrogen (16α-OH-E), respectively. These metabolites can have stronger or weaker estrogenic activity and may often determine the mutagenic or carcinogenic potential of an estrogen, and thus increase a woman’s risk of breast, uterine and other cancers.
The assumption that specific hydroxyl estrogens may have important biological activities, favoring breast cancer onset and/or progression, has been repeatedly reported and debated [5-8]. On the basis of the concept that 2-OH-E and 16α-OH-E represent mutually major pathways of estrogen metabolism and that the resulting hydroxylated metabolites may, respectively, behave as estrogen antagonists and agonists [9], much attention has been devoted in the last two decades to define the potential role of an EM ratio of 2- to 16-EMs in relation to breast cancer [10-14]. Recently, some studies have begun to focus on the C-4 pathway EMs or their ratio. 4-Hydroxyestradiol (4-OH-E2) with a catechol structure readily undergoes oxidation to electrophilic estradiol-3,4-quinone that can react with DNA to form depurinating adducts. The reactive quinone derived from 4-OH-E2 may induce DNA damage on specific genes involved in carcinogenesis or induce microsatellite instability [15-20].
Sex steroid metabolism depends on several factors, including a person’s genetic makeup, lifestyle, diet and environment. Understanding estrogen metabolism and the things that affect it offers significant opportunities to reduce cancer risks, particularly of breast cancer. Although some studies have analyzed the relationship of 2-OH-E1 and 16α-OH-E1 with breast cancer risk in humans [21-24], other individual metabolites, estrogen metabolism pathways and other sex steroids have not been evaluated systematically in human populations. Particularly, little attention has been given to 4-OH-E in epidemiological studies and molecular mechanisms. A high-performance gas chromatography/tandem mass spectrometry (GC/MS-MS) assay was developed to measure concurrently estrogens and EMs, progesterone and metabolites, and testosterone and metabolites in urine with high sensitivity, specificity, accuracy and reproducibility. In this study, associations between urinary sexual metabolite levels and breast cancer risk among premenopausal women of Mainland China were evaluated. The molecular metabolism of the cancer-related metabolites based on the clinical data was also studied.
Materials and methods
Study population and urine collection
All experiments and all methods were approved by the Ethics Committee of The First Affiliated Hospital of Nanjing Medical University (2012-SR-159). All patients with breast cancer were diagnosed in The First Affiliated Hospital of Nanjing Medical University during July 2012 and December 2014. Normal controls were the nurses working in The First Affiliated Hospital of Nanjing Medical University. The exclusion criteria were as follows: women with a history of irregular menstruation or bilateral oophorectomy, those with chronic or acute liver disease, and those on hormone therapy within 3 months before recruitment into the study. The BMI was calculated as weight in kilograms divided by height in meters squared.
Informed consent was obtained from all participants. All women participating in the study were asked to collect the first-morning urine using a standard testing paper provided by the hospital. The testing papers were hung at room temperature till they were totally dried. Dried urine samples were stored at -20°C.
Laboratory analysis
The dried urine strips were extracted with both water and methanol and cleaned up using C18 solid phase extraction cartridges. Eluents were dried down and enzymatically hydrolyzed. Liquid extracts of the hydrolysis mixture were dried down and derivatized to trimethylsilyl ethers using MSTFA:NH4I:ethanethiol. Then, 2 mL of the final product was injected onto an Agilent 7890A Gas Chromatograph, coupled to an Agilent 7000B Triple Quadrupole Mass Spectrometer, and all the results were analyzed using the MassHunter software.
Statistical method
The Student t test was used for comparing the levels of EMs in the breast cancer and control groups. Unconditional logistic regression models were used to estimate ORs and 95% CIs for individual EM, pathway EM, and the ratios of the pathways. The progesterone metabolites and male hormone metabolites were also estimated in the same way. The results were adjusted by age (20-30, 30-40, ≥40 years) and BMI (<18, 18-25, >25 kg/m2). Multivariate logistic regression analysis was performed for assessing the risk of different EMs. All the EMs were entered using forward (pe = 0.99, pr = 0.999) stepwise approach. The quartile-specific ORs and 95% CIs were used trend Chi square test in additional analyses for individual EM, pathway EM, and the ratios of the pathways.
For all analyses, all tests of significance were two-tailed, and probability values of <0.05 were considered statistically significant. Analyses and forest plots were performed using Stata, version 11.1 (StatCorp Inc., TX, USA).
Cell culture
Human MCF-7 breast cancer cells and human MCF 10A breast cells were obtained from the American Type Culture Collection. The MDA-MB-231-ESR1 cell line was kindly presented by Dr. Ding Qiang. MCF-7 and MDA-MB-231-ESR1 cells were precultured in the phenol red-free Dulbecco’s modified Eagle’s medium (DMEM) containing 5% charcoal-stripped fetal bovine serum (FBS) for 4 days before further treatments. After adding different concentrations of 2-OH-E2 with or without 17β-E2 (10-9 M), 2-OH-E2 (10-7 M) combined with Tamoxifen (10-6 M), and 17β-E2 (10-9 M) combined with Tamoxifen (10-6 M) for 24 h, the cells were collected for assays of estrogenic/antiestrogenic effects.
MCF-10A cells were routinely cultured in phenol red-free DMEM media supplemented with 10% FBS and penicillin-streptomycin (100 mg/mL each) and incubated at 37°C in a humidified atmosphere containing 5% CO2. The cell culture media, serum, and antibiotics were purchased from Gibco. The cells were treated with 10-8 M 4-OH-E2 for 8 weeks (MCF10A-H) or with 10-8 M E2 for 8 weeks (MCF10A-E).
2-OH-E2, 4-OH-E2, 17β-E2 and 4-OH-Tamoxifen (TAM) were purchased from Sigma-Aldrich. These drugs were first diluted in dimethylsulfoxide and then in a growth medium. The final dimethylsulfoxide concentration was below 0.01%.
Estrogen-responsive luciferase reporter assay
MCF-7 and MDA-MB-231-ESR1 were transiently transfected with a firefly luciferase reporter plasmid construct (pERE4-Luc) containing four copies of the ERE. A Renilla luciferase reporter plasmid, pRL-SV40-Luc, was used as an internal control for transfection efficiency. The Lipofectamine 3000 (Invitrogen) reagent was used for transfection. Luciferase activities were measured using the Dual Luciferase Assay System. Absolute ERE promoter firefly luciferase activity was normalized against the Renilla luciferase activity to correct for transfection efficiency. Triplicate wells were performed for each transfection condition, and data were collected from at least three independent experiments.
Wound-healing assay
Breast cancer cells were seeded into six-well plates and allowed to grow until 100% confluence was reached. Then, the cell layer was gently scratched through the central axis using a sterile plastic tip, and the loose cells were washed away. The cell motility was quantified by measuring the distance between the invading fronts of cells in three randomly selected microscopic fields (200×). The cells were fed for another 24 h in a serum-free medium. The cells were photographed before and after the serum-free culture period using light microscopy, and the rate of wound area was calculated using Image J.
Colony formation assay
The effect of 4-OH-E2 and E2 on breast cancer colony formation was tested. The breast cancer cells were plated in six-well plates at a density of 500 cells/well for 2 weeks with or without 4-OH-E2 treatment (10-8 M). Then, the medium was removed and the cells were washed twice with phosphate-buffered saline (PBS) and stained for 15 min using the Giemsa solution, rinsed with tap water, and dried at room temperature. The colonies in each well were counted, and all cell colonies contained 50 or more cells.
Cell migration assay
The cells growing in the log phase were trypsinized, resuspended in a serum-free medium, and seeded into Boyden chambers (8 mM pore size with polycarbonate membrane). The chambers were then inserted into the Transwell apparatus (Costar, MA, USA). The medium with 10% FBS (600 ml) was added to the lower chamber. After incubation for 24 h, the cells on the top surface of the insert were removed by wiping with a cotton swab. The cells that migrated to the bottom surface of the insert were stained in 0.1% crystal violet for 30 min, rinsed in PBS, and then subjected to microscopic inspection. Values of migration were obtained by counting five high-power fields (100×) per membrane, and the average of three independent experiments was taken.
Tumorigenesis in nude mice
MCF10A, MCF10A-E and MCF10A-H cells (1×106 cells in 0.1 mL of PBS) were subcutaneously orthotopically injected into 2 sides of the mammary fat pads of 10 female nude BALB/C mice (4-6 weeks’ old, weighing 18-22 g). The mice were randomly divided into two groups. Two kinds of cells were injected into each side of the mice. For group I, MCF10A-H was injected on the right and MCF10A on the left, while for group II, MCF10A-H was injected on the right and MCF10A-E on the left. E2 valerate (Progynova, Bayer), 0.125 mg/week, was injected on the back skin of each mouse.
The growth of tumors was followed up for 4 weeks. The tumor volume was measured weekly using a caliper and calculated as (tumor length × width2)/2. After 4 weeks, the mice were sacrificed and checked for the final tumor size. Mouse studies were conducted according to the Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Nanjing Agricultural University. All the samples were collected according to the ethical guidelines of the Declaration of Helsinki and approved by the ethics and research committee of the First Affiliated Hospital of Nanjing Medical University.
Microarray data analysis
Cellular RNA from MCF10A, MCF10A-E and MCF10A-H cells was extracted using the TRIzol reagent (Invitrogen) and quality controlled as directed in the Affymetrix Expression Technical Manual. RNA (50 ng) was used to produce biotin-labeled cDNA, which was hybridized to Affymetrix GeneChip Human Transcriptome Array 2.0. The gene intensities between MCF10A, MCF10A-E and MCF10A-H cells were compared. The genes were considered significantly differentially expressed when the fold change was ≥2. The slides were scanned using the GeneChip Scanner 3000 (Affymetrix, CA, USA) and Command Console Software 3.1 (Affymetrix) with default settings. Raw data were normalized using the Expression Console.
Immunofluorescence staining assay
MCF10A and MCF10A-H cells were grown in six-well plates with cover slides, treated or untreated with docetaxel (an anti-tubulin drug work on SAC). IF-IC staining was performed as described in the cell signal technology protocol. Primary antibodies used were Phosphor-Histone H3 (Ser10) (D2C8) XP Rabbit (Cell Signaling, 3377) (1:800). The MI refers to percentage of the number of cells in the mitosis phase against the total number of cells. In this study, the cells showed fluorescence of pH3 (Ser10), indicating entry in the mitosis phase. Then, 10 high-power fields with light microscope slices were randomly selected. The fluorescent cells were counted, and the MI was calculated.
Transgenic mice
A plasmid containing the full coding region of the murine CYP1B1 gene was prepared, following the proximal promoter of the murine EF1α gene, to generate the C57BL/6J-CYP1B1 transgenic mouse. The mice were backcrossed with C57BL/6J mice for more than six generations. Genotyping involved extracting genomic DNA from mice tails and carrying out PCR with the following primers: forward, 5’-TTTCTCTTCATCTCCATCCTCGCTCA-3’, and reverse, 5’-CAAACGCACACCGGCCTTATTCCA-3’. A PCR amplification was performed with Taq polymerase from Qiagen under the following conditions: 30 cycles of 94°C for 3 min, 94°C for 30 s, 62°C for 30 s, 72°C for 1 min 30 s, and 72°C for 10 min. The PCR products were visualized on a 1% agarose gel. C57BL/6J mice were obtained from the Vital River Laboratory Animal Technology Co. (Beijing, China). All animal experiments were undertaken in accordance with the United States National Institutes of Health Guidelines for Care and Use of Laboratory Animals, with the approval by the Institutional Animal Care and Use Committee of Nanjing Medical University (ID: 1601172). The mice were maintained in the Animal Core Facility of Nanjing Medical University under specific-pathogen-free conditions and housed on a 12-h light/dark cycle with food and water available ad libitum.
Experimental animals and treatment
For animal studies, the mice were earmarked before grouping and then randomly separated into groups by an independent person. Then, female mice of 6-7 weeks were randomized into four groups (n = 4-5/group): C57BL/6J mice (controls), C57BL/6J mice implanted subcutaneously with 90-day slow-release 17β-E2 pellets, C57BL/6J-CYP1B1 mice (controls), and C57BJ/6J-CYP1B1 mice implanted subcutaneously with 90-day slow-release E2 pellets (0.25 mg, Innovative Research America, FL, USA). After 90 days of treatment, E2-implanted mice were euthanized with corresponding controls. The breast, uterus, ovaries, lung, and liver were removed for further analysis.
Analysis of histological features
Breast, uterus, ovaries, lung and liver samples were fixed in 10% buffered formalin, embedded in paraffin, sectioned and stained with H&E.
Immunohistochemistry analysis
Deparaffinized, rehydrated and acid-treated mice breast sections (5-μm thick) were treated with H2O2, trypsin, and blocked with normal goat serum. The sections were incubated with specific mouse monoclonal antibodies against ER, PR, HER-2 and Ki67 (American Diagnostica Inc.) at 25°C for 30 min, followed by incubation with the biotin-conjugated secondary anti-mouse antibodies (Dako, Glostrup, Denmark). The colorimetric detection was performed by a standard indirect streptavidin-biotin immunoreaction method using Dako’s Universal LSAB Kit according to the manufacturers’ protocols. Using images taken at 100× magnification, a total of eight random images per time point (n = 8), were used for image analysis. The negative cases were confirmed with two independent experiments. All stained sections were reviewed by two pathologists. Five fields of view were randomly assessed for each group. Positive immunostaining was defined as >20% of tumor cells showing immunoreactivity to the antibody.
Results
This study included 42 patients with breast cancer and 37 healthy women. All the participants were premenopausal. The breast cancer risk factors, including age, body mass index (BMI), age at menarche, smoke and alcohol, were analyzed. On average, age was 39.03 ± 3.48 years and 39.43 ± 5.39 years and BMI was 21.89 ± 1.74 kg/m2 and 22.52 ± 2.86 kg/m2 for healthy women and women with breast cancer, respectively. None of the participants smoked or consumed alcohol.
The results for individual EMs of E1, E2 and E3, and nine EMs in three hydroxylation pathways of estrogen metabolism are presented in Table 1. The parent estrogen (E1, E2 and E3) levels were not significantly higher in the breast cancer group compared with the control group. Three EMs in the 2-hydroxylation pathways were higher in patients with breast cancer than in healthy women. However, no significant difference was found in 16-hydroxylation pathways of the two groups. The levels of 4-OH-E1 increased in the breast cancer group, but the level of 4-methoxy-E2 did not significantly increase (Table 1).
Table 1.
Estrogen metabolites of premenopausal patients with breast cancer and normal controls
| Control | Breast cancer | P | OR (95% CI)* | P * | |
|---|---|---|---|---|---|
| Estrone (E1) | 3.09 ± 2.18 | 3.94 ± 3.41 | 0.23 | 1.47 (0.84-2.57) | 0.18 |
| Estradiol (E2) | 1.24 ± 0.81 | 1.49 ± 0.94 | 0.23 | 1.33 (0.83-2.15) | 0.24 |
| Estriol (E3) | 1.75 ± 1.14 | 1.55 ± 0.86 | 0.39 | 0.8 (0.5-1.26) | 0.34 |
| 2-OH-E1 | 0.89 ± 0.68 | 1.54 ± 1.08 | 0.01 | 2.31 (1.27-4.2) | 0.01 |
| 2-OH-E2 | 0.25 ± 0.22 | 0.25 ± 0.15 | 0.99 | 1.04 (0.66-1.63) | 0.88 |
| 2-Methoxy-E1 | 0.29 ± 0.19 | 0.73 ± 0.53 | <0.01 | 6.97 (2.26-21.52) | <0.01 |
| 2-Methoxy-E2 | 0.04 ± 0.03 | 0.07 ± 0.05 | <0.01 | 3.29 (1.66-6.52) | <0.01 |
| 4-OH-E1 | 0.18 ± 0.13 | 0.59 ± 0.41 | <0.01 | 17.88 (4.05-78.9) | <0.01 |
| 4-OH-E2 | 0.09 ± 0.04 | 0.15 ± 0.09 | <0.01 | 3.36 (1.6-7.07) | <0.01 |
| 4-Methoxy-E1 | 0.03 ± 0.02 | 0.05 ± 0.04 | 0.01 | 3.06 (1.35-6.95) | 0.01 |
| 4-Methoxy-E2 | 0.02 ± 0.02 | 0.03 ± 0.02 | 0.22 | 1.39 (0.84-2.28) | 0.2 |
| 16α-OH-E1 | 0.96 ± 0.58 | 0.87 ± 0.55 | 0.51 | 0.89 (0.56-1.4) | 0.61 |
EMs are the estimated standardized regression coefficients.
P and OR (95% CI) were adjusted by age and BMI.
As a result of the changes in EM levels, the ratio of 4-hydroxylation metabolites of 4-OH-E (4-OH-E1 + 4-OH-E2):E (E1 + E2) showed a significant rise in the patient group versus the control group. In the C-2 pathway, 2-OH-E (2-OH-E1 + 2-OH-E2):E (E1 + E2) ratio was statistically significant. Interestingly, compared with the previous reports [14,25-27], the 2-OH-E1: 16α-OH-E1 ratio was higher in the patient group versus the control group, while the ratio of 2-OH-E (2-OH-E1 + 2-OH-E2):4-OH-E (4-OH-E1 + 4-OH-E2) was lower in the patient group versus control group (Table 2).
Table 2.
Estrogen metabolite ratios of patients with breast cancer and normal controls
| Control | Breast cancer | P | OR (95% CI)* | P * | |
|---|---|---|---|---|---|
| 2-OH-E:E | 0.27 ± 0.12 | 0.35 ± 0.16 | 0.01 | 1.494 (0.936-2.385) | 0.092 |
| 4-OH-E:E | 0.08 ± 0.05 | 0.15 ± 0.06 | <0.001 | 6.43 (2.1-19.63) | <0.001 |
| 16α-OH-E1:E1 | 0.36 ± 0.18 | 0.25 ± 0.13 | 0.01 | 0.453 (0.27-0.759) | 0.003 |
| 2-OH-E1:16α-OH-E1 | 1.08 ± 0.81 | 1.97 ± 1.28 | <0.001 | 2.81 (1.45-5.45) | 0.003 |
| 2-OH-E:4-OH-E | 4.26 ± 2.4 | 2.45 ± 0.85 | <0.001 | 0.17 (0.06-0.49) | <0.001 |
P and OR (95% CI) were adjusted by age and BMI.
The stepwise regression analysis revealed 4-OH-E1 to be the most important factor of breast cancer risk. At subsequent steps of the forward-stepping procedure, E1 (P = 0.0023), 2-OH-E2, and 2-methoxy-E1 were also found to be independent and statistically significant factors (Table 3).
Table 3.
Associated factors identified in forward stepwise logistic regression model
| EM | OR | Std. Err. | Z | P | 95% CI |
|---|---|---|---|---|---|
| 4-OH-E1 | 156.52 | 239.84 | 3.30 | 0.0001 | 7.77-3154.37 |
| E1 | 0.08 | 0.07 | -2.97 | 0.0023 | 0.01-0.42 |
| 2-OH-E2 | 0.13 | 0.11 | -2.44 | 0.0162 | 0.03-0.67 |
| 2-Methoxy-E1 | 50.35 | 86.77 | 2.27 | 0.0229 | 1.72-1474.96 |
Variables entered into the model: estrone (E1), estradiol (E2), estriol (E3), 2-OH-E1, 4-OH-E1, 16α-OH-E1, 2-methoxy-E1, 4-methoxy-E1, 2-OH-E2, 4-OH-E2, 2-methoxy-E2 and 4-methoxy-E2. EMs are the estimated standardized regression coefficients. OR indicates breast cancer risk.
We tested the progesterone metabolites and male hormone metabolites in the urine samples of 29 patients with breast cancer and 22 normal healthy women (Table 4). Among many metabolites, the levels of one progesterone metabolite and two male hormone metabolites significantly increased in the breast cancer group compared with the normal control group.
Table 4.
Androgen and progesterone metabolites of patients with breast cancer and normal controls
| Control | Breast cancer | P | OR (95% CI)* | P * | |
|---|---|---|---|---|---|
| Dehydroepiandrosterone | 70.827 ± 87.698 | 105.461 ± 149.062 | 0.346 | 1.004 (0.998-1.009) | 0.229 |
| Androstenedione | 9.625 ± 10.045 | 15.979 ± 16.009 | 0.116 | 1.04 (0.985-1.097) | 0.155 |
| Androsterone | 489.995 ± 258.941 | 629.159 ± 347.064 | 0.129 | 1.001 (0.999-1.003) | 0.203 |
| Etiocholanolone | 637.655 ± 315.028 | 607.3 ± 338.631 | 0.75 | 1 (0.998-1.001) | 0.637 |
| Testosterone | 0.888 ± 1.104 | 3.566 ± 4.719 | 0.014 | 1.491 (1.001-2.219) | 0.049 |
| 5α-Dihydrotestosterone | 0.519 ± 0.662 | 0.616 ± 0.571 | 0.586 | 1.244 (0.479-3.233) | 0.654 |
| 5α-Androstanediol | 6.531 ± 7.231 | 7.709 ± 4.319 | 0.482 | 1.021 (0.904-1.153) | 0.742 |
| 5β-Dihydrotestosterone | 0.41 ± 0.943 | 0.211 ± 0.343 | 0.309 | 0.58 (0.212-1.588) | 0.289 |
| 5β-Androstanediol | 7.004 ± 6.435 | 6.27 ± 4.115 | 0.629 | 0.968 (0.868-1.08) | 0.562 |
| Pregnanediol | 856.489 ± 1400.928 | 791.032 ± 716.645 | 0.832 | 1 (0.999-1) | 0.892 |
| Allopregnanediol | 37.876 ± 50.431 | 30.621 ± 31.011 | 0.538 | 0.997 (0.983-1.011) | 0.641 |
| Allopregnanolone | 6.936 ± 11.501 | 5.186 ± 6.273 | 0.499 | 0.981 (0.919-1.047) | 0.564 |
| 3α-OH-Progesterone | 1.069 ± 0.984 | 1.037 ± 0.636 | 0.891 | 0.954 (0.478-1.905) | 0.893 |
| 20α-OH-Progesterone | 3.928 ± 4.485 | 8.678 ± 7.521 | 0.013 | 1.13 (1.016-1.256) | 0.024 |
| 5α-Dihydroprogesterone | 1.347 ± 1.204 | 1.237 ± 0.809 | 0.703 | 0.923 (0.525-1.624) | 0.782 |
| Progesterone | 0.81 ± 0.728 | 1.277 ± 0.75 | 0.034 | 2.372 (1.002-5.616) | 0.049 |
P and OR (95% CI) were adjusted by age and BMI.
Figure 1 shows forest plots of odd ratios for breast cancer by quartiles. When examining each hydroxylation pathway as a ratio to the parent estrogens, an elevated ratio of the 4-hydroxylation pathway to parent estrogens, 4-OH-E1 + 4-OH-E2:(E1 + E2), was found to be significantly associated with an increment in risk (P-trend <0.001). No significant trend was observed for the ratio of 2-hydroxylation estrogens to parent estrogens, while the ratio of 2-hydroxylation estrogens to 2-methoxy estrogens negatively correlated with breast cancer risk (P-trend <0.001). The ratio of 16α-OH-E1 to E1 was inversely related to the risk (P-trend = 0.003). EMs were also examined as ratios based on metabolic pathways. As the levels of 4-pathway EMs of patients with breast cancer increased far more steeply than the levels of 2-pathway EMs, the ratios of 2-pathway to the 4-pathway was inversely related to risk (P-trend <0.001). Compared with previous studies, this study indicated that breast cancer risk increased with increasing levels of 2-OHE1:16α-OHE1 ratio (P-trend <0.001) (Figure 1).
Figure 1.
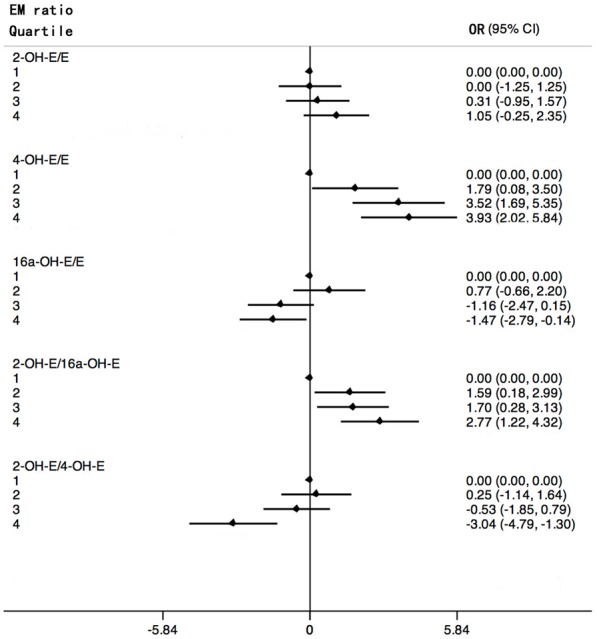
Forest plots of odd ratios (in natural logarithm) for breast cancer and 95% confidence intervals (in natural logarithm) by quartiles of estrogen metabolites ratios. ORs (black rhombus) are shown on a In scale. Black lines represent 95% CIs. Quartiles are abbreviated 1, 2, 3, 4.
Estrogenic activity of 4-OH-E2
Estrogen response is mediated by two closely related members of the nuclear receptor family of transcription factors, ER-α and ER-β. The modulatory effect of 4-OH-E2, 2-OH-E2 and E2 on the transcriptional activity of ER-α in human breast cancer cells indicated that ER-α was the major ER in mammary epithelia (Figure 2A). The ER-positive MCF-7 cells and ER-α transiently transfected MDA-MB-231-ESR1 were treated with 2-OH-E2, 4-OH-E2 and E2. Treating both cells with 4-OH-E2 and E2 resulted in a significant increase in the estrogen-responsive reporter ERE4-Luc activity relative to basal levels in control cells. 4-OH-E2 and E2 increased the ER-α reporter activity by fourfold compared with the control group (MCF-7:E2, 2.843 ± 0.746, 4-OHE2, 4.017 ± 0.231, 2-OHE2, 12.603 ± 0.315; 231-ESR1:E2, 3.657 ± 0.335, 2-OH-E2, 1.005 ± 0.112, 4-OH-E2, 4.035 ± 0.050). The results indicated that although 2-hydroxy metabolite 2-OHE2 had limited estrogenic activity, 4-hydroxy metabolite 4-OHE2 had much stronger estrogenic activity, which was even more potent than that of E2 in MCF-7 cells.
Figure 2.
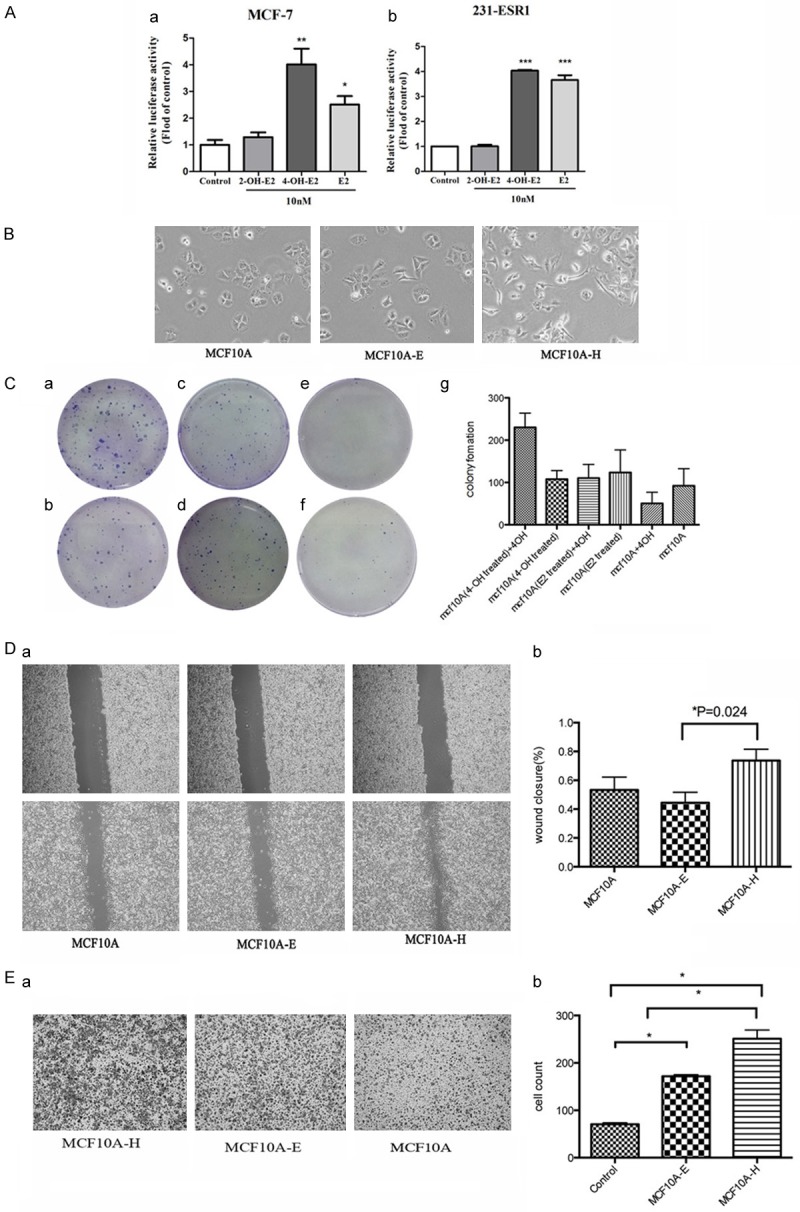
(A) Estrogen-responsive luciferase reporter assay showed that 4-OH-E2 had estrogen activity. We treated MCF-7 and MDA-MB-231-ESR1 cells with 4-OH-E2, 2-OH-E2 and E2. 4-OH-E2 and E2 increased ER-α reporter activity about 4 fold over the control group. We treated MCF10A cells with 4-OH E2 (MCF10A-H) and E2 (MCF10A-E) both at 10-8 M for 2 months. (B) The effects of 4-OH E2 (MCF10A-H) and E2 (MCF10A-E) on MCF10A cells. Microscope images show that 4-OH E2 induced cell-to-cell contact and led to a higher spreading with more formation of filo podia by comparison with the E2 and control groups. (C) The cell transformation was carried out by colony formation. MCF-10A-H cells (a, b), MCF10A-E cells (c, d) and MCF10A cells (e, f) were seeded in six-well plates at a density of 500 cells/well. After 24 hours of seeding, cells were exposed to 4OH E2 (a, c, e) or not (b, d, f), The control groups were cultured in red-free DMEM media supplemented with 10% fetal bovine serum. The 4-OH E2 group was treated with 10-8 M 4-OH E2. Colony efficiency was determined by a count of the number of Colonies (more than 50 cells). The MCF10A-H cells in the culture medium with 10-8 M 4-OH E2 have much more colonies than other groups (P = 0.0013). 4-OH E2 may inhibit the growth of the MCF10A and MCF10A-E (g). (D) Cell migration was measured by using the Culture-Inserts Wound healing assay. Images of wound repair were taken at 0, 24 h after wound. The distance of wound closure is shown by area at 24 h. Representative photographs and quantification are shown, original magnification, ×200 (a), Cell migration (%) was quantified by calculating the wound width (b). (E) Transwell migration assay. Representative photographs and quantification are shown. Columns: average of three independent experiments, original magnification, ×200.
4-OH-E2 induced transformed phenotype in breast epithelial cells
The normal mammary epithelial cells MCF10A were treated with 4-OH-E2 (MCF10A-H) and E2 (MCF10A-E) for 8 weeks. As shown in Figure 2B, treatment with 4-OH-E2 induced morphological changes with a higher spreading and increased formation of filopodia compared with control cells or cells treated with E2.
The effect of 4-OH-E2 on anchorage-independent growth was also investigated in this study. Soft agar colony assays demonstrated that the anchorage-independent growth of MCF10A cells was stimulated by 4-OH-E2 but not by E2 (Figure 2C).
The proliferation of cells was examined using colony formation assays. The MCF10A cell colony formation was analyzed (Figure 2Ca-Cf), and the colony formations were calculated (Figure 2Cg). MCF-10A-H cells (MCF10A treated with 4-OH-E2 for 8 weeks) (a, b), MCF10A-E cells (MCF10A treated with E2 for 8 weeks) (c and d), and MCF10A cells (e and f) were seeded in six-well plates at a density of 500 cells/well. After 24 h of seeding, the cells were exposed to 10-8 M 4-OH-E2 (a, c and e) or not (b, d and f) for 2 weeks. In the culture medium with 10-8 M 4-OH-E2, MCF-10A-H grew 2.14 times faster (230 ± 19.6 vs 107.7 ± 11.86, P = 0.0059). However, 4-OH-E2 did not accelerate the growth of MCF10A-E (110.3 ± 18.66 vs 123.7 ± 30.66, P = 0.7291) or MCF10A (50.3 ± 15.3 vs 92 ± 23.43, P = 0.2107).
The migration rate was measured using wound-healing and transwell migration assays. As shown in Figure 2D and 2E, the wound closure of MCF10A treated with 4-OH-E2 was 1.39-fold higher than that of MCF10A. However, the wound closure of MCF10A was 1.17-fold higher than that of MCF10A treated with E2 (the wound closure: MCF10A-H, 0.74 ± 0.04; MCF10A, 0.53 ± 0.06; MCF10A-E, 0.45 ± 0.05) (Figure 2D). The effect of 4-OH-E2 and E2 on the MCF10A cell migration behavior was also investigated using a transwell migration assay (Figure 2E). The cell migration significantly increased in MCF10A-H and MCF10A-E cells. The number of migrated MCF10A-H cells increased by nearly threefold compared with that of MCF10A (P<0.05), while that of MCF10A-E increased by twofold. (the cells through the Boyden chambers: MCF10A, 70.63 ± 3.00; MCF10A-E, 171.8 ± 3.41; MCF10A-H, 251 ± 18.37, P<0.05).
4-OH-E2 induced tumorigenesis
The physiological and pathological effects of 4-OH-E2 on the mammary glands and development of breast cancer were analyzed in two separate experimental models. The tumorigenic effect of 4-OH-E2 was first evaluated in athymic nude mice models (Figure 3). The tumor growth of 4-OH-E2-treated MCF10A-H cells versus parental nontreated MCF10A cells was compared. As expected, while mice injected with MCF10A cells into the mammary fat pad of nude mice did not form tumors, four out of five mice (80%) injected with MCF10A-H cells formed tumors after 2 weeks. Next, the tumor growth of MCF10A-H cells versus E2-treated MCF10A-E cells was compared. While 60% (3/5) of mice injected with MCF10A-E cells were capable of forming tumors by 2 weeks after injection, all mice (5/5) injected with MCF10A-H cells formed tumors. Furthermore, MCF10A-E cells formed much smaller tumors compared with those formed by MCF10A-H cells (P<0.01) (Figure 3C).
Figure 3.
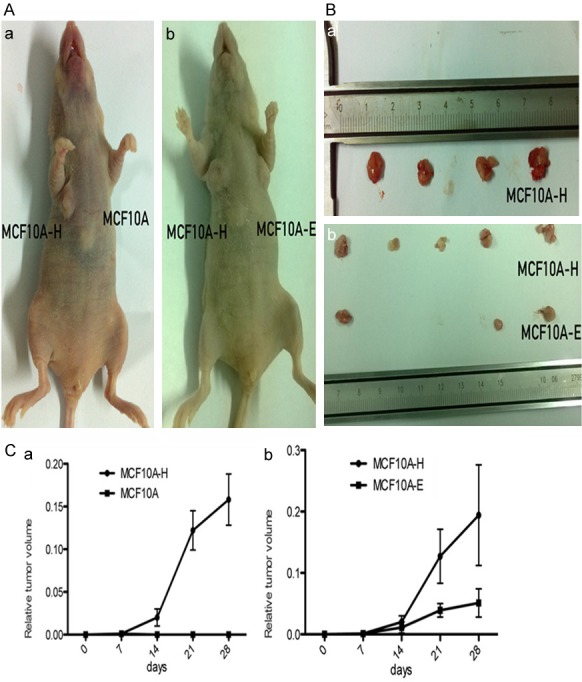
4-OH E2 suppressed tumor growth in nude mice. The MCF10A-H cells MCF10A-E cells and MCF10A cells were subcutaneously orthotopically injected into mammary two sides of fat pads of BALB/C female nude mice. (A, a) group I MCF10A-H cells and MCF10A cells were injected into two sides of fat pads of 5 mice. 4 mice have MCF10A-H cells tumor on one side. MCF10A cells could not form tumor. (b) group II MCF10A-H cells and MCF10A-E cells were injected into two sides of fat pads of 5 mice. All mice have MCF10A-H cells tumor on one side, and MCF10A-E cells formed smaller tumor volume in 3 mice on the other side. (B) Photographs of tumors from mice. (a) group I, (b) group II. (C) Relative tumor volume of mice. (a) group I, (b) group II.
The effects of 4-OH-E2 on the mammary glands and development of mammary tumors were evaluated in a more relevant C57BL/6J-CYP1B1 transgenic mice model expressing transgene CYP1B1, which is a cytochrome P450 superfamily enzyme converting E2 into 4-OH-E2. Female mice were supplemented with or without E2 pellets (0.25 mg/90-day slow release). The effect of transgene expression on the mammary glands was assayed by morphological analyses of the gland after 90 days of E2 exposure (Figure 4). Histological evaluation of hematoxylin & eosin (H&E)-stained mammary sections revealed a normal monolayer and similar morphology in the glands from the virgin transgenic mice and the control littermate, indicating that the expression of transgene did not alter the mammary gland development. However, a significant alteration of the gland morphology with the appearance of mammary carcinoma in the transgenic mouse was observed when the mice were supplemented with E2 (Figure 4B). The epithelium of transgenic glands exhibited a disorganized structure with respect to the ordinate arrangement of the wild-type epithelium. Furthermore, while a normal mammary gland had a single layer of epithelial cells, the glands from transgenic mouse displayed a highly proliferative stage characterized by areas of multilayers. The duct lumen was narrowed and blocked by the hyperplastic glandular epithelial cells in the mammary glands of C57BL/6J-CYP1B1 + E2 mice, indicating a highly proliferative capability of the cells in the gland. Indeed, a strong immunohistochemical staining of Ki67 was only observed in the C57BL/6J-CYP1B1 + E2 group (Figure 4B), whereas Ki67 signal was undetectable in the mammary glands from other groups (Figure 4A). No morphological differences were observed in the tissues of liver and lung among all groups of mice. Taken together, multiple mammary tumors were only present in mice of the C57BL/6J-CYP1B1 + E2 group, but not in mice of other groups.
Figure 4.
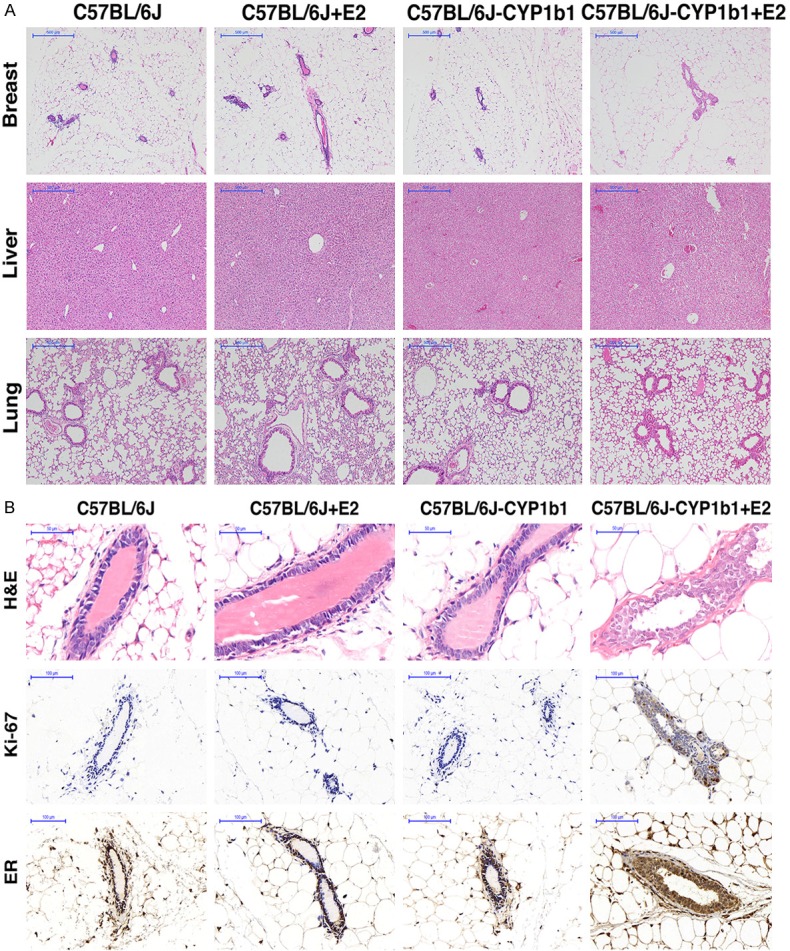
A. Immunohistochemical analysis of four groups mice. H&E staining of breast in mice from C57BL/6J group, C57BL/6J + E2 group, C57BL/6J-CYP1B1 group and C57BL/6J-CYP1B1 + E2 group. 400×, bar = 50 μm. B. Representative images for H&E staining of breast, liver and lung samples from four groups. B. Expression of ki67 and ER protein in mammary gland of four groups mice. Cells stained brown indicate positive expression. Mammary gland from C57BL/6J group, C57BL/6J + E2 group, C57BL/6J-CYP1B1 group and C57BL/6J-CYP1B1 + E2 group mice were stained for Ki67 and ERα.
4-OH-E2 attenuated the SAC function
The MCF-10A cell line was treated with 10-8 M 4-OH-E2 or E2 for 8 weeks, and the total RNA was extracted. Microarray experiments were conducted using Affymetrix GeneChip Human Transcriptome Array 2.0 to determine the changes in the specific genes encoding proteins related to pathological alterations in intracellular signaling pathways in response to 4-OH-E2 and E2.
After E2 treatment, 18 genes in MCF10A cells were differentially expressed, while 4-OH-E2 treatments induced changes in 440 genes by at least twofold, compared with controls. In this study, 955 genes changed in MCF10A-H compared with MCF 10A-E. Among them, two genes were significantly downregulated. Centromere protein E (CENPE), whose gene expression changed the most, was an important protein in the spindle-assembly checkpoint (SAC). CENPE was downregulated by at least ninefold after 4-OH-E2 treatment compared with MCF10A-E and MCF10A cells. TTK was another gene downregulated by fourfold after treatment (MCF10A vs MCF10A-H, 4.09; MCF10A-E vs MCF10A-H, 5.69). TTK encoded a dual-specificity protein kinase that was found to be a critical mitotic checkpoint protein for accurate segregation of chromosomes during mitosis. The results were verified using real-time polymerase chain reaction (PCR) (Figure 5A).
Figure 5.
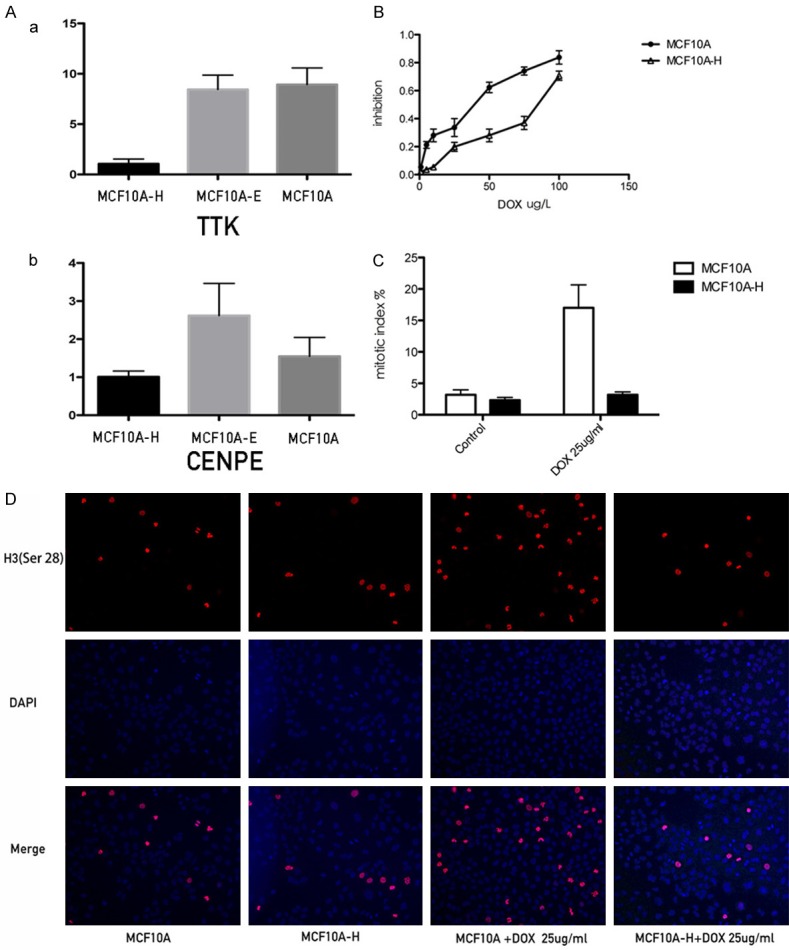
4-OH E2 renders resistance to antimicrotubule drugs. Docetaxel inhibits the proliferation of the cells by blocking the cell cycle to G2/M phase, and develops its function by inducing cells apoptosis. The blocking was far more effective in MCF10A cells than MCF10A-H cells. A. The realtime results of CENPE and TTK in MCF-10A, MCF-10AE, MCF10AH cells. B. The inhibition of MCF10A and MCF10A-H cell; C. Mitotic index of MCF10A and MCF10A-H cells treated by Doxetaxel 25 ug/l for 48 hours or not; D. Phosphor-Histone H3 (Ser10) immunofluorescence staining of MCF10A-H and MCF10A cells treated by Doxetaxel 25 ug/l for 48 hours or not.
Docetaxel is a class of anti-tubulin drugs. It works on SAC, incurs cell cycle arrest in the G2/M phase, and induces apoptosis. Figure 5B shows the inhibitory effect of docetaxel on MCF10A and MCF10A-H. The half maximal inhibitory concentration of MCF-10A was higher than that of MCF10A-H (MCF-10A, 66.76 ± 14.20; MCF10A-H, 23.44 ± 3.82, P<0.05). The mitotic index (MI) of MCF10A and MCF10A-H cells was also measured. As shown, MCF10A mitotic index was 3.167 ± 1.941 and MCF10A-H mitotic index was 2.333 ± 1. 033. MCF10A and MCF10A-H cells were treated with DOX with a concentration of 25 mg/mL. The MCF10A mitotic index was four times higher than before (17 ± 8.165), while the mitotic index of MCF10A-H slightly increased (3.167 ± 0.477). The MI of MCF10A-H did not rise as high as that of MCF10A (P = 0.0037) after treatment with DOX (Figure 5C and 5D).
Discussion
The continuing controversy over the health benefits and risk of estrogen, particularly its metabolites, is a complex and evolving story. This was the first comprehensive study to evaluate sex hormone metabonomics in premenopausal women with breast cancer versus age-matched healthy women, and the effects of some metabolites on mammary epithelial and breast cancer cells. First, a comprehensive set of sex hormones and their metabolites were analyzed in urine samples of premenopausal women with or without breast cancer. This study compared the levels of 12 estrogens and EMs, 9 androgens and androgen metabolites, and 7 progestogens and progestogen metabolites in the urine of premenopausal women with breast cancer and normal health women using a quantitative GC/MS-MS method. While no significant change was found in androgen and progesterone metabolites, the most significant alteration was an increase in 4-hydroxy metabolites relative to other metabolites in EMs in patients with breast cancer. Second, the biological relevance of increased 4-OH-E metabolism to breast cancer development was investigated in mammary epithelial cells and in vivo experimental models. It was found that 4-OH-E2 not only induced malignant transformation of breast epithelial cells in vitro but also stimulated tumor growth in the xenograft model and induced mammary carcinomas in the transgenic mice model expressing CYP1B1, a key enzyme of 4-hydroxy metabolites. Third, the molecular mechanisms underlying 4-OH-E metabolites induced malignant transformation. At the molecular level, 4-OH-E2 compromised the function of SAC and thus rendered genome instability.
Although E2 implication in mammary tumor promotion and progression has been based mostly on evidence for ER-mediated mitogenic effects, no significant difference was observed in the urine samples of patients with breast cancer and normal controls in the present study, which was consistent with previous studies [28]. Much attention has been devoted in the last two decades to define the potential role of an imbalance between 2- and 16α-hydroxylation activities in relation to breast cancer based on the concept that C-2 and C-16α hydroxylations represent major pathways of estrogen metabolism and that the resulting hydroxylated metabolites may, respectively, behave as estrogen antagonists and agonists. The lower ratio of 2-OHE1:16α-OHE1 had been hypothesized to be associated with breast cancer risk. Compared with previous findings [14,25-27], a significant higher urinary 2/16 ratio was observed in patients with breast cancer in this study. 2-OH-E2 was found to increase in the breast cancer group. The levels of 16α-OH-E1 did not change significantly in the breast cancer group versus normal controls. It was interesting to note that the levels of 2-methylated metabolites were also significantly elevated in the breast cancer group, while the 4-hydroxy-methylated metabolites were not. The 2-hydroxy metabolites were more likely to be methylated, thereby losing the weak estrogenic effect and protective role in preventing the onset and progression of breast cancer.
Among many alterations of EMs in the breast cancer group, the most significant one in this study was an increase in 4-hydroxy metabolites. Urine 4-OH-E1 in the patients with breast cancer was three times higher than that in healthy women, while other EMs changed less. The best indicator that reflected the risk of breast cancer was the ratio of 4-hydroxy metabolites to total estrogen. The findings of this study were consistent with the assumption that the metabolic conversion of E2 to 4-OH-E2 and its additional activation to reactive semiquinone/quinone intermediates may be genotoxic, leading to various types of DNA damage, and have a direct role as tumor initiators [15-20].
Consistent with the increased 4-OH-E in patients with breast cancer, 4-OH-E2 could neoplastically transform MCF10A mammary epithelial cells as indicated by in vitro increased colony-forming capacity in soft agar and cell migration and in vivo tumorigenesis in nude mice. The results indicated that 4-OH-E2 might function as an initiator for the malignant transformation of mammary glands [29]. A transgenic mice model expressing CYP1B1 was established to investigate whether 4OH-E could initiate the malignant transformation of mammary glands. CYP1B1 is highly expressed in a variety of cancers. Recent studies showed that the cytochrome P450 CYP1B1 variant increased the production of 4-OH-E2, and that a polymorphism in CYP1B1 increased the risk of endometrial carcinoma [30]. Using these transgenic mice expressing high levels of endogenous 4-OH-E, it was demonstrated that 4OH-E induced highly proliferative phenotype of mammary epithelial cells and mammary carcinomas, while the transgene had no effect on the ER levels. The findings of the present study were in contrast to an earlier report showing that administering 4-OH-E2 failed to induce mammary tumorigenesis in the animal model [31]. The reason for this difference might be that the transgenic model presented in this study was more relevant to in vivo physiological or pathological situations in which it provided sustainable high levels of endogenous 4-OH-E.
Microarray analyses were performed to study 4-OH-E-regulated genes so as to investigate the molecular mechanisms underlying the 4-OH-E-induced malignant transformation. Among many differentially regulated genes by 4-OH-E2 versus E2, the two most varied gene expressions were CENPE and TTK, which were downregulated by more than nine- and fourfold upon treatment of MCF10A cells with 4-OH-E2, respectively. Both CENPE and TTK were mitotic genes involved in the integrity of microtubules. CENPE is one of the key elements of SAC. At this checkpoint, the integrity of microtubules and the completion of chromosome alignment with spindle microtubules were monitored. Because of the presence of this surveillance mechanism, normal cells proceeded to anaphase as long as chromosomes remained unattached to mitotic spindles. It was anticipated that the significant downregulation of CENPE compromised the checkpoint function of SAC and led to genomic instability. One of the major mechanisms underlying anti-microtubule drug resistance involved acquired inactivation of SAC.
To prove the hypothesis, the cells were treated with docetaxel, an antitumor drug acting in SAC. It was found that the mitotic checkpoint was attenuated in 4-OH-E2-treated cells (Figure 1). Previous studies reported that the proportion of patients with ovarian cancer having high expression of CYP1B1 was more likely due to paclitaxel resistance [32]. These patients with high expression of CYP1B1 had higher levels of endogenous 4-OH-E. The present study provided a mechanistic link between 4-OH-E and docetaxel resistance.
Conclusions
In summary, our study is the most comprehensive test in the world until now. We tested over 30 metabolites of estrogen, progesterone and androgen metabolic pathways. In this comprehensive study of sexual hormone metabolism and risk of breast cancer in premenopausal women, we consider the ratio of 2-OHE1:16α-OHE1 is not a clear marker of breast cancer risk in premenopausal women. However, the ratio of 4-hydroxy metabolites to total estrogen is the best indicator reflects the risk of breast cancer. Our study found 4-OH-E2 induced carcinogenesis by destroying the SAC and induced the abnormal mitosis. The malignant cells transformed by 4-OH-E2 was hard to kill by antitumor drug. Thus, the relative proportions of 4-hydroxy estrogenic metabolites and 4-hydroxy estrogens are predictors of breast cancer risk and are also important factors in the prognosis of breast cancer patients and the choice of treatment.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81572907 and 81702830), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_1483).
Disclosure of conflict of interest
None.
References
- 1.Cavalieri E, Rogan E. Catechol quinones of estrogens in the initiation of breast, prostate, and other human cancers: keynote lecture. Ann N Y Acad Sci. 2006;1089:286–301. doi: 10.1196/annals.1386.042. [DOI] [PubMed] [Google Scholar]
- 2.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 3.Chang M. Dual roles of estrogen metabolism in mammary carcinogenesis. Bmb Rep. 2011;44:423–434. doi: 10.5483/BMBRep.2011.44.7.423. [DOI] [PubMed] [Google Scholar]
- 4.Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuhrman BJ, Schairer C, Gail MH, Boyd-Morin J, Xu X, Sue LY, Buys SS, Isaacs C, Keefer LK, Veenstra TD, Berg CD, Hoover RN, Ziegler RG. Estrogen metabolism and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2012;104:326–339. doi: 10.1093/jnci/djr531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eliassen AH, Spiegelman D, Xu X, Keefer LK, Veenstra TD, Barbieri RL, Willett WC, Hankinson SE, Ziegler RG. Urinary estrogens and estrogen metabolites and subsequent risk of breast cancer among premenopausal women. Cancer Res. 2012;72:696–706. doi: 10.1158/0008-5472.CAN-11-2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obi N, Vrieling A, Heinz J, Chang-Claude J. Estrogen metabolite ratio: is the 2-hydroxyestrone to 16 alpha-hydroxyestrone ratio predictive for breast cancer? Int J Womens Health. 2011;3:37–51. doi: 10.2147/IJWH.S7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falk RT, Brinton LA, Dorgan JF, Fuhrman BJ, Veenstra TD, Xu X, Gierach GL. Relationship of serum estrogens and estrogen metabolites to postmenopausal breast cancer risk: a nested case-control study. Breast Cancer Res. 2013;15:R34. doi: 10.1186/bcr3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fishman J. Aromatic hydroxylation of estrogens. Annu Rev Physiol. 1983;45:61–72. doi: 10.1146/annurev.ph.45.030183.000425. [DOI] [PubMed] [Google Scholar]
- 10.Bradlow HL, Hershcopf RJ, Martucci CP, Fishman J. Estradiol 16 alpha-hydroxylation in the mouse correlates with mammary tumor incidence and presence of murine mammary tumor virus: a possible model for the hormonal etiology of breast cancer in humans. Proc Natl Acad Sci U S A. 1985;82:6295–6299. doi: 10.1073/pnas.82.18.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telang NT, Bradlow HL, Kurihara H, Osborne MP. In vitro biotransformation of estradiol by explant cultures of murine mammary tissues. Breast Cancer Res Treat. 1989;13:173–181. doi: 10.1007/BF01806529. [DOI] [PubMed] [Google Scholar]
- 12.Muti P, Bradlow HL, Micheli A, Krogh V, Freudenheim JL, Schunemann HJ, Stanulla M, Yang J, Sepkovic DW, Trevisan M, Berrino F. Estrogen metabolism and risk of breast cancer: a prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology. 2000;11:635–640. doi: 10.1097/00001648-200011000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Bradlow HL, Telang NT, Sepkovic DW, Osborne MP. 2-hydroxyestrone: the ‘good’ estrogen. J Endocrinol. 1996;150(Suppl):S259–S265. [PubMed] [Google Scholar]
- 14.Meilahn EN, De Stavola B, Allen DS, Fentiman I, Bradlow HL, Sepkovic DW, Kuller LH. Do urinary oestrogen metabolites predict breast cancer? Guernsey III cohort follow-up. Br J Cancer. 1998;78:1250–1255. doi: 10.1038/bjc.1998.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF. Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res. 2007;67:812–817. doi: 10.1158/0008-5472.CAN-06-2133. [DOI] [PubMed] [Google Scholar]
- 16.Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D. Estrogens as endogenous genotoxic agents--DNA adducts and mutations. J Natl Cancer Inst Monogr. 2000:75–93. doi: 10.1093/oxfordjournals.jncimonographs.a024247. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Z, Kosinska W, Khmelnitsky M, Cavalieri EL, Rogan EG, Chakravarti D, Sacks PG, Guttenplan JB. Mutagenic activity of 4-hydroxyestradiol, but not 2-hydroxyestradiol, in BB rat2 embryonic cells, and the mutational spectrum of 4-hydroxyestradiol. Chem Res Toxicol. 2006;19:475–479. doi: 10.1021/tx0502645. [DOI] [PubMed] [Google Scholar]
- 18.Zahid M, Kohli E, Saeed M, Rogan E, Cavalieri E. The greater reactivity of estradiol-3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: implications for tumor-initiating activity. Chem Res Toxicol. 2006;19:164–172. doi: 10.1021/tx050229y. [DOI] [PubMed] [Google Scholar]
- 19.Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R, Sutter T. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci U S A. 1997;94:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellejus A, Olsen A, Tjonneland A, Thomsen BL, Overvad K, Loft S. Urinary hydroxyestrogens and breast cancer risk among postmenopausal women: a prospective study. Cancer Epidemiol Biomarkers Prev. 2005;14:2137–2142. doi: 10.1158/1055-9965.EPI-04-0934. [DOI] [PubMed] [Google Scholar]
- 22.Cauley JA, Zmuda JM, Danielson ME, Ljung BM, Bauer DC, Cummings SR, Kuller LH. Estrogen metabolites and the risk of breast cancer in older women. Epidemiology. 2003;14:740–744. doi: 10.1097/01.ede.0000091607.77374.74. [DOI] [PubMed] [Google Scholar]
- 23.Eliassen AH, Missmer SA, Tworoger SS, Hankinson SE. Circulating 2-hydroxy- and 16alpha-hydroxy estrone levels and risk of breast cancer among postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2008;17:2029–2035. doi: 10.1158/1055-9965.EPI-08-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arslan AA, Shore RE, Afanasyeva Y, Koenig KL, Toniolo P, Zeleniuch-Jacquotte A. Circulating estrogen metabolites and risk for breast cancer in premenopausal women. Cancer Epidemiol Biomarkers Prev. 2009;18:2273–2279. doi: 10.1158/1055-9965.EPI-09-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho GH, Luo XW, Ji CY, Foo SC, Ng EH. Urinary 2/16 alpha-hydroxyestrone ratio: correlation with serum insulin-like growth factor binding protein-3 and a potential biomarker of breast cancer risk. Ann Acad Med Singapore. 1998;27:294–299. [PubMed] [Google Scholar]
- 26.Kabat GC, O’Leary ES, Gammon MD, Sepkovic DW, Teitelbaum SL, Britton JA, Terry MB, Neugut AI, Bradlow HL. Estrogen metabolism and breast cancer. Epidemiology. 2006;17:80–88. doi: 10.1097/01.ede.0000190543.40801.75. [DOI] [PubMed] [Google Scholar]
- 27.Fowke JH, Qi D, Bradlow HL, Shu XO, Gao YT, Cheng JR, Jin F, Zheng W. Urinary estrogen metabolites and breast cancer: differential pattern of risk found with pre-versus post-treatment collection. Steroids. 2003;68:65–72. doi: 10.1016/s0039-128x(02)00116-2. [DOI] [PubMed] [Google Scholar]
- 28.Key TJ, Appleby PN, Reeves GK, Travis RC, Alberg AJ, Barricarte A, Berrino F, Krogh V, Sieri S, Brinton LA, Dorgan JF, Dossus L, Dowsett M, Eliassen AH, Fortner RT, Hankinson SE, Helzlsouer KJ, Hoff MJ, Comstock GW, Kaaks R, Kahle LL, Muti P, Overvad K, Peeters PH, Riboli E, Rinaldi S, Rollison DE, Stanczyk FZ, Trichopoulos D, Tworoger SS, Vineis P. Sex hormones and risk of breast cancer in premenopausal women: a collaborative reanalysis of individual participant data from seven prospective studies. Lancet Oncol. 2013;14:1009–1019. doi: 10.1016/S1470-2045(13)70301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liehr JG, Ricci MJ. 4-Hydroxylation of estrogens as marker of human mammary tumors. Proc Natl Acad Sci U S A. 1996;93:3294–3296. doi: 10.1073/pnas.93.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teng Y, He C, Zuo X, Li X. Catechol-O-methyltransferase and cytochrome P-450 1B1 polymorphisms and endometrial cancer risk: a meta-analysis. Int J Gynecol Cancer. 2013;23:422–430. doi: 10.1097/IGC.0b013e3182849e0d. [DOI] [PubMed] [Google Scholar]
- 31.Turan VK, Sanchez RI, Li JJ, Li SA, Reuhl KR, Thomas PE, Conney AH, Gallo MA, Kauffman FC, Mesia-Vela S. The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17beta-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16alpha-hydroxyestradiol, and 4-hydroxyestrone. J Endocrinol. 2004;183:91–99. doi: 10.1677/joe.1.05802. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Z, Mu Y, Qi C, Wang J, Xi G, Guo J, Mi R, Zhao F. CYP1B1 enhances the resistance of epithelial ovarian cancer cells to paclitaxel in vivo and in vitro. Int J Mol Med. 2015;35:340–348. doi: 10.3892/ijmm.2014.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]