

Key Points
Inflammatory agent exposure causes c-Abl to interact with and phosphorylate PARP1.
Tyrosine phosphorylation of PARP1 is crucial for NF-κB activation and gene expression.
Visual Abstract
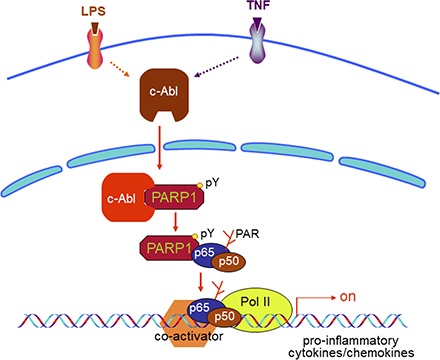
Abstract
Poly(ADP-ribosyl)ation is a rapid and transient posttranslational protein modification mostly catalyzed by poly(ADP-ribose) polymerase-1 (PARP1). Fundamental roles of activated PARP1 in DNA damage repair and cellular response pathways are well established; however, the precise mechanisms by which PARP1 is activated independent of DNA damage, and thereby playing a role in expression of inflammatory genes, remain poorly understood. In this study, we show that, in response to LPS or TNF-α exposure, the nonreceptor tyrosine kinase c-Abl undergoes nuclear translocation and interacts with and phosphorylates PARP1 at the conserved Y829 site. Tyrosine-phosphorylated PARP1 is required for protein poly(ADP-ribosyl)ation of RelA/p65 and NF-κB–dependent expression of proinflammatory genes in murine RAW 264.7 macrophages, human monocytic THP1 cells, or mouse lungs. Furthermore, LPS-induced airway lung inflammation was reduced by inhibition of c-Abl activity. The present study elucidated a novel signaling pathway to activate PARP1 and regulate gene expression, suggesting that blocking the interaction of c-Abl with PARP1 or pharmaceutical inhibition of c-Abl may improve the outcomes of PARP1 activation-mediated inflammatory diseases.
Introduction
Poly(ADP-ribose) polymerase-1 (PARP1) is an abundant nuclear enzyme, catalyzing the formation of poly(ADP-ribose) (PAR) on specific acceptor proteins by consuming substrate NAD+. This enzymatic process is designated as poly(ADP-ribosyl)ation (PARylation), one of the essential and conserved posttranslational protein modifications in eukaryotic cells (1, 2). PARP1’s crucial roles in DNA transactions, such as chromatin replication, DNA damage repair, and gene transcription, as well as its roles in cell death have been solidly established (1, 3). Traditionally, PARP1 was documented to be activated by DNA damage, including strand breaks and apurinic/apyrimidinic (AP) sites and was thus functionally linked to DNA damage detection and repair as well as cell death (4–6). Once PARP1 engages DNA, the interaction with DNA damage organizes PARP1 domains into a collapsed conformation that can explain the strong activation of PARP1 (7). Under such conditions, the primary target for PARP1-mediated PARylation usually is PARP1 itself, which is called PARP1 automodification. Besides this, under conditions lacking abundant DNA breaks, binding to nondamage DNA structures (e.g., hairpin, cruciform, stem-loop, as well as other specific double-stranded sequences) has been considered as effective determinants of PARP1 activation (8–10). In addition, recent studies indicate that posttranslational modifications such as phosphorylation, acetylation, and methylation are alternative mechanisms for the regulation of PARP1 activity (11).
In recent years, roles of PARP1 activation have been well acknowledged as to be involved in regulation of proinflammatory gene transcription in infectious and noninfectious as well as allergic disorders (12–14). Proinflammatory agents (e.g., LPS, IL-1, and TNF-α) that may not introduce severe damage to genomic DNA increase the level of protein PARylation in various cell types (15–18). However, signaling mechanisms eliciting PARP1 activation remain poorly elucidated. Studies have suggested that PARP1 activity may be regulated through phosphorylation by kinases that are parts of important regulatory pathways (19). Among them, serine/threonine phosphorylation imposed by ERKs represents a major alternative pathway to stimulate PARP1 (15, 20–22). Likewise, other serine/threonine kinases, such as AMP-activated protein kinase (AMPK) and protein kinase C (PKC) (23–25) have also been demonstrated to phosphorylate PARP1 and regulate its functions. PARP1 activation may be translated into PARylation of transcription factors (e.g., NF-κB, Sp1, or FOXP3) (15, 26) and thereby transcriptional modulation of target genes positively or negatively.
In leukocytes, the roles of nonreceptor tyrosine kinases in the transmission of membrane receptor-transduced immune signals have drawn increasing attention (27, 28). However, to date, whether tyrosine phosphorylation constitutes a regulatory mechanism in PARP1-mediated inflammatory gene transcription has not been investigated. Our previous studies have addressed that the Abelson (ABL) family of nonreceptor tyrosine kinases c-Abl, spleen tyrosine kinase (Syk), as well as lymphocyte cell kinase (Lck) mediate signals transduced from the leukocyte-specific adhesion molecules (including L-selectin, P-selectin glycoprotein ligand-1, and β2-integrin), directing the polymerization of actin and thus regulating leukocytes’ adhesion and migration (29–33) or stimulating proinflammatory cytokine expression in human leukemic Jurkat T cells (34–36). Abl tyrosine kinase has also been shown to be activated by TCR engagement, which is required for maximal TCR signaling for T cell development and function of mature T cells (28). c-Abl tyrosine kinase has been reported to be involved in LPS-mediated activation of macrophages, prompting an innate host response (37).
Our previous work documented an LPS-induced activation of PARP1 in murine macrophages (15, 38). Thus, we questioned whether c-Abl functions as an activator of PARP1. In the current study, we demonstrated that exposure to inflammatory agents LPS and TNF-α increased both protein tyrosine phosphorylation and PARylation levels in murine macrophages and mouse lungs. Inflammatory stimulation induced c-Abl nuclear translocation and interaction with PARP1, leading to tyrosine phosphorylation of the latter. Pharmacological inhibition or genetic depletion of c-Abl prevented PARP1 activation and decreased PARP1–NF-κB–dependent inflammatory gene expression. We identified that the conserved 829 tyrosine residue of PARP1 might be the major site to be phosphorylated by c-Abl. Pharmacological inhibition of c-Abl also reduced airway lung inflammation induced by LPS. Given the central role of PARP1 in inflammation, these results suggest that c-Abl tyrosine kinase–mediated phosphorylation is required for optimal PARP1 activation. c-Abl inhibitors may thereby have clinical use to treat inflammation-related diseases.
Materials and Methods
Abs and reagents
Mouse mAb against PARP1 (1:2000, B-10, sc-74470) and rabbit polyclonal Ab against c-Abl (1:3000, K-12, sc-131) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–β-tubulin (1:8000, HC101) and anti–β-actin (1:8000, HC201) mouse mAbs were purchased from TRANS (Beijing, China). Mouse mAb against PAR (1:2000, ALX-804-220) was purchased from Alexis (San Diego, CA). Mouse mAb against FLAG (1:8000, F1804) was purchased from Sigma-Aldrich (Saint Louis, MO). The mouse anti-phosphotyrosine Ab pY20 (1:3000, ab10321) was purchased from Abcam (Cambridge, U.K.). LPS (L2630) and TNF-α was purchased from PeproTech (Rocky Hill, NJ).Tyrosine kinase inhibitor imatinib mesylate STI571 (CGP-57148B) was purchased from BioVision (Milpitas, CA).
Cell culture and treatment
Murine RAW 264.7 macrophages were cultured in DMEM (Invitrogen) supplemented with 10% (v/v) FBS and antibiotics. Human monocytic THP1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/L glucose, penicillin (100 U/ml), and streptomycin (100 μg/ml) (Invitrogen, Carlsbad, CA). For the immune challenge, the dose of LPS was 200 ng/ml and the dose of c-Abl inhibitor STI571 was 10 nM. Human embryonic kidney 293/hTLR4A-MD2-CD14 (HEK 293) cells stably transfected with the human TLR4, MD2, and CD14 genes (InvivoGen, San Diego, CA) were used in transfection experiments as described previously (38).
Reverse transcription and real-time PCR
Total RNA was extracted from RAW 264.7 cells using TRIzol reagent (Invitrogen), and 1 μg of purified RNA from each sample was transcribed to cDNA. The cDNA was used as a template for real-time PCR. Primers for real-time PCR included the following: mCxcl2: forward: 5′-TCAATGCCTGAAGACCC-3′, reverse: 5′-TGGTTCTTCCGTTGAGG-3′; mTNF-α: forward: 5′-AACTCTCAAGCTGCTCTGCC-3′, reverse: 5′-CAAGGAATCTCCTCCCCGTC-3′; mIL-1β: forward: 5′-AAGGAAGTGCGTGTCTCTCC-3′, reverse: 5′-TCAAGGGGTGGCAGATAGTG-3′, and mβ-actin: forward: 5′-AACAGTCCGCCTAGAAGCAC-3′, reverse: 5′-CGATGACATCCGTAAAGACC-3′. Real-time PCR was performed in combination with the SYBR Green qPCR Master Mix (catalog no. 638320; TaKaRa) in an ABI 7000 thermal cycler. Relative expression levels of target genes were calculated by the ΔΔ cycle threshold method.
Immunoprecipitation and Western blotting
RAW 264.7 cells (1 × 107 per sample) were stimulated as described above and lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Nonidet P-40, 2.5 mM sodium pyrophosphate, 1 mM glycerophosphate, 1 mM Na3VO4, 1 mM NaF, and 20 μg/ml aprotin/leupeptin/PMSF). Lysates were centrifuged at 4°C, 13,000 × g for 30 min, and the supernatants were incubated with 30 μl of protein G Sepharose (Amersham Bioscience) at 4°C for 2 h. The precleared supernatants were incubated with the indicated Abs for 12 h and protein G Sepharose for 2 h with continuous rotation. Immunoprecipitates were then washed with lysis buffer and resolved by SDS-PAGE. After the proteins were transferred to nitrocellulose membranes, the membranes were washed with TBST and blocked with 5% nonfat dry milk and then incubated for 1 h each with the indicated primary Ab and HRP-conjugated secondary Ab. Signal was detected using ECL Plus chemiluminescent detection system (Amersham).
rDNA constructs
To constitute GST-fused PARP1 domain mutant plasmids, DBD, automodification domain (AMD), and CD domains of murine PARP1 were amplified by PCR using the murine cDNA from RAW 264.7 macrophages as a template and cloned into the vector pGEX-4T-2. The open reading frame of the inserted DNA was confirmed by sequencing, and the expression of the GST-fused proteins was verified by SDS-PAGE. To construct eukaryotic expression plasmids expressing wild-type (WT) Flag-tagged human PARP1 (h-PARP1), as well as Y775F, Y829F, and Y907F mutants, GFP-PARP1 plasmid (a gift from Professor G. G. Poirier from Laval University, QC, Canada) was used as template and amplified by PCR using flag-tagged PARP1 primers. The PCR-amplified product and p3× Flag CMV-10 plasmid vector were digested with restriction enzymes Nhe1 and Xba1. Purified DNA fragment of PARP1 (flag tagged) was cloned into plasmid vector (p3× Flag CMV-10) by T4 ligase enzyme to form rDNA. Construction of Flag-PARP1 recombinant plasmid was confirmed by DNA sequencing.
GST pulldown assay
GST and GST-fused proteins (GST-PARP1-DBD, GST-PARP1-AMD, and GST-PARP1-CD) were expressed in Escherichia coli strain BL21. The induction was performed by adding 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to the culture with OD 1.0 at 37°C for 3 h. Whole bacteria lysates were applied to glutathione Sepharose (GE Healthcare Life Science), and GST-tagged proteins were purified according to the manufacturer’s instructions. For pulldown experiments, GST and GST-fused protein immobilized on 40 μl of glutathione Sepharose 4B were incubated with 1 ml of cell extracts at 4°C for 3 h. After three washes with Nonidet P-40 lysis buffer, the bound proteins were analyzed by Western blotting.
Immunofluorescence microscopy
RAW 264.7 cells were cultured on cover slips in 12-well plates and incubated with LPS at 200 ng/ml for 1 h. Then the cells were fixed with 10% formaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 5 min, and blocked by incubation with 10% FBS for 30 min. For the detection of c-Abl, cells were sequentially incubated for 1 h each with anti–c-Abl rabbit polyclonal Ab (1:200) and TRITC-conjugated secondary Ab (1:200). The nuclei of the cells were stained with DAPI dye for 5 min, and cells were visualized by using a confocal microscope (Nikon, Tokyo, Japan).
Small interfering RNA
Small interfering RNAs (siRNAs) targeting murine c-Abl (1] GACUUAGAUUGAAGAAACU, 2] CCUUUGAUG CUUACAAACU) were used at 100 pM. Cells were seeded in plates, incubated in growth medium without antibiotics overnight, and then transiently transfected with RNA oligos using Lipofectamine 2000 following the manufacturer’s instructions. At 4–6 h posttransfection, cells were replaced with complete medium to promote recovery.
Mouse work
Six- to eight-week-old female C57BL/6 mice (20–25 g) were purchased from Jilin University (Changchun, Jilin, China). Mice were housed in a specific pathogen-free facility at Northeast Normal University (Changchun, Jilin, China) and allowed unlimited access to sterilized feed and water. They were maintained at 23 ± 1°C and kept under a 12-h light/dark cycle. All experiments were conducted in accordance with the Chinese Council on Animal Care Guidelines. Mice were anesthetized with pelltobarbitalumnatricum (6.5 mg·kg−1) and then randomized to be challenged with LPS (20 ng per mouse in 60 μl of saline) or 60 μl of pH-balanced saline solution (pH = 7.4) using the intranasal route (39), with or without an i.p. pretreatment of STI571 (100 mg·kg−1 in a 200-μl volume without anesthesia) or vehicle only (5% DMSO, 10% Tween 80 in saline) 60 min before the LPS challenge (40). One hour later, mice lungs were collected, and homogenates were prepared for RNA detection.
Evaluation of airway inflammation
Evaluation of mouse lung airway inflammation was performed as described (41). Briefly, tracheae were cannulated, and lungs were lavaged by two instillations of 0.6 ml of ice-cold PBS after LPS challenge for 16 h. Bronchoalveolar lavage fluid (BALF) samples were centrifuged (800 × g for 5 min at 4°C), and the supernatants were stored at −80°C for further analysis. The pellet included total cells, for which cell counts in the BALF were determined from an aliquot of the cell suspension using a hemocytometer. Differential cell counts were performed on centrifuge preparations (Shandon CytospinR 4 Cytocentrifuge; Thermo Fisher Scientific, Waltham, MA). Cells were stained with modified Wright–Giemsa using HEMA-TEK 2000 Slide Stainer (Protocol) for differential cell counts. Differential cell counts were performed in a blind fashion by two independent researchers counting 1000 cells from each animal (each animal was counted near 16 different fields). Randomly selected fields were photographed using an OLYMPUS Microscope System BX53P microscope with a built-in digital charge-coupled device color camera, DP73WDR. In parallel experiments, lung tissue sections were processed for staining with H&E to identify neutrophils in lung sections as we published previously (39). Randomly, five fields of each sample were captured at a magnification of ×160 using a CoolSNAP HQ2 camera mounted on a Nikon Eclipse Ti microscope.
Statistical analysis
Results were tested for statistical significance using one-way ANOVA to analyze changes in mRNA levels. All data values are presented as mean ± SD with significance set at *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Increase in protein PARylation induced by inflammatory stimuli is regulated by c-Abl
Murine RAW 264.7 macrophages and human monocytic THP1 cells were incubated with LPS at a concentration of 200 ng/ml for different time intervals (0, 0.5, 1, 3, and 5 h). Western blot analysis using Abs against PAR showed that the level of PARylated proteins notably increased by 0.5 h and reached the maximum level at 1 h after LPS stimulation (Fig. 1A, Supplemental Fig. 1A). To support these results, RAW 264.7 macrophages were also challenged by another immune challenger, TNF-α, at concentration of 10 ng/ml for the same time intervals. The level of PARylated proteins notably increased from 0.5 h on and reached the maximum level at 3 h postexposure with TNF-α (Fig. 1B). The increase in protein PARylation induced by both inflammatory stimuli (LPS and TNF-α) was diminished when c-Abl tyrosine kinase inhibitor STI571 was supplied (Fig. 1C, 1D, Supplemental Fig. 1B). To further verify that c-Abl acts as an upstream modulator of PARP1 activation, we used siRNA targeting c-Abl. Knockdown of c-Abl greatly inhibited LPS-induced increase in PARylated protein content and also notably lowered the basal protein PARylation level in cells without LPS stimulation (Fig. 1E).
FIGURE 1.

Inflammatory agent-stimulated protein PARylation is regulated by c-Abl. (A and B) Inflammatory agents’ (LPS or TNF-α) stimulation promotes protein PARylation. RAW 264.7 cells were challenged with LPS or TNF-α for various lengths of time. Western blotting was performed to detect protein PARylation levels in whole-cell lysates. (C and D) c-Abl activity is required for inflammatory agent-stimulated protein PARylation. RAW 264.7 cells were incubated with or without LPS for 1 h or TNF-α for 3 h in the presence or absence of c-Abl inhibitor (STI571). Cell extracts were subjected to Western blotting to detect the protein PARylation. (E) siRNA of c-Abl blocked protein PARylation. RAW 264.7 cells were transfected with siRNA targeting c-Abl or the control for 48 h and then challenged with LPS for 1 h. Western blotting was performed to detect protein PARylation levels. (F) LPS induces protein PARylation in mice lungs. Mice were challenged with LPS (20 μg per mouse) through the intranasal route for different time intervals, and then the lungs were excised and homogenized. Western blotting was performed to detect the PARylation levels of each protein. (G) LPS-induced PARP1 activation in mice lung is dependent on c-Abl. Mice were challenged with LPS for 1 h with or without i.p. pretreatment of STI571(30 mg·kg−1). Lung homogenates were prepared, and Western blotting was performed to detect protein PARylation levels. Similar results were obtained from at least three independent experiments.
To further explore the role of c-Abl in PARP1 activation, a mouse lung inflammation model was used as described previously (38). Mice were challenged with LPS via the intranasal route for various lengths of time (0, 0.5, 1, 3, and 5 h). Western blot analysis showed that protein PARylation notably increased at 0.5 h and reached a maximum level at 1 h post LPS exposure in mice lungs (Fig. 1F). Pretreatment of mice with STI571 hampered LPS-induced enhancement of protein PARylation (Fig. 1G). Similar results were observed after TNF-α challenges (data not shown). The combined data suggested that inflammatory stimuli-induced activation of nuclear enzyme PARP1 is a downstream event of nonreceptor tyrosine kinase c-Abl signaling.
Inflammatory stimuli induce the association of c-Abl with PARP1
To investigate the mechanism by which c-Abl increases protein PARylation, we first examined whether c-Abl and PARP1 interact upon LPS stimulation. Immunoprecipitates were obtained by using PARP1 Ab, and Western blots showed a weak association of c-Abl with PARP1 in the nontreated cells, which was markedly enhanced by LPS stimulation from 0.5 h on and reached a maximum level at 1 h (Fig. 2A). Likewise, the enhancement of the interaction between c-Abl and PARP1 (from 0.5 h on and reached a maximum at 3 h) was also verified in the cells with TNF-α stimulation (Fig. 2B). The interaction of the two proteins was eliminated by treatment with c-Abl inhibitor STI571 (Fig. 2C–E). The presence of DNase in the lysis buffer did not abolish the association of the two molecules, implying the formation of the complex is not mediated by DNA (data not shown). Next, we asked what domain(s) of PARP1 interact(s) with c-Abl. PARP1 has three domains through which it interacts with partner proteins. (Fig. 2F, upper). GST-PARP1 domain expression plasmids were constructed (Fig. 2F, lower). A GST pulldown assay using GST-fused PARP1 domains further revealed that the AMD of PARP1 enables its binding with c-Abl (Fig. 2G). Furthermore, a question spontaneously arises as to whether LPS induces c-Abl nuclear translocation. In unstimulated cells, inactivated c-Abl is mainly located in the cytoplasm and shuttles between cytoplasmic and nuclear compartments upon induction, whereas PARP1 mainly resides in the nucleus. RAW 264.7 cells were challenged with LPS for various lengths of time (0, 0.5, 1, 3, and 5 h), and the subcellular distribution of c-Abl was determined. Western blot analysis showed c-Abl was notably translocated to nuclei at 0.5 h and reached the maximum level at 1 h after LPS stimulation (Fig. 3A). Nuclear translocation of c-Abl was further verified by immunofluorescence staining (Fig. 3B).
FIGURE 2.

c-Abl interacts with PARP1 in response to exposure to inflammatory agents. (A and B) Exposure to inflammatory agents increases the association of c-Abl with PARP1. RAW 264.7 cells were exposed to LPS (A) or TNF-α (B) for various lengths of time. Whole-cell extracts (WEs) were prepared, and immunoprecipitates were obtained using Ab recognizing PARP1. The association of c-Abl with PARP1 was detected by Western blotting. (C–E) STI571 blocked the association of c-Abl with PARP1. RAW 264.7 cells were mock treated or exposed to LPS (±STI571) for 1 h (C and E) and TNF-α (±STI571) for 3 h (D). WEs were prepared, and immunoprecipitates were obtained using Ab recognizing PARP1 or c-Abl. The blockage in association of c-Abl with PARP1 or vice versa was detected by Western blotting. (F) Diagram of the domains in PARP1 and the schematics of the GST-PARP1 domains expression plasmids. (G) AMD mediates the association of PARP1 with c-Abl. GST and GST-PARP1 domains were incubated with equal amounts of WEs from LPS-treated cells. Levels of pulled-down c-Abl were detected by Western blotting. Similar results were obtained from at least three independent experiments.
FIGURE 3.

LPS stimulation induces c-Abl’s nuclear transportation. (A) LPS stimulation induces nuclear import of c-Abl. RAW 264.7 cells were challenged with LPS for various lengths of time. Cell lysates from nuclear and cytoplasmic fractions were subjected to Western blotting to determine the subcellular distribution of c-Abl. (B) Immune-fluorescence staining verifies LPS-induced nuclear import of c-Abl. RAW 264.7 cells were mock treated or LPS exposed (±STI571) for 1 h, and then cells were fixed and permeabilized and incubated with anti–c-Abl rabbit polyclonal Ab and TRITC-conjugated secondary Ab. The nuclei of the cells were stained with DAPI. Similar results were obtained from at least three independent experiments. Original magnification ×180.
c-Abl is involved in inflammatory stimuli-induced tyrosine phosphorylation of PARP1
Because LPS and TNF-α stimulation enhanced the interaction between c-Abl and PARP1, we predicted c-Abl played a role in the phosphorylation of PARP1. Immunoprecipitation assay showed that tyrosine phosphorylation level of PARP1 in RAW 264.7 cells was increased in response to LPS exposure (Fig. 4A), coinciding with the kinetics of its interaction with c-Abl (Fig. 2A). Similar results were obtained when THP1 cells were exposed to LPS (Supplemental Fig. 2A) and when RAW 264.7 cells were exposed to TNF-α (Fig. 4B). To determine the role of c-Abl in tyrosine phosphorylation of PARP1, STI571 was applied, and LPS-induced tyrosine phosphorylation of PARP1 in RAW 264.7 cells was inhibited (Fig. 4C). Similar results were obtained when THP1 cells were exposed to STI571 with LPS (Supplemental Fig. 2B) and when RAW 264.7 cells were exposed to STI571 with TNF-α (Fig. 4D). To further specify the role of c-Abl in tyrosine phosphorylation of PARP1, we used siRNA targeting c-Abl, and the result showed that LPS stimulation failed to induce significant tyrosine phosphorylation of PARP1 in c-Abl–silenced cells (Fig. 4E).
FIGURE 4.

c-Abl is required for tyrosine phosphorylation of PARP1 in response to inflammatory agents. (A and B) Exposure of inflammatory agents increases the levels of tyrosine phosphorylation (pTyr) of PARP1. RAW 264.7 cells were exposed to LPS (A) or TNF-α (B) for various lengths of time. Whole-cell extracts (WEs) were prepared, and immunoprecipitates were obtained using Ab recognizing PARP1. The levels of pTyr of PARP1 were detected by Western blotting. (C and D) Inhibition of c-Abl activity eliminates the inflammatory agent-induced increase in pTyr of PARP1. RAW 264.7 cells were mock treated or exposed to LPS (±STI571) for 1 h (C) or TNF-α exposed (±STI571) for 3 h (D). WEs were prepared, and immunoprecipitates were obtained using Ab recognizing PARP1. The levels of pTyr of PARP1 were detected by Western blotting. (E) siRNA of c-Abl interferes with LPS-induced PARP1 tyrosine phosphorylation. RAW 264.7 cells were transfected with siRNA targeting c-Abl or a control for 48 h and then challenged with LPS for 1 h. WEs were prepared, and immunoprecipitates were obtained using Ab recognizing PARP1. The levels of pTyr of PARP1 in the presence or absence of c-Abl were detected by Western blotting. Similar results were obtained from at least three independent experiments.
To further unveil the molecular mechanism of tyrosine phosphorylation of PARP1 by c-Abl, we constructed eukaryotic expression plasmids expressing Flag-tagged WT PARP1 and tyrosine site mutants. We analyzed the tyrosine site(s) of h-PARP1 to consider the potential translational significance of h-PARP1 tyrosine phosphorylation. Online software (http://kinasephos.mbc.nctu.edu.tw/predict.php) predicted that Tyr775 (Y775), Tyr829 (Y829), and Tyr907 (Y907) are potential sites of phosphorylation for h-PARP1. All three tyrosine sites are conserved across phyla from invertebrates (Drosophila) to mammals (humans) (Fig. 5A). In light of the high transfection efficacy, HEK 293 cells were used. Cells were transfected with WT and mutant plasmids and later were stimulated with LPS.
FIGURE 5.

PARP1 Y829 might be the site phosphorylated by c-Abl in response to LPS stimulation. (A) Conservative comparison of tyrosine residues of h-PARP1 in segment 751–930 with those relevant in other species through Drosophila to primate. Tyrosine residues that are conserved across all sequences are highlighted with an asterisk (*) and tyrosine residues that are not conserved with a line (—). (B) Y775 and Y829 sites of PARP1 undergo phosphorylation in LPS-exposed cells. HEK 293 cells were transfected with WT Flag-PARP1 as well as Y775F, Y829F, and Y907F mutant plasmids and then challenged with LPS for 1 h. Immunoprecipitates were prepared using flag Ab and then subjected to Western blotting to detect phosphorylation level. (C) The Y829 site of PARP1 might be phosphorylated by c-Abl in response to LPS exposure. WT Flag-PARP1 as well as both Y775F and Y829F mutants were transfected in HEK 293 cells, and then the cells were stimulated with LPS (±STI571) for 1 h. Immunoprecipitates were prepared using Flag Ab and then subjected to Western blotting to detect phosphorylation levels. (D) Y829F mutation does not affect the interaction of c-Abl and PARP1. HEK 293 cells were transfected with WT Flag-PARP1 and Y829F mutant plasmids and then challenged with LPS or left untreated for 1 h. Immunoprecipitates were prepared using Flag Ab and then subjected to Western blotting to detect the interaction with c-Abl. Similar results were obtained from at least three independent experiments.
Immunoprecipitation assay using Ab recognizing the Flag tag revealed that phosphorylation of WT Flag-PARP1 and Y907F mutant was markedly increased in LPS-exposed cells, whereas that of Y775F and Y829F mutants were not (Fig. 5B). This result suggested that Y907 does not undergo phosphorylation in response to LPS challenge. To pinpoint which tyrosine site of PARP1 is the target of c-Abl, WT as well as both Y775F and Y829F mutants were transfected into HEK 293 cells, and then the cells were stimulated with LPS in the presence or absence of STI571. Immunoprecipitation assay revealed that STI571 was able to lower the levels of tyrosine phosphorylation from WT Flag-PARP1 and the Y775F mutant, but not Y829F, implying Y829 is the site undergoing c-Abl–mediated tyrosine phosphorylation (Fig. 5C). To exclude the possibility that the lack of response of the Y829F mutant to STI571 was due to the mutation disrupting the interaction between c-Abl and PARP1, we compared the coimmunoprecipitated c-Abl with WT-PARP1 or Y829F mutant, and the result showed the Y829F mutation did not affect the interaction of PARP1 with c-Abl (Fig. 5D).
c-Abl signals the function of PARP1 on NF-κB in response to LPS stimulation
Our previous study demonstrated an LPS-induced PARP1–NF-κB signal axis is a key for NF-κB–dependent proinflammatory gene expression (15). To address the significance of c-Abl–mediated PARP1 phosphorylation, we first examined the role of c-Abl in the induced association of PARP1 with RelA/p65 (the transcription activation subunit of NF-κB). Upon LPS stimulation, the amount of RelA/p65 in the immunoprecipitate obtained by Ab against PARP1 was increased (Fig. 6A) and vice versa PARP1 in the immunoprecipitate obtained by Ab against RelA/p65 (Fig. 6B). Accordingly, the PARylation level of RelA/p65 was also enhanced (Fig. 6C) in line with our previous results. Notably, pretreatment of cells with c-Abl tyrosine kinase inhibitor STI571 diminished the LPS-induced interaction of PARP1 with RelA/p65 as well as PARylation of the latter (Fig. 6A–C). To specify c-Abl’s role in RelA/p65 PARylation, siRNA targeting c-Abl was applied, and data indicated that in cells deficient of c-Abl, RelA/p65 PARylation was markedly impaired (Fig. 6D), underlining the upstream-activating effect of c-Abl on the activation of PARP1–NF-κB signal axis.
FIGURE 6.

c-Abl activity promotes RelA/p65 binding with PARP1 and PARyaltion (A and B) c-Abl activity promotes RelA/p65 binding with PARP1. RAW 264.7 cells were mock treated or exposed to LPS (±STI571) for 1 h. Immunoprecipitation was performed, and Ab against PARP1 (A) or RelA/p65 (B) was used to get precipitate complexes. Western blotting was performed using Abs specific for p65 or PARP1 in the complexes, respectively. (C and D) c-Abl activity promotes RelA/p65 PARyaltion. (C) RAW 264.7 cells were treated as described above. Immunoprecipitates were obtained by using Ab against RelA/p65; PARylation levels of RelA/p65 were determined by Western blotting using Ab recognizing PAR. (D) RAW 264.7 cells were transfected with siRNA targeting c-Abl or the control for 48 h and then challenged with LPS for 1 h. Immunoprecipitates were obtained using Ab recognizing RelA/p65. PARylation levels of RelA/p65 in the presence or absence of c-Abl were detected by Western blotting. Similar results were obtained from at least three independent experiments.
c-Abl is required for proinflammatory gene upregulation and inflammation
To further elucidate the significance of c-Abl–mediated activation of PARP1–NF-κB signaling pathway, expression of selected NF-κB–driven genes was examined. RAW 264.7 or THP1 cells were incubated with LPS for increasing time intervals (0, 0.5, 1, 3, and 5 h). Real-time PCR showed that mRNA levels of TNF-α and IL-1β increased and persisted at the maximum level during 1 h, revealing an activation of cells in response to immune challenge (Fig. 7A, Supplemental Fig. 3A). Likewise, incubation of RAW 264.7 macrophages with TNF-α resulted in a similar pattern of Cxcl2 and IL-1β mRNA expression, which reached a maximum at 3 h (Fig. 7B). To determine the role of c-Abl in proinflammatory gene expression, siRNA targeting c-Abl was used. c-Abl–deficient RAW 264.7 cells were incubated with LPS for 1 h or not; real-time PCR showed that LPS-induced expression of proinflammatory genes (TNF-α and IL-1β) was decreased (Fig. 7C). When c-Abl–deficient RAW 264.7 cells were incubated with TNF-α for 3 h, upregulation of proinflammatory genes (Cxcl2 and IL-1β) was blocked too (Fig. 7D). In THP1 cells, STI571 was supplied along with LPS for 1 h; real-time PCR showed that LPS-induced expression of proinflammatory genes (TNF-α and IL-1β) was decreased by STI571 (Supplemental Fig. 3B). Moreover, mouse lungs were challenged with LPS for various lengths of time (0, 0.5, 1, 3, and 5 h); RT-PCR showed that mRNA levels of TNF-α and IL-1β exhibited similar kinetics as that of RAW 264.7 macrophages (Fig. 7E). Pretreatment with STI571 prevented the upregulation of the inflammatory genes induced by LPS in mouse lungs (Fig. 7F).
FIGURE 7.

Inflammatory agent-induced proinflammatory gene expression is enhanced by c-Abl. (A and B) Inflammatory agents stimulate inflammatory gene expression in murine macrophages. RAW 264.7 cells were incubated with LPS for various lengths of time. Real-time PCR was performed to detect the mRNA expression of TNF-α and IL-1β (n = 5) (A). RAW 264.7 cells were incubated with TNF-α for various lengths of time. Real-time PCR was performed to detect the mRNA expression of Cxcl2 and IL-1β (n = 5) (B). (C and D) c-Abl knockdown blocks upregulation of inflammatory genes. RAW 264.7 cells were subjected to siRNA targeting c-Abl and then mock treated or exposed to LPS for 1 h; real-time PCR was performed to detect the mRNA expression of TNF-α and IL-1β. Inset, Western blotting shows efficacy of c-Abl knockdown (n = 5) (C); c-Abl–deficient RAW 264.7 cells were mock treated or exposed to TNF-α for 3 h, and real-time PCR was performed to detect the mRNA expression of Cxcl2 and IL-1β (n = 5) (D), **p < 0.01. (E) LPS stimulates inflammatory gene expression in mice lungs. Mice were exposed to LPS through the intranasal route for various lengths of time. Mice lungs were collected, and homogenates were prepared. RNA was extracted, and RT-PCR was performed to detect mRNA expression of TNF-α and IL-1β (n = 5). Data were expressed as mean ± SD. Difference significance was analyzed by one-way ANOVA. (F) c-Abl inhibition blocks upregulation of inflammatory genes in mice lungs. Mice were exposed to LPS through the intranasal route for 1 h with or without an i.p. pretreatment of STI571. Mice lungs were collected, and homogenates were prepared. RNA was extracted, and RT-PCR was performed to detect the mRNA expression of TNF-α and IL-1β. Similar results were obtained from at least three independent experiments.
In other experiments, mice were euthanized 16 h after LPS challenge, the lungs were lavaged, and the cell numbers in BALF were determined. Although challenge with LPS induced the robust recruitment of neutrophils to the airways (∼5 × 105 per ml), the number of neutrophils in the samples from STI571-treated animals decreased to 1.5 × 105 per ml. STI571 inhibits LPS-induced airway inflammation ∼70% (Fig. 8A, 8B). In parallel experiments, lung tissue sections were processed for staining with H&E to examine the subepithelium accumulation of leukocytes in lung tissues. The result showed that the administration of STI571 markedly blocked LPS-induced airway lung inflammation (Fig. 8C).
FIGURE 8.

c-Abl activity is responsible for LPS-induced lung inflammation. (A and B) Visual depiction and quantification of cells in BALF. Mice were mock treated or challenged with LPS in the presence or absence of pretreatment with STI571. After 16 h, mice were euthanized, lungs were lavaged, and the cell numbers in BALF were determined. Ten to sixteen randomly selected fields of view per cytospin slide were photographed (A). Differential cell counts were performed after modified Wright–Giemsa staining. Data are average ± SD, representative of six experimental animals from one experimental run. (C) STI571 administration blocked LPS-induced subepithelium accumulation of leukocytes in lung tissues. Mice were treated as described above. Lung tissue sections were processed for staining with H&E to examine the subepithelium accumulation of leukocytes in lung tissues. Similar results were obtained from at least three independent experiments. Original magnification ×200.
To dissect the significance and function of PARP1’s Y829 site in transcriptional activation of NF-κB targets in an intracellular context, we transfected WT, Y829F, and Y907F PARP1-expressing plasmids in endogenous PARP1-silenced Raw 264.7 cells. Proinflammatory gene expression (TNF-α and IL-1β) in Y829F mutant–expressing cells was strongly blocked compared with WT-PARP1–transfected cells, but that in Y907F mutant–expressing cells was not (Fig. 9A, 9B).
FIGURE 9.

Proinflammatory genes’ mRNA expression is impaired in Y829F PARP1-expressing cells. Endogenous PARP1 in Raw 264.7 cells was silenced using siRNA targeting a distinguished sequence of PARP1, and human WT PARP1, Y829F PARP1, and Y907F PARP1 expressional plasmids were transfected and then the cells were stimulated with LPS or not for 1 h. Immunoblotting was performed to detect the interference of endogenous PARP1 as well as the ectopic expression of Flag-tagged WT, Y829F, and Y907F PARP1 (A). Raw 264.7 cells as described above were used; proinflammatory genes’ mRNA levels were detected by RT-PCR and electrophoresis (upper) or real-time PCR (lower) (B). Data were expressed as mean ± SD. Difference significance was analyzed by one-way ANOVA. **p < 0.01.
Discussion
PARP1 is the most abundant and ubiquitous member of the PARP family enzymes (42). Previous studies have explored a large number of signaling pathways leading to PARP1 activation independent of DNA damage under various inflammatory conditions (16, 43–46); however, the precise mechanisms by which PARP1 is activated and regulates the inflammatory genes expression remain not comprehensively understood. In the present work, we investigated the role of nonreceptor tyrosine kinase c-Abl in PARP1 activation, gene expression regulation, and inflammation mechanism in murine RAW 264.7 macrophages, human THP1 monocytic cells, or mouse lungs exposed to proinflammatory agents LPS or TNF-α. Results from cell culture showed that the increase in protein PARylation induced by LPS or TNF-α was diminished by c-Abl inhibitor STI571 or siRNA-mediated c-Abl knockdown (Fig. 1A–E, Supplemental Fig. 1A, 1B). Results from mouse lungs also confirmed that the increase in protein PARylation induced by LPS was diminished by the c-Abl inhibitor STI571 (Fig. 1F, 1G). Moreover, LPS- or TNF-α–induced expression of proinflammatory cytokines/chemokines was blocked due to expression knockdown or inhibition of the activation of c-Abl (Fig. 7A–D, Supplemental Fig. 3A, 3B) or was inhibited by preadministration of STI571 in mouse lung tissues (Fig. 7E, 7F). Furthermore, LPS-induced mouse airway lung inflammation is inhibited by STI571 administration (Fig. 8A–C). These data point to a unique activation mechanism for PARP1 that is not associated with DNA damage and is downstream of the activation of the nonreceptor tyrosine kinase c-Abl.
In the current study, we observed the induced interaction between c-Abl and PARP1, which accounts for PARP1 tyrosine phosphorylation and thereby the enhancement of PARP1’s catalytic activity (Fig. 2A–E). We identified that the conserved 829 tyrosine residue of PARP1 might be the major site to be phosphorylated by c-Abl (Fig. 5). PARP1–c-Abl interaction has not, to our knowledge, been reported before. Proteomic studies announced a large number of proteins interacting with or serving as substrates of c-Abl, including tyrosine kinases (both receptor and nonreceptor types), cytoskeleton-regulating proteins, signal adaptors, and DNA damage response proteins (47), yet PARP1 was not on these lists. Our previous study documented how activated c-Abl kinase interacts with transcription factor AP-1 to form a complex in the CSF-1 promoter region to regulate gene transcription in the L-selectin ligation-activated leukocytes (34). In the current study, whether interaction between PARP1 and c-Abl occurs on target gene promoter regions was not investigated. c-Abl is able to physically bind with PARP1 through the AMD of PARP1 (Fig. 2F, 2G), which is in line with a report showing that the AMD of PARP1 is prone to mediating PARP1’s association with its partners (48). PARP1 is a nuclear enzyme, whereas c-Abl resides in both the cytoplasm and the nucleus and shuttling between two compartments, which is based on its three nuclear localization signals and single nuclear export signal. In nucleus, c-Abl regulates transcription and chromatin, which may involve three high-mobility group–like boxes (HLB) that bind to DNA (49). Studies documented DNA damage agents that induced c-Abl nuclear translocation in which c-Abl modified DNA damage response proteins or apoptotic proteins, participating in DNA damage repair or cell death (50, 51). In the current study, we observed that c-Abl is primarily located in cytoplasm and undergoes nuclear translocation in response to LPS stimulation (Fig. 3), suggesting a potent membrane receptor-mediated signal-induced redistribution of c-Abl. However, whether nuclear translocation of c-Abl is indispensable for the increased interaction of c-Abl and PARP1 needs further elucidation.
Our previous studies and those of others elucidated ERK1/2-mediated serine phosphorylation of PARP1. ERK1/2 promotes PARP1 activation through direct phosphorylation of PARP1 at serine 372 and threonine 373 (15, 20, 21). In addition, phosphorylation of other serine sites of PARP1 has also been reported to result in its DNA-independent activation (52). The hormone-activated kinase CDK2, in complex with Cyclin E and progesterone receptor, phosphorylates Ser785 and Ser786 of PARP1, which are essential for the enhanced activity of PARP1, shown by remarkable increases in PAR and PARP1 levels in breast cancer cells upon progestin stimulation (52). More recently, it was reported that the receptor tyrosine kinase c-Met translocated into the nucleus upon H2O2 and sodium arsenite treatment and phosphorylated PARP1 at Tyr 907. Phosphorylation of Y907 can increase PARP1 enzymatic activity and reduce binding to a PARP inhibitor, thereby rendering cancer cells resistant to PARP inhibition (53). Our present study did not observe Y907 undergoing phosphorylation in cells exposed to LPS, indicating different tyrosine kinases may be activated under various stimulation conditions that direct PARP1 phosphorylation at different sites. However, both serine and tyrosine phosphorylations are observed in cells upon LPS stimulation (15, 54), which opens a question as to how these two modifications cross-talk. The role of nonreceptor tyrosine kinases in PARP1 phosphorylation and activation has not drawn much attention. An early study documented that Txk, a member of Tec family tyrosine kinases, is involved in the expression of Th1 cytokines. PARP1 and elongation factor 1 α (EF-1α) were identified as Txk-associated molecules that bound to the Txk-responsive element of IFN-γ gene promoter (55); however, whether Txk phosphorylates PARP1 in vivo was not addressed. In this study, we explored if the interaction between nonreceptor tyrosine kinase c-Abl and nuclear enzyme PARP1 resulted in the latter’s tyrosine phosphorylation and enzymatic activation (Fig. 4, Supplemental Fig. 2).
The software program (KinasePhos) (56) predicted that Y775, Y829, and Y907 are tyrosine phosphorylation sites. Site mutation analyses showed the weakened PARP1 tyrosine phosphorylation on mutants of Y775 and Y829 but not Y907, excluding a c-Met–mediated PARP1 phosphorylation in response to LPS exposure (Fig. 5B). Out of the remaining two sites, Y829 was identified as the substrate that is subjected to c-Abl–mediated modification because administration of STI571 could further reduce the extent of tyrosine phosphorylation of PARP1 Y775F mutant upon LPS stimulation (Fig. 5C). Y829 is adjacent to “the PARP signature,” a sequence of 50 aa (residues 859–908) that shows 100% homology among vertebrates and possesses PARP1 catalytic activity (57, 58). Phosphorylation of Y829 may lead to a conformational change in PARP1 that leaves its active site exposed. So far, three generic anti-pY Abs (4G10, pY20, and p-TYR-100) have been commonly used for phosphoproteomic approaches, and they display a sequence preference (59). The Ab used in the current study is pY20, which may omit other phosphorylated sites. Nevertheless, analysis of 119 separately validated ABL1, ABL2, and BCR-ABL1 substrates revealed an enrichment for acidic residues (D or E) at position +1 (47), supporting Y829 as the substrate of c-Abl in the current study.
Stimulation of macrophages with LPS endotoxin, a component of the outer membrane of Gram-negative bacteria, results in the production of various cytokines, including TNF-α and IL-1β (60). The critical role of PARP1 in NF-κB–dependent immunity gene activation is conserved in both mammals and Drosophila (61, 62). In this study, we observed the increase in the association of PARP1 with the transcription activation subunit of NF-κB (RelA/p65) as well as the enhanced PARylation level of RelA/p65, which is in line with our previous results (15). Notably, pretreatment of cells with STI571 diminished LPS-induced interaction of PARP1with RelA/p65 (Fig. 6A, 6B). Moreover, RelA/p65 PARylation was blocked by STI571 or c-Abl siRNA (Fig. 6C, 6D). Consequently, the upregulation of TNF-α, Cxcl2, and IL-1β was abolished in the cells subjected to pharmacological inhibition or expressional deficiency of c-Abl (Fig. 7, Supplemental Fig. 3) as well as in the cells expressing Y829F PARP1 (Fig. 9). Importantly, LPS-induced airway lung inflammation was reduced by administration of STI571 (Fig. 8). Combined data are strengthening the crucial role of c-Abl in activation of PARP1–NF-κB signal axis.
In summary, this study demonstrated that in response to immune stimuli, binding of c-Abl with PARP1 and tyrosine phosphorylation of PARP1 are crucial for PARP1–NF-κB signaling pathway-mediated upregulation of proinflammatory genes and lung inflammation. Our data also suggest a potential strategy to treat inflammation-related disorders through the inhibition of c-Abl–mediated tyrosine phosphorylation of PARP1.
Supplementary Material
Acknowledgments
We thank Professor Guy G. Poirier (Laval University, Quebec, Canada) for providing GFP-PARP1 plasmid and members of Dr. Ba's laboratory for technical help and useful discussions.
This work was supported by the National Natural Science Foundation of China (Grants 31371293 and 31571339 to X.B.), the Program for Introducing Talent to Universities (Grant B07017 to X.B.), the Natural Science Foundation of Jilin, China (Grant 20180101236JC to X.B.), the National Natural Science Foundation of China (Grant 31801182 to Y.K.), the National Institute of Environmental Health and Sciences (Grant R01 ES018948 to I.B.), and the National Institute of Allergic and Infectious Diseases (Grant AI062885 to I.B.).
The online version of this article contains supplemental material.
- ABL
- Abelson
- AMD
- automodification domain
- BALF
- bronchoalveolar lavage fluid
- HEK 293
- human embryonic kidney 293/hTLR4A-MD2-CD14
- h-PARP1
- human PARP1
- PAR
- poly(ADP-ribose)
- PARP1
- poly(ADP-ribose) polymerase-1
- PARylation
- poly(ADP-ribosyl)ation
- siRNA
- small interfering RNA
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Schreiber V., Dantzer F., Ame J. C., de Murcia G. 2006. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 7: 517–528. [DOI] [PubMed] [Google Scholar]
- 2.Gupte R., Liu Z., Kraus W. L. 2017. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31: 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erdélyi K., Bakondi E., Gergely P., Szabó C., Virág L. 2005. Pathophysiologic role of oxidative stress-induced poly(ADP-ribose) polymerase-1 activation: focus on cell death and transcriptional regulation. Cell. Mol. Life Sci. 62: 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malanga M., Althaus F. R. 2005. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem. Cell Biol. 83: 354–364. [DOI] [PubMed] [Google Scholar]
- 5.Haince J. F., McDonald D., Rodrigue A., Déry U., Masson J. Y., Hendzel M. J., Poirier G. G. 2008. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283: 1197–1208. [DOI] [PubMed] [Google Scholar]
- 6.Bonicalzi M. E., Haince J. F., Droit A., Poirier G. G. 2005. Regulation of poly(ADP-ribose) metabolism by poly(ADP-ribose) glycohydrolase: where and when? Cell. Mol. Life Sci. 62: 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langelier M. F., Planck J. L., Roy S., Pascal J. M. 2012. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336: 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lonskaya I., Potaman V. N., Shlyakhtenko L. S., Oussatcheva E. A., Lyubchenko Y. L., Soldatenkov V. A. 2005. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J. Biol. Chem. 280: 17076–17083. [DOI] [PubMed] [Google Scholar]
- 9.Potaman V. N., Shlyakhtenko L. S., Oussatcheva E. A., Lyubchenko Y. L., Soldatenkov V. A. 2005. Specific binding of poly(ADP-ribose) polymerase-1 to cruciform hairpins. J. Mol. Biol. 348: 609–615. [DOI] [PubMed] [Google Scholar]
- 10.Kun E., Kirsten E., Ordahl C. P. 2002. Coenzymatic activity of randomly broken or intact double-stranded DNAs in auto and histone H1 trans-poly(ADP-ribosylation), catalyzed by poly(ADP-ribose) polymerase (PARP I). J. Biol. Chem. 277: 39066–39069. [DOI] [PubMed] [Google Scholar]
- 11.Piao L., Fujioka K., Nakakido M., Hamamoto R. 2018. Regulation of poly(ADP-Ribose) polymerase 1 functions by post-translational modifications. Front. Biosci. 23: 13–26. [DOI] [PubMed] [Google Scholar]
- 12.Ba X., Garg N. J. 2011. Signaling mechanism of poly(ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am. J. Pathol. 178: 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraus W. L., Lis J. T. 2003. PARP goes transcription. Cell 113: 677–683. [DOI] [PubMed] [Google Scholar]
- 14.Virág L. 2005. Poly(ADP-ribosyl)ation in asthma and other lung diseases. Pharmacol. Res. 52: 83–92. [DOI] [PubMed] [Google Scholar]
- 15.Liu L., Ke Y., Jiang X., He F., Pan L., Xu L., Zeng X., Ba X. 2012. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-κB transcription in murine macrophages. Hum. Immunol. 73: 439–447. [DOI] [PubMed] [Google Scholar]
- 16.Veres B., Gallyas F., Jr., Varbiro G., Berente Z., Osz E., Szekeres G., Szabo C., Sumegi B. 2003. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem. Pharmacol. 65: 1373–1382. [DOI] [PubMed] [Google Scholar]
- 17.Andreone T. L., O’Connor M., Denenberg A., Hake P. W., Zingarelli B. 2003. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J. Immunol. 170: 2113–2120. [DOI] [PubMed] [Google Scholar]
- 18.Li H. Y., Pan L., Ke Y. S., Batnasan E., Jin X. Q., Liu Z. Y., Ba X. Q. 2014. Daidzein suppresses pro-inflammatory chemokine Cxcl2 transcription in TNF-α-stimulated murine lung epithelial cells via depressing PARP-1 activity. Acta Pharmacol. Sin. 35: 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gagné J. P., Moreel X., Gagné P., Labelle Y., Droit A., Chevalier-Paré M., Bourassa S., McDonald D., Hendzel M. J., Prigent C., Poirier G. G. 2009. Proteomic investigation of phosphorylation sites in poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase. J. Proteome Res. 8: 1014–1029. [DOI] [PubMed] [Google Scholar]
- 20.Kauppinen T. M., Chan W. Y., Suh S. W., Wiggins A. K., Huang E. J., Swanson R. A. 2006. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 103: 7136–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen-Armon M., Visochek L., Rozensal D., Kalal A., Geistrikh I., Klein R., Bendetz-Nezer S., Yao Z., Seger R. 2007. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol. Cell 25: 297–308. [DOI] [PubMed] [Google Scholar]
- 22.Vuong B., Hogan-Cann A. D., Alano C. C., Stevenson M., Chan W. Y., Anderson C. M., Swanson R. A., Kauppinen T. M. 2015. NF-κB transcriptional activation by TNFα requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. J. Neuroinflammation 12: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gongol B., Marin T., Peng I. C., Woo B., Martin M., King S., Sun W., Johnson D. A., Chien S., Shyy J. Y. 2013. AMPKα2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc. Natl. Acad. Sci. USA 110: 3161–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shang F., Zhang J., Li Z., Zhang J., Yin Y., Wang Y., Marin T. L., Gongol B., Xiao H., Zhang Y. Y., et al. 2016. Cardiovascular protective effect of metformin and telmisartan: reduction of PARP1 activity via the AMPK-PARP1 cascade. PLoS One 11: e0151845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegedus C., Lakatos P., Oláh G., Tóth B. I., Gergely S., Szabó E., Bíró T., Szabó C., Virág L. 2008. Protein kinase C protects from DNA damage-induced necrotic cell death by inhibiting poly(ADP-ribose) polymerase-1. FEBS Lett. 582: 1672–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo X., Nie J., Wang S., Chen Z., Chen W., Li D., Hu H., Li B. 2015. Poly(ADP-ribosyl)ation of FOXP3 protein mediated by PARP-1 protein regulates the function of regulatory T cells. [Published erratum appears in 2016 J. Biol. Chem. 291: 1201.] J. Biol. Chem. 290: 28675–28682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh D., Tsokos G. C. 2010. Spleen tyrosine kinase: an Src family of non-receptor kinase has multiple functions and represents a valuable therapeutic target in the treatment of autoimmune and inflammatory diseases. Autoimmunity 43: 48–55. [DOI] [PubMed] [Google Scholar]
- 28.Gu J. J., Ryu J. R., Pendergast A. M. 2009. Abl tyrosine kinases in T-cell signaling. Immunol. Rev. 228: 170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui L., Chen C., Xu T., Zhang J., Shang X., Luo J., Chen L., Ba X., Zeng X. 2009. c-Abl kinase is required for beta 2 integrin-mediated neutrophil adhesion. J. Immunol. 182: 3233–3242. [DOI] [PubMed] [Google Scholar]
- 30.Chen C., Ba X., Xu T., Cui L., Hao S., Zeng X. 2006. c-Abl is involved in the F-actin assembly triggered by L-selectin crosslinking. J. Biochem. 140: 229–235. [DOI] [PubMed] [Google Scholar]
- 31.Ba X., Chen C., Gao Y., Zeng X. 2005. Signaling function of PSGL-1 in neutrophil: tyrosine-phosphorylation-dependent and c-Abl-involved alteration in the F-actin-based cytoskeleton. J. Cell. Biochem. 94: 365–373. [DOI] [PubMed] [Google Scholar]
- 32.Tong H., Zhao B., Shi H., Ba X., Wang X., Jiang Y., Zeng X. 2013. c-Abl tyrosine kinase plays a critical role in β2 integrin-dependent neutrophil migration by regulating Vav1 activity. J. Leukoc. Biol. 93: 611–622. [DOI] [PubMed] [Google Scholar]
- 33.Luo J., Xu T., Li C., Ba X., Wang X., Jiang Y., Zeng X. 2013. p85-RhoGDI2, a novel complex, is required for PSGL-1-induced β1 integrin-mediated lymphocyte adhesion to VCAM-1. Int. J. Biochem. Cell Biol. 45: 2764–2773. [DOI] [PubMed] [Google Scholar]
- 34.Chen C., Shang X., Cui L., Xu T., Luo J., Ba X., Zeng X. 2008. L-selectin ligation-induced CSF-1 gene transcription is regulated by AP-1 in a c-Abl kinase-dependent manner. Hum. Immunol. 69: 501–509. [DOI] [PubMed] [Google Scholar]
- 35.Ba X. Q., Chen C. X., Xu T., Cui L. L., Gao Y. G., Zeng X. L. 2005. Engagement of PSGL-1 upregulates CSF-1 transcription via a mechanism that may involve Syk. Cell. Immunol. 237: 1–6. [DOI] [PubMed] [Google Scholar]
- 36.Chen C., Shang X., Xu T., Cui L., Luo J., Ba X., Hao S., Zeng X. 2007. c-Abl is required for the signaling transduction induced by L-selectin ligation. Eur. J. Immunol. 37: 3246–3258. [DOI] [PubMed] [Google Scholar]
- 37.Le Q., Daniel R., Chung S. W., Kang A. D., Eisenstein T. K., Sultzer B. M., Simpkins H., Wong P. M. 1998. Involvement of C-Abl tyrosine kinase in lipopolysaccharide-induced macrophage activation. J. Immunol. 160: 3330–3336. [PubMed] [Google Scholar]
- 38.Ke Y., Han Y., Guo X., Wen J., Wang K., Jiang X., Tian X., Ba X., Boldogh I., Zeng X. 2017. Erratum: PARP1 promotes gene expression at the post-transcriptional level by modulating the RNA-binding protein HuR. Nat. Commun. 8: 15191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visnes T., Cázares-Körner A., Hao W., Wallner O., Masuyer G., Loseva O., Mortusewicz O., Wiita E., Sarno A., Manoilov A., et al. 2018. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 362: 834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim I. K., Rhee C. K., Yeo C. D., Kang H. H., Lee D. G., Lee S. H., Kim J. W. 2013. Effect of tyrosine kinase inhibitors, imatinib and nilotinib, in murine lipopolysaccharide-induced acute lung injury during neutropenia recovery. Crit. Care 17: R114–R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aguilera-Aguirre L., Bacsi A., Radak Z., Hazra T. K., Mitra S., Sur S., Brasier A. R., Ba X., Boldogh I. 2014. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway. J. Immunol. 193: 4643–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo X., Kraus W. L. 2012. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 26: 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ba X., Gupta S., Davidson M., Garg N. J. 2010. Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes. J. Biol. Chem. 285: 11596–11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jog N. R., Dinnall J. A., Gallucci S., Madaio M. P., Caricchio R. 2009. Poly(ADP-ribose) polymerase-1 regulates the progression of autoimmune nephritis in males by inducing necrotic cell death and modulating inflammation. J. Immunol. 182: 7297–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galbis-Martínez M., Saenz L., Ramírez P., Parrilla P., Yélamos J. 2010. Poly(ADP-ribose) polymerase-1 modulates interferon-gamma-inducible protein (IP)-10 expression in murine embryonic fibroblasts by stabilizing IP-10 mRNA. Mol. Immunol. 47: 1492–1499. [DOI] [PubMed] [Google Scholar]
- 46.Kaplin A. I., Deshpande D. M., Scott E., Krishnan C., Carmen J. S., Shats I., Martinez T., Drummond J., Dike S., Pletnikov M., et al. 2005. IL-6 induces regionally selective spinal cord injury in patients with the neuroinflammatory disorder transverse myelitis. J. Clin. Invest. 115: 2731–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colicelli J. 2010. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 3: re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaalishvili G., Margiani D., Kutalia K., Suladze S., Zaalishvili T. 2010. Automodification of PARP-1 mediates its tight binding to the nuclear matrix. Biochem. Biophys. Res. Commun. 393: 123–125. [DOI] [PubMed] [Google Scholar]
- 49.Hantschel O. 2012. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer 3: 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maiani E., Diederich M., Gonfloni S. 2011. DNA damage response: the emerging role of c-Abl as a regulatory switch? Biochem. Pharmacol. 82: 1269–1276. [DOI] [PubMed] [Google Scholar]
- 51.Wang J. Y. 2014. The capable ABL: what is its biological function? Mol. Cell. Biol. 34: 1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright R. H., Castellano G., Bonet J., Le Dily F., Font-Mateu J., Ballaré C., Nacht A. S., Soronellas D., Oliva B., Beato M. 2012. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev. 26: 1972–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du Y., Yamaguchi H., Wei Y., Hsu J. L., Wang H. L., Hsu Y. H., Lin W. C., Yu W. H., Leonard P. G., Lee G. R., IV, et al. 2016. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. [Published erratum appears in 2016 Nat. Med. 22: 1192.] Nat. Med. 22: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu P., Usatyuk P. V., Lele A., Harijith A., Gregorio C. C., Garcia J. G., Salgia R., Natarajan V. 2015. c-Abl mediated tyrosine phosphorylation of paxillin regulates LPS-induced endothelial dysfunction and lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 308: L1025–L1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maruyama T., Nara K., Yoshikawa H., Suzuki N. 2007. Txk, a member of the non-receptor tyrosine kinase of the Tec family, forms a complex with poly(ADP-ribose) polymerase 1 and elongation factor 1alpha and regulates interferon-gamma gene transcription in Th1 cells. Clin. Exp. Immunol. 147: 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang H. D., Lee T. Y., Tzeng S. W., Horng J. T. 2005. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 33(Web Server issue): W226–W229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nguewa P. A., Fuertes M. A., Valladares B., Alonso C., Pérez J. M. 2005. Poly(ADP-ribose) polymerases: homology, structural domains and functions. Novel therapeutical applications. Prog. Biophys. Mol. Biol. 88: 143–172. [DOI] [PubMed] [Google Scholar]
- 58.Virág L., Szabó C. 2002. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 54: 375–429. [DOI] [PubMed] [Google Scholar]
- 59.Tinti M., Nardozza A. P., Ferrari E., Sacco F., Corallino S., Castagnoli L., Cesareni G. 2012. The 4G10, pY20 and p-TYR-100 antibody specificity: profiling by peptide microarrays. N. Biotechnol. 29: 571–577. [DOI] [PubMed] [Google Scholar]
- 60.Parrillo J. E. 1993. Pathogenetic mechanisms of septic shock. N. Engl. J. Med. 328: 1471–1477. [DOI] [PubMed] [Google Scholar]
- 61.Hassa P. O., Hottiger M. O. 2002. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell. Mol. Life Sci. 59: 1534–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tulin A., Spradling A. 2003. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299: 560–562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.