

Abstract
Background
Uncovering the evolutionary principles of gene coexpression network is important for our understanding of the network topological property of new genes. However, most existing evolutionary models only considered the evolution of duplication genes and only based on the degree of genes, ignoring the other key topological properties. The evolutionary mechanism by which how are new genes integrated into the ancestral networks are not yet to be comprehensively characterized. Herein, based on the human ribonucleic acid-sequencing (RNA-seq) data, we develop a new evolutionary model of gene coexpression network which considers the evolutionary process of both duplication genes and de novo genes.
Results
Based on the human RNA-seq data, we construct a gene coexpression network consisting of 8061 genes and 638624 links. We find that there are 1394 duplication genes and 126 de novo genes in the network. Then based on human gene age data, we reproduce the evolutionary process of this gene coexpression network and develop a new evolutionary model. We find that the generation rates of duplication genes and de novo genes are approximately 3.58/Myr (Myr=Million year) and 0.31/Myr, respectively. Based on the average degree and coreness of parent genes, we find that the gene duplication is a random process. Eventually duplication genes only inherit 12.89% connections from their parent genes and the retained connections have a smaller edge betweenness. Moreover, we find that both duplication genes and de novo genes prefer to develop new interactions with genes which have a large degree and a large coreness. Our proposed model can generate an evolutionary network when the number of newly added genes or the length of evolutionary time is known.
Conclusions
Gene duplication and de novo genes are two dominant evolutionary forces in shaping the coexpression network. Both duplication genes and de novo genes develop new interactions through a “rich-gets-richer" mechanism in terms of degree and coreness. This mechanism leads to the scale-free property and hierarchical architecture of biomolecular network. The proposed model is able to construct a gene coexpression network with comprehensive biological characteristics.
Electronic supplementary material
The online version of this article (10.1186/s12859-019-3035-7) contains supplementary material, which is available to authorized users.
Keywords: Genome, Network evolution, Network biology, Duplication genes, De novo genes
Background
Gene coexpression networks are of biological interest because coexpressed genes are controlled by the same transcriptional regulatory program, functionally related, or members of the same pathway. The development of an evolutionary model which can construct a gene coexpression network with comprehensive features is helpful for us to further understand the real gene regulation network [1]. It is also important for us to understand the network topological property of new genes and reveal the molecular mechanism underlying the connection between genotype and phenotype.
Gene coexpression network, as a basic biological network, exhibits the inherent characteristics and commonly evolutionary mechanism of biological networks. From the perspective of natural selection and biological evolution, duplication and mutation are the basic causes of evolution of biological molecular networks. The adding of duplication genes and de novo genes is the main reason why biomolecular networks are different from general social networks [2, 3]. Recent studies have proposed some general properties of biomolecular networks. These properties can be summarized as follows: (1) The degree distribution of genes in the network obeys to the power law distribution [4] and the power exponent is between 1 and 2 [5–7]. (2) These networks have a hierarchical structure. The average clustering coefficient of genes with degree k obeys that c(k)∼k−1 [8]. (3) These networks have a small world characteristic [9]. The mean pathlength of the network is small and the average clustering coefficient is relatively large [10–12]. (4) These networks are sparse, which means the average number of edges connected to the gene is small [13].
Based on the topological properties of gene networks, researchers speculated the original mechanisms of the network and proposed some evolutionary models of the network. Most of these evolutionary models only considered duplication genes, thus many gene duplication models were proposed. According to these models, some properties of biomolecular networks can be reproduced. In order to generate a scale-free network, a randomly evolutionary model was developed [14]. In this model, new nodes were added into the network and they connected to nodes with large degree in the network. However, this model cannot generate the network with power exponent smaller than 2 [10]. This may be due to the growth of biomolecular network only caused by gene duplication [15–19]. Hence, other evolutionary models with duplication genes were proposed. Owing to pure duplication model cannot support scale-free distribution, models contained duplication process and a second event were developed [20]. One new evolutionary model was based on gene duplication and re-wiring [21]. In the model, a parent gene was chosen randomly from the network. Next, edges from its parent genes would be deleted with fixed probability and then new connections would be created. Different from other models, the degree of duplication gene depended on the degree of its parent gene in this model. Networks simulated from this model showed small-world property and obeyed to scale-free distribution. Moreover, a mixed model which considered gene duplication and rewiring process was presented [22]. In the process of gene duplication, nodes were duplicated randomly from the network, and the connections were reserved. In rewiring process, new genes which the duplication genes connected to were chosen randomly. In this model, both gene duplication and rewiring process were randomly and would occur with the probability of one half. Other duplication-divergence models for evolution of biological network had also been proposed [23–25], such as duplication-divergence model with only one parameter [26] and duplication-divergence-heterodimerization model [27]. These models can fit to the scale-free, small-world and sparse properties of biomolecular network.
Particularly, some studies have considered the evolution of human gene coexpression network [28, 29]. These studies find that new genes (deoxyribonucleic acid-based (DNA-based) duplication genes, ribonucleic acid-based (RNA-based) duplication genes and de novo genes) showed preferential attachment in developing new links with other genes [28]. They tended to connect to genes with high topological centralities [28]. Interestingly, it is also found that the genes with large degree evolved more slowly and were more conserved than genes with small degree [29]. Hence, when choosing new partners for new genes, both the degree and other topological properties of the node should be taken into consideration.
For most of the previous evolutionary models, the parameter selections were not closely related to the biological experiment results. In addition, the existing evolutionary models were generally focused on the average degree, degree distribution and small-world properties of networks, rather than on the comprehensive topological properties of the biological network. They only considered the evolutionary process of duplication genes and only based on the degree of genes, ignoring the other important topological properties, such as the edge betweenness and coreness of genes. However, the coreness of gene can measure the depth of this gene in the network. Genes with large coreness means they are close to the center of this network. This property may constrain the growth of genes with large degree but less important. Both the edge betweenness and coreness of genes play an important role in determining how are new genes integrated into the ancestral network. Therefore, in this paper, based on the comprehensive topological properties of genes, we propose a novel evolutionary model for constructing the human gene coexpression network. The model we developed is different from previous models which only considered the duplication genes during evolutionary process, our model also considers the effect of de novo genes. For duplication genes, our model considers the duplication process and rewiring process. For de novo genes, we consider the process of new interactions. We analyze the specific mechanism of these evolutionary processes. According to this model, we can construct a gene coexpression network with comprehensive biological characteristics.
Results
Through analyzing the network 0-6 (see Methods), we reproduce the evolutionary process of human gene coexpression network. The generation rates of duplication genes and de novo genes can be estimated and the mechanism of these genes integrated into the network will be inferred. Finally, we propose a novel evolutionary model of human gene coexpression network and predict the growth of new network which is evolved from this original network after a period of time.
Rate of gene generation
Based on the gene origination mechanism data (see Additional file 1) [30], we obtain Table 1. From Table 1, we can see that gene duplication and de novo are two dominant evolutionary forces in shaping the gene coexpression network. By counting the number of duplication genes and de novo genes that added to each of the network, we can estimate the average rate of producing duplication genes and de novo genes. Assuming that the network 0 which is consisted of parent genes is the initial network. The network evolved from the original network by adding new genes is the new network.
Table 1.
Number of newly added genes in each branch
| New branch | Number of new genes | Number of duplication genes | Number of de novo genes |
|---|---|---|---|
| 1 | 780 | 708 | 72 |
| 2 | 168 | 150 | 18 |
| 3 | 210 | 185 | 25 |
| 4 | 271 | 260 | 11 |
| 5 | 22 | 22 | 0 |
| 6 | 69 | 69 | 0 |
This table shows number of genes (new genes, duplication genes and de novo genes) that added into each network
If the generation rates of duplication genes, de novo genes and newly added genes (new genes consist of duplication genes and de novo genes) are assumed to be a constant, then the average generation rates of these kinds of genes during the network evolution are calculated as the mean value of generation rates from network 0 to network 6. By direct calculation, we obtain that the generation rates of duplication genes, de novo genes and newly added genes are 3.58/Myr, 0.31/Myr and 3.89/Myr, respectively (Fig. 1, Table 1).
Fig. 1.
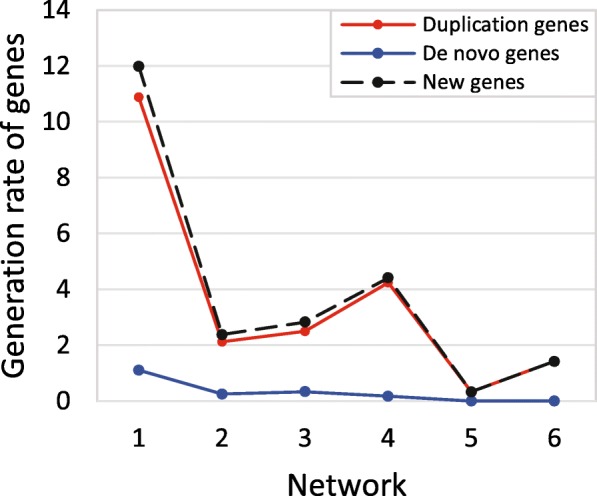
The generation rates of genes with different mechanisms. By calculating the average generation rates of duplication genes, de novo genes and all newly added genes, we obtain that the generation rates of duplication genes, de novo genes and newly added genes are 3.58/Myr, 0.31/Myr and 3.89/Myr, respectively
Random gene duplication
Assuming that the newly added duplication genes in new network is vc. The corresponding parent in the original network is vp. The degree vector and coreness vector of parent gene vp in the original network is and , respectively. Then the mean value of all elements in the degree vector dp is the average degree of parent genes in the initial network. Similarly, the average coreness of parent genes in the initial network is calculated in the same way.
In our model, we propose that parent genes are randomly chosen from the original network. From Fig. 2, we can see that the degree and coreness of parent genes are always fluctuating around the corresponding mean value of the network. Furthermore, we obtain that from network 0 to network 5 the proportions of parent genes whose degree are larger than the median of network are 52.52%, 58.11%, 50.85%, 59.42%, 20% and 45%, respectively. Similarly, the proportions of parent genes whose degree are larger than the median of network are 53.05%, 59.46%, 47.46%, 59.42%, 20% and 45% from network 0 to network 5. Therefore, we discover that gene duplication is a random process.
Fig. 2.

Random selection of parent genes. a The average degree of network genes and parent genes. b The average coreness of network genes and parent genes. From network 0 to network 6, the degree and coreness of parent genes are fluctuating near the average value of corresponding network
In addition, we also take the KEGG enrichment analysis for duplication genes at pathway level. However, there are no significant enrichment for duplication genes at pathway. For duplication genes of branch 3, there are sixteen genes enriched in 5 pathways. And for duplication genes of branch 1, branch 2 and branch 6, there are no enrichment gene sets. For duplication genes in branch 4 and branch 5, they both have only one enrichment set, which consisted of four and three genes, respectively. Hence, gene duplication is also a random process at the pathway level.
In summary, we conclude that gene duplication is a random process in which a parent gene is randomly selected from the original network to duplicate. Shortly after a gene duplication, the parent gene and duplication gene will interact with the same genes.
Reservation rate of interaction
Shortly after gene duplication, duplication genes will inherit all connections of parent genes. From Fig. 3, we can see that the duplication genes will eventually lose common connections with larger edge betweenness and reserve the connections with smaller edge betweenness. The edge with larger edge betweenness in the network plays an important role in the information propagation of the network. The deletion of these edges can reduce the redundancy of the network and the remaining edges and rewiring edges will establish new important pathways of the network.
Fig. 3.
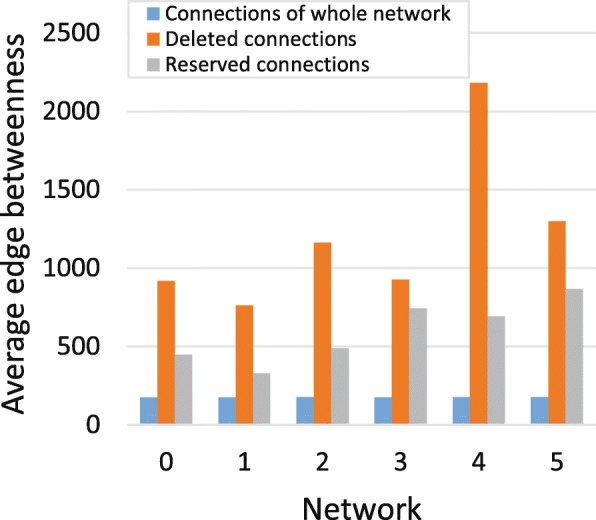
Edge betweenness of deleted edges and reserved edges for duplication genes. This figure shows that duplication genes will lose common edges with larger edge betweenness and reserve the connections with smaller edge betweenness
To simulate the evolution of network, we have to calculate the average reservation rate of edges of duplication genes. Assuming that the degree of parent gene is dp, the number of reserved edges of duplication gene is dpub, then the reservation rate α of edges of this duplication gene is defined as:
| 1 |
We find that the average reservation rates from network 1 to network 6 are 4.83%, 6.56%, 10.45%, 7.85%, 5.90% and 41.75%, so the average reservation rate of edges of duplication genes during the evolution of network is 12.89%.
Rewiring rate of duplication genes
Duplication genes will build new interactions with the remaining nodes in the network after loss of common interactions. From Fig. 4, we can see that the duplication genes prefer to connect to nodes with large degree and large coreness. Assuming that the duplication genes newly added in the network are denoted as , then the total number of reconnected nodes for vector vc in the initial network is nre, then the reconnected vector corresponding to the vector vc is . The degree vector of reconnected vector vc is . The average degree of reconnected vector is calculated as the mean value of all elements in vector dre, and the average coreness of reconnected vector is calculated in the same way. Form network 0 to network 5, the proportions of reconnected genes whose degree larger than the mean value of network are 93.45%, 87.75%, 96.31%, 90.02%, 83.16% and 92.20%, respectively. Similarly, the proportions of reconnected genes whose coreness larger than the mean value of network are 93.41%, 87.22%, 96.37%, 89.06%, 82.28% and 92.92%, respectively. Hence, we conclude that duplication genes will connect to nodes in the network with large degree and large coreness (Fig. 4).
Fig. 4.

Rewiring mechanism of duplication genes. a The average degree of whole network genes and the genes that establish new connections to duplication genes. b The average coreness of whole network genes and reconnected genes. The genes that the duplication genes establish newly connections are defined as rewiring genes. This figure indicates that average degree and average coreness of rewiring genes are much larger than the average value of corresponding networks
In other words, during the rewiring process, duplication genes develop new interactions through a “rich-gets-richer" mechanism [11]. However, this mechanism here means they prefer to connect with nodes whose degree and coreness are both large at the same time. Degree shows the number of its neighbours and coreness indicates importance of this node. Therefore, the more important and more connected a node is, the more possible this node will be connected by duplication genes. As a result, genes with larger degree and larger coreness will acquire new neighbours faster than other genes. Finally, the scale-free distribution of degree will be formed.
In order to simulate the evolution of network, we need to estimate the average rewiring rate of duplication genes. The rewiring rate β of duplication gene i is defined as:
| 2 |
where is the number of rewiring nodes for duplication gene i. The average rewiring rates for network 1 to network 6 are 102.60%, 58.72%, 148.37%, 80.61%, 43.65% and 44.66%. Therefore, the average rewiring rate for duplication gene during network’s evolution is 79.77%.
In summary, gene duplication is a random process and shortly after gene duplication, duplication genes inherit all common interactions of their parent genes. Then they will delete edges with large edge betweenness and reconnect to the remaining nodes in the network with large degree and large coreness.
Connection rate of de novo genes
The addition of de novo genes is another important factor that promotes the evolution of network. Firstly, we estimate the average connection rate of de novo genes. This rate ζ is calculated as:
| 3 |
where is the degree of de novo gene i and 〈d〉 is the mean degree of the original network. Owing to there are no de novo genes added in the network 5 and network 6, we only calculate the average connection rate for network 1 to network 4. From network 1 to network 4, the average connection rates are 126.02%, 78.03%, 100.97% and 92.12%, respectively. Thus, the average connection rate for de novo genes under the evolution of network is 99.29%.
For de novo genes, we find that they also prefer to connect with nodes whose degree and coreness are both large in the initial network. Next, we will show that de novo genes will connect to nodes with large degree and large coreness.
The de novo genes that added in the initial network are noted as a vector . Assuming that the total number of de novo genes’ neighbours is nco, and they constitute the vector . The degree vector corresponding to is , and the corresponding coreness vector is . In Fig. 5, the average degree and coreness of de novo genes’ neighbours are much larger than that mean value of the original network. From network 0 to network 3, the proportions for neighbours whose degree are larger than the median of the original are 94.66%, 89.70%, 94.31% and 91.95%, respectively. Similarly, the proportions for neighbours whose coreness are larger than the median of initial network are 94.61%, 90.83%, 95.90% and 91.22%, respectively.
Fig. 5.

Connection mechanism of de novo genes. a The average degree of network genes and genes that connect to de novo genes. b The average coreness of network genes and genes that connect to de novo genes. From network 0 to network 3, the average degree and coreness of neighbours of de novo genes are much larger than the mean value of corresponding networks
Therefore, de novo genes will establish connections with genes whose degree and coreness are both large in the initial network. This mechanism also cause genes with many neighbours and more importance in the network connect to new neighbours faster than other genes. So the genes in the center of network will grow rapidly. Genes will connect to the de novo genes until the degree of de novo gene is around the average degree of the original network.
Two novel evolutionary models for constructing gene coexpression networks
This section, we will introduce our evolutionary models for constructing gene expression networks. The flow charts of the traditional model and our novel model are shown in Fig. 6. Supposing that the number of genes in the initial network is N. The degree of node i is di and its coreness is ksi. Based on the above network evolution parameters and evolutionary principles of genes, we obtain the schematic diagram of evolutionary model for constructing a gene coexpression network, which is shown in Fig. 7. The new network is being formed step by step. Here we assume that it will take six steps to form a new network. In each step, the reservation rate and rewiring rate of duplication gene is α and β, respectively. The connection rate of de novo genes is γ. In this paper, we have estimated that α=12.89%,β=79.77% and γ=99.29%. Moreover, the model we proposed is suitable for two situations. In situation 1, we assume that for each step, the number of newly added duplication genes and de novo gene is known. In situation 2, we assume that the number of newly added genes is unknown, but the length of evolutionary time in each step is known. The detailed simulation processes of these two models are described as follows.
Fig. 6.

Flow chart of the traditional evolutionary model and our novel evolutionary model. a The flow chart of the traditional evolutionary model. Moreover, the probability δ of edges to be removed sets to 0.8711 in our simulation. b The flow chart of our novel evolutionary models for constructing gene coexpression networks. The novel model considers the adding of duplication genes and de novo genes at the same time while the traditional model only contains duplication genes
Fig. 7.

Schematic diagram of the evolutionary model for constructing a gene coexpression network. Process (a) is the evolutionary process of duplication genes. Gene 7 is a duplication gene of gene 3. Besides, the probability that gene 3 chosen as a parent gene is . Process (b) is the evolutionary process of adding de novo genes into the network. Gene 8 is a de novo gene. Process (c) integrates the results of process (a) and process (b), and produces the final evolutionary network
Evolutionary model 1: when the number of new genes in each step is known
In this situation, we assume that it will take six steps to form a final network. For each step, the number of newly added genes n=n1+n2 is known, where n1 and n2 are respectively the number of newly added duplication genes and de novo genes. We start from the network 0 which has N=6541 genes, and in each step we perform the following operations (The R code for running this model is appended in Additional file 2,, and the support data are in Additional files 3, 4 and 5): (a) Evolutionary process of duplication genes (a-1) Gene dupication The probability that the gene i(i=1,2,...,N) is chosen as a parent gene vp is
| 4 |
The duplication gene of gene i is denoted as vc and it reserves all interactions of vp. (a-2) Edge deletion process The degree of parent gene vp is dp and the edge betweenness of parent gene’s connections are Bij(j=1,2,...,dp). The edge i−k (gene k is a neighbour gene of the parent gene) is reserved with the probability
| 5 |
Finally, the number of reserved edge is [αdp] and the other edges will be deleted. (a-3) Rewiring process The probability that the remaining gene i(i=1,2,...,N−dparent) in the initial network is chosen to connect with duplication gene vc is
| 6 |
where ksi is the coreness of gene i and di is the degree of gene i. The number of reconnected nodes is [〈dold〉×β], where 〈dold〉 is the average degree of the initial network. (b) Evolutionary process of de novo genes Process (a) and process (b) are parallel process, they can be produced at the same time from the original network. (b-1) De novo gene The de novo gene generates and added to the original network. (b-2) New connection For de novo gene vnovo, the probability that node i(i=1,2,...,N) in the initial network connected to this de novo gene is
| 7 |
where ksi is the coreness of gene i and di is the degree of gene i. De novo genes will establish connections with genes in the initial network until the degree of de novo genes is [〈dold〉×γ]. (c) Combination of process (a) and (b) Assuming that there are n1 duplication genes and n2 de novo genes added into the original network. Then the original network will evolve until all these n=n1+n2 new genes are added into the network. Combining the results of the process (a) and process (b), we obtain a new network.
Evolutionary model 2: when the length of evolutionary time in each step is known
In this situation, we assume that the number of newly added genes in each step is unknown, but the length of evolutionary time t(Myr) is known. Different from the evolutionary model 1, the number of new genes are unknown in model 2. Therefore, model 2 will be more effective in the prediction problem where only the evolutionary time is known. We will take six steps to form a final network. According to the above analysis (Fig. 1), we obtain that the average generation rates of duplication genes and de novo genes are respectively 3.58/Myr and 0.31/Myr. Hence, in each step, the number of newly added duplication genes and de novo genes are 3.58ti and 0.31ti (i=0,1,...,5), respectively. Here, ti is the evolutionary time from branch i to the next branch (i+1). The initial network and evolutionary process (a) to process (c) is the same as that in situation 1. However, the terminating condition of this model is different from that in situation 1. In this situation, the new network evolves by adding 3.58ti duplication genes and 0.31ti de novo genes into the original network. (The R code for running this model is appended in Additional file 6, and the support data are in Additional files 3, 4, 5, and 7).
Simulation results
Given initial network 0, based on the above evolutionary model, we simulate the evolutionary process of gene coexpression network under two situations. In situation 1, the number of genes that newly added into the network is known. In situation 2, the length of evolutionary time in each step is known. The relative errors of topological properties of gene coexpression network (such as average degree, clustering coefficient, pathlength, node betweenness, edge betweenness) are defined as:
| 8 |
where a is the simulated value and A is the real value of gene coexpression network. Comparing σ based on our improved model with that corresponding traditional model, we can test the accuracy of our model. For traditional model, all newly added genes are considered as duplication genes, so the generation rate of genes are set as 3.89/Myr, which is the average generation rate of all newly added genes. Moreover, in the traditional model the probability δ that how many edges of duplication gene will be removed is unknown. Here we set the probability δ equals to 1−α, where α is the reservation rate and can be estimated from the real network.
By comparing the simulation result of traditional model with our improved model in situation 1 (Fig. 8) and situation 2 (Fig. 9), we find that the topological properties of simulation network obtained by our improved model are more similar to that of the real gene coexpression network. Specifically, when the number of newly added genes is known, by using our improved model 1, we obtain evolutionary networks 1-6. We find that the average relative errors of networks 1-6 in average degree, transitivity, pathlength, node betweenness and edge betweenness are 0.63%, 3.96%, 6.81%, 4.78% and 4.83%, respectively. However, under traditional model, the average relative errors of these five topological properties are 0.63%,7.43%, 13.20%, 9.36% and 9.43%, respectively, which are much bigger than that of our improved model 1. Moreover, when the length of evolutionary time in each step is given, by using our improved model 2 we obtain the evolutionary networks 1-6. We find that the average relative errors of these five properties are 2.23%, 4.01%, 7.75%, 5.51% and 5.41%, respectively. Yet for traditional model, the corresponding relative errors are 2.18%, 7.50%, 15.62%, 11.16% and 11.08%, respectively, which are much bigger than that of our improved model 2. Comparing with the evolutionary models which only consider the degree property of genes, we can see that the simulation networks obtained by our improved model are more similar to the real gene coexpression network in the five basic topological properties. Hence, the comprehensive characteristics (such as average degree, transitivity, pathlength, node betweenness and edge betweenness) of the evolutionary network obtained by our improved model are more closer to that of the real network.
Fig. 8.

Topological properties of simulation network in situation 1. In situation 1, the number of newly added duplication genes and de novo genes are given. We obtain two simulation networks based on the traditional model and our improved model and compare the topological properties of these two kinds of simulation networks from (a) Average degree, (b) Transitivity, (c) Pathlength, (d) Node betweeness and (e) Edge betweenness. We take the reservation rate and rewiring rate of duplication gene, and the connection rate of de novo genes be α=12.89%,β=79.77% and γ=99.29%, respectively
Fig. 9.

Topological properties of simulation network in situation 2. In situation 2, the number of newly added duplication genes and de novo genes are unknown. For given length of evolutionary time, the number of newly added genes are estimated according to the generation rates of genes. We obtain two simulation networks based on the traditional model and our improved model and compare the topological properties (a Average degree, b Transitivity, c Pathlength, d Node betweeness and e Edge betweenness) of these two kinds of simulation networks. We take the average generation rates of duplication genes and de novo genes are respectively 3.58/Myr and 0.31/Myr
Evolution of network topological properties of new genes
Based on the gene coexpression network and gene age, we analyze how the network topological properties of new genes in branch 1 evolve. We assume that the network 0 is the initial network. This coexpression network will gradually grow with the continual addition of new genes. In branch 1 there are 780 new genes which are generated from 361.2 to 324.5 million years ago. In particular, we analyze how the average degree, transitivity, pathlength, node betweenness and edge betweenness of these new genes in branch 1 change over the evolutionary time (Fig. 10). We find that by continually adding of new genes into the coexpression network, the new genes in branch 1 will gain more and more neighbors, their average degree will become larger and larger. The new connections between the new genes in branch 1 and their new neighbours facilitate the formulation of the shortest paths through these new genes in branch 1. Therefore, the average node betweenness and edge betweenness contain these new genes in branch 1 will be gradually increased. During the evolutionary process, the newly added genes which connect to the new genes in branch 1 will establish new connections with their neighbours, then the average transitivity of these new genes in branch 1 will substantially retain. The pathlength of new genes in branch 1 will also be stable because these new genes may own the same topological location as their neighbours. In other words, during the evolution of genetic networks, the hierarchical architecture and overall navigability of new genes will remain the same.
Fig. 10.

Evolution of network topological properties of new genes in branch 1. Along with the new genes added into the network, the network topological properties of new genes in branch 1 will change. a Average degree, b Transitivity, c Pathlength, d Node betweeness and e Edge betweenness of the new genes in branch 1
Results of sensitivity analysis
In our analysis, due to the evolutionary processes of each branch are the same in two situations, we only consider the network evolved from branch 0 when the number of added genes is known by our novel model. The parameter variables we considered are the generation rate, the retention rate, and the rewiring rate of duplication genes and the generation rate, the connection rate of de novo genes. The topological properties we have considered are the average degree, transitivity, pathlength, node betweenness, and edge betweenness of the network. The standard value of index A is the corresponds values of topological features in simulation network 1 (the number of new genes is known, Fig. 8). To calculate the sensitivity coefficients of the evolutionary network, we take the |△F/F|=20%. That is to say, the change in the value of the parameter variable eauqals to 0.2F, where F is the corresponding value in our evolutionary model 1. Based on Fig. 11, we discovered that the rewiring rate of duplication genes is the most sensitive parameter variable to the topological features of evolutionary network. The sensitivity coefficient of the rewiring rate of duplication genes to the edge betweenness is 0.182. The change of the rewiring rate of duplication genes has a significant effect on the five properties of the network, especially on the edge betweenness and the average degree of this network. In additions, edge betweenness and average degree will be affected easier by all five parameters than other topological properties in general.
Fig. 11.

Sensitivity coefficients of the topological properties of network 1. The topological property in Figs a, b, c, d and e is degree, transitivity, pathlength, node betweenness and edge betweenness, respectively. We calculated the sensitivity coefficients of the five parameter variables(the generation rate, the retention rate, and the rewiring rate of duplication genes, and the generation rate and the connection rate of de novo genes) to the five topological properties(degree, transitivity, shortest pathlength, node betweenness and edge betweenness) respectively. We take the change of the parameter variables to be 20%.
Discussion
Understanding the evolutionary mechanism and topological properties of human gene coexpression network is important for identifying the function and evolution of new genes [3, 21]. In this paper, based on the human RNA-seq data, we construct a human gene coexpression network. Then according to the human gene age data, we reproduce the evolutionary process of this network and develop a new evolutionary model of gene coexpression network, which help us discover the evolutionary mechanism of biomolecular networks.
Moreover, the previous evolutionary models [22, 31] only focus on the duplication genes and ignore the role of de novo genes. However, based on the human gene origination mechanism data, we find that gene duplication and de novo are two dominant evolutionary forces in shaping this gene coexpression network. Therefore, except for the duplication genes, we need to consider how many de novo genes are produced at a given time and how are de novo genes integrated into the initial network and change the topological properties of gene-gene interaction network.
Our improved evolutionary model contains the evolutionary processes of both duplication genes and de novo genes. For the duplication model, we analyze how do the duplication genes choose the neighbour genes to connect. Traditional models find that duplication genes choose neighbours according to the degree of genes [32, 33]. In our study, we find that both the coreness of genes and the edge betweenness of connections will affect how the duplication genes choose their neighbours. In particular, shortly after gene duplication, duplication genes will reserve all connections of parent genes and eventually edges with larger edge betweenness will be deleted. Edge betweenness measures the ability of this edge in delivering information. Deleting edges with large edge betweenness and building new interactions with other genes can reduce network redundancy and enable this network to have new biological pathways. This can improve the efficiency of information transmission in the network and make this network have a small world property. After the duplication genes lose the common interactions, we find that they will connect to genes with large degree and large coreness. The larger the degree and coreness of genes, the more neighbours they will have in the network. The coreness of gene measures the depth of this gene in the network. Genes with large coreness mean that they are close to the center of this network. This evolutionary mechanism not only satisfies the traditional “rich-gets-richer" mechanism for degree, but also constrains the growth of genes with large degree but less important. This mechanism makes the evolutionary networks generating from our model are more similar to the real gene coexpression network. For de novo genes, we also find that they prefer to connect genes with large degree and large coreness. This preference attachment mechanism fundamentally explains why the biomolecular network shows the property of scale-free and hierarchical architecture. In addition, we analyze how the network topological properties of new genes in branch 1 evolve. Through the evolutionary analysis step by step, we find that the hierarchical structure and overall navigability of new genes will remain the same with the increase of time. The sensitivity analysis of the evolutionary network also has been done in this paper. The change of the rewiring rate of duplication genes has a tremendous effect on the five properties of the network, and edge betweenness and degree will be affected easier than other topological properties.
In general, the main novelty of this study is reflected in the following three aspects. First, when we develop the evolutionary model we consider the evolutionary process of both duplication genes and de novo genes. Second, in order to determine how are new genes integrated into the network, we not only consider the average degree of genes, but also take coreness and edge betweenness into consideration. In other words, in order to develop an evolutionary model with comprehensive biological characteristics, we should consider the different topological properties of genes and the underlying universal principle of gene-gene interactions. Third, the network evolution parameters and evolutionary principles of genes are inferred from the real network constructed with actual gene expression data. However, the observations made here stimulate a multitude of questions regarding their evolutionary significance. Is the change in network structure driven by neutral evolution or by natural selection for advantageous interaction patterns? Does the change in network configurations over time reflect different environments or different adaptations that the organism evolves at different times? These problems require further study from the theoretical and experimental perspectives.
This study has several limitations. Firstly, in this gene coexpression network the strength of connections is ignored, that is, the network is unweighted. But for a real gene coexpression network, the strength of connections is different and the correlation between genes may be positive or negative. Secondly, when we consider the evolutionary process of gene coexpression network, we assume that the parental genes and other existing genes in the network do not lose their interactions during evolution. Furthermore, we only considered the generation of new genes in our evolutionary model rather than the deletion of the original genes. More realistically, it is also likely that a certain amount of “rewiring" takes place during the network growth for the parental genes and other existing genes. Thirdly, if we can collect more human RNA-seq data and update the dataset of gene age and gene origination mechanism, we will be able to more accurately reveal the evolutionary mechanism of gene coexpression network. Finally, in this study we only consider the evolutionary model of human gene coexpression network. It is interesting to further investigate the evolutionary model of gene coexpression network in other species and compare the difference with this model.
Conclusions
In conclusion, based on the human RNA-seq data and human gene origination mechanism data, this study develops a new evolutionary model of gene-gene interaction network which considers the evolutionary process of both duplication genes and de novo genes. For the evolutionary process of duplication genes, based on the average degree and coreness of parent genes, we find that gene replication is a random process in which a parent gene is randomly selected from the original network to duplicate. Shortly after gene duplication, duplication genes will inherit all connections from their parent genes, but they eventually lose the common connections with larger edge betweenness and only reserve the connections with smaller edge betweenness. Moreover, we find that duplication genes prefer to develop new interactions with genes which have a large degree and a large coreness. In other words, the duplication genes develop new interactions through a “rich-gets-richer" mechanism. For de novo genes, we also find that they prefer to connect genes with large degree and large coreness. The mechanism of preferential gene attachment fundamentally leads to the scale-free property and hierarchical architecture of biomolecular network. In particular, we find that the topological properties of evolutionary network obtained by our improved model are more similar to that of the real gene coexpression network. During the evolutionary process, the hierarchical structure and overall navigability of new genes will remain the same. Our newly developed evolutionary model reveals the potential evolutionary mechanism of biomolecular networks and can generate evolutionary networks with comprehensive biological characteristics.
Methods
RNA-seq data of human
RNA-seq technology [34, 35] is a recently developed approach to transcriptome profiling that uses deep-sequencing technologies. RNA-seq technology provides a far more precise measurement of levels of transcripts and their isoforms than other methods [36]. In this study, we use the gene expression data from RNA-seq experiments which measured 14341 different genes in 71 tissues of human, which is downloaded from reference [28] (http://longlab.uchicago.edu/?q=SD_GB) (see Additional file 8). Since the expression data of some genes are missing in most organs, this data needs to be preprocessed. In order to construct a gene coexpression network, we need to choose the organs with the most abundant gene expression data and the genes with relatively strong expression level. Finally we select 26 different tissues in which all of 8237 genes have expression data (see Additional file 9).
Construction of a human gene-coexpression network
The gene coexpression network we construct is an undirected and unweighted network. For each differently expressed human gene, the expression data in each of the 26 organs are used to construct a vector as the expression data of this gene [29]. Different genes represent different nodes in the network and they are connected when their Pearson correlation coefficient is larger than a specific threshold. Because a biological network generally obeys to a power law distribution and has a hierarchical structure [22], based on the two criterions we calculate Pearson correlation coefficient between all pairs of selected genes to determine the threshold of our network. Network with scale-free property means that the degree distribution of the network exhibits a power tail with an exponent γ. And for the biological network, the exponent γ is often between 1 and 2 [7, 12]. For biological networks, it also reports that the clustering coefficient c(k)∼k−1, here k is the degree of the node [37].
When the threshold of Pearson correlation coefficient is set to 0.6, the exponent of degree distribution γ equals to 0.93, the network does not approximate to a power law distribution completely. When the threshold is set to 0.7 and 0.8, both of the networks approximate to a power law distribution and have the hierarchical structure. However, the network with threshold 0.7 includes more genes and links (Fig. 12). Hence, we set the threshold of Pearson correlation coefficient to 0.7. In this case, the network has 8061 genes and 638624 links (see Additional file 10).
Fig. 12.

Degree distribution and clustering coefficient of gene coexpression networks with different thresholds of Pearson correlation coefficient. a ∼ c The degree distribution of networks when the threshold of Pearson correlation coefficient is 0.6, 0.7 and 0.8, respectively. The value of γ in the figure is the slope of fitted line. d ∼ f The clustering coefficient distribution of networks when the threshold of Pearson correlation coefficient is 0.6, 0.7 and 0.8, respectively. Similarly, we calculate the slope γ of the fitted line
Topological properties of gene networks
The basic topological properties of biological networks are average degree, node betweenness, edge betweenness, clustering coefficient of node, coreness of node and the shortest pathlength. We use these topological properties to derive the evolutionary model and compare our simulated networks with real networks. (i) Average degree Assuming that the undirected and unweighted network has N nodes and M edges, the degree di of the node i is defined as the number of nodes directly connecting to node i. The average degree 〈d〉 of network is defined as the average degree of all nodes in the network, which is given by
| 9 |
(ii) Clustering coefficient The clustering coefficient ci of a node measures how closely its neighbours are connected. The clustering coefficient is between 0 and 1 and is calculated as follows
| 10 |
where Ei is the number of edges among the neighbours of node i, di is the degree of node i. ci equals to 0 when node i has at most one neighbour or its neighbours have no connections between each other. The average clustering coefficient 〈c〉 of network is defined as
| 11 |
Moreover, c(k) is defined as the average of clustering coefficients of all nodes with k links. If c(k)∼k−1, then the network may have a hierarchical architecture. (iii) Shortest pathlength The shortest pathlength of the network, also called geodesic path is defined as a path with the least number of edges connected to two nodes. The length of the shortest path is the number of the edges in this path. The average shortest pathlength 〈l〉 of the network is defined as
| 12 |
where lij is the shortest pathlength between node i and node j. In particular, the average pathlength of an undirected network is defined as the average distance between nodes with connected paths. (iv) Node betweenness Node betweenness measures the node’s ability in controlling information flow of the network. Node betweenness is defined as the ratio of the number of shortest paths passing through this node to the total number of shortest paths [38]. Assuming that the number of the shortest paths between node j and node k is njk and the number of edges passing through node i is njk(i), then the node betweenness bi of this node is defined as
| 13 |
and the average node betweenness 〈b〉 of the network is defined as
| 14 |
(v) Edge betweenness Edge betweenness is defined as the ratio of the number of shortest paths passing through this edge E to the total number of shortest paths [39, 40]. The edge betweenness BE of edge E is calculated as follows
| 15 |
where njk(E) is the number of shortest paths which connect node j and node k and pass through edge E at the same time. The average edge betweenness of a network, 〈B〉, is defined as the average of BE over all edges E.
From the perspective of information spreading, both node betweenness and edge betweenness can characterize the importance of this node or edge in the network. The higher the betweenness is, the greater the impact it has on the transmission of network information. (vi) Node coreness The average degree indicates the number of neighbours of each gene, but it cannot measure the significance of nodes in the network. Node betweenness can estimate the essentiality of nodes in message passing. However, it could not judge the location of nodes. We use k-shell decomposition [41, 42] to weigh the location and importance of nodes [43]. Nodes near the center of the network will have more importance than nodes at the border of the network. The k-shell decomposition can be actualized as follows: firstly, all isolated nodes in the network will be identified. These isolated nodes constitute 0- shell of the network with coreness of 0, and then they will be removed from the network. Secondly, removing the nodes with degree of 1 and simultaneously deleting their edges. In this way we obtain a new network. Continue to remove the nodes with degree of 1 and their connections from the new network until the degree of all nodes in the network is greater than 1. All nodes removed in this step constitute network’s 1-shell and their coreness is 1. Thirdly, continue to conduct shell decomposition until the degree of nodes in the remaining network is at least equal to 3. Thus, the network’s 2-shell is obtained and the coreness of them is 2. The decomposition will be continue until no node is remained in the network. Ultimately, each node in the network corresponds to a unique k- shell [44]. The coreness of node i is ks(i) and the average coreness 〈ks〉 of a network with N nodes is
| 16 |
Obviously, the greater the coreness of a node, the more important this node is.
Gene age and origination mechanism data
In order to study the evolutionary process of this gene coexpression network, we need to reproduce the evolution of network based on human gene age data. Evolutionary time tree of life is essentially needed in exploring the evolution of gene coexpression network. Data from http://gentree.ioz.ac.cn/index.php [30] shows us the gene age and origination mechanism from the perspective of human (see Additional files 1 and 11). All human genes are divided into 14 branches (branch 0 to branch 13). The younger the gene is, the greater number it is assigned to. New genes are mainly produced by two mechanisms which are gene duplication and de novo origination, and these new genes will lead to the evolution of the network. Each branch has its own splitting time, and age of genes in each branch is defined as the midpoint of the splitting time of this branch and the next branch [30]. Concretely, genes are divided into 14 branches by their age as listed in Table 2.
Table 2.
Gene age and numbers of each branch
| Branch | Number of genes in the network | Splitting time (Myr) | Gene age (Myr) | Time interval (Myr) |
|---|---|---|---|---|
| 0 | 6541 | -454.6 | -407.9 | 0 |
| 1 | 780 | -361.2 | -342.85 | 65.05 |
| 2 | 168 | -324. | -272.35 | 70.5 |
| 3 | 210 | -220.2 | -198.15 | 74.2 |
| 4 | 142 | -176.1 | -140.4 | 57.75 |
| 5 | 129 | -104.7 | -101.05 | 39.35 |
| 6 | 16 | -97.4 | -94.2 | 6.85 |
| 7 | 6 | -91 | -67.6 | 26.6 |
| 8 | 20 | -44.2 | -36.9 | 30.7 |
| 9 | 10 | -29.6 | -24.2 | 12.7 |
| 10 | 9 | -18.8 | -16.95 | 7.25 |
| 11 | 11 | -15.1 | -10.6 | 6.35 |
| 12 | 11 | -6.1 | -3.05 | 7.55 |
| 13 | 8 | 0 | 0 | 3.05 |
This table exhibits the branch spitting time, gene generation time and the generation interval of genes between adjacent branches from branch 0 to branch 13
To analyze the evolution of network, we have to study changes of network properties in similar time interval. However, from the right-hand side of the Table 2, we can see that the time interval of gene generation is nonuniform. Moreover, when the age is divided in a precise way, the number of newly added genes in every network is very small, so the law of evolution will be not obvious. Hence, accurate partitions of gene age are not suitable for us to study the network evolution. Therefore it is necessary to regroup the 0-13 branches. In the new division, we retain branch 0 to branch 3 of accurate division, and then combine branch 4 and branch 5 into the branch 4 of the new division. Branch 6 and branch 7 are merged into the branch 5 in the new division. Meanwhile, branch 8 to branch 13 are merged into the new branch 6. Hence, in this way, genes are redivided into seven branches (branch 0 to branch 6) under new division method. As a result, the time intervals of gene generation become more uniform as exhibited in Table 3.
Table 3.
Uniform repartition of gene age
| New branch | Number of genes in the network | Splitting time (Myr) | Gene age (Myr) | Time interval (Myr) | Components of the new branch |
|---|---|---|---|---|---|
| 0 | 6541 | -454.6 | -407.9 | 0 | Original branch 0 |
| 1 | 780 | -361.2 | -342.85 | 65.05 | Original branch 1 |
| 2 | 168 | -324.5 | -272.35 | 70.5 | Original branch 2 |
| 3 | 210 | -220.2 | -198.15 | 74.2 | Original branch 3 |
| 4 | 271 | -176.1 | -136.75 | 61.4 | Original branch 4-5 |
| 5 | 22 | -97.4 | -70.8 | 65.95 | Original branch 6-7 |
| 6 | 69 | -44.2 | -22.1 | 48.7 | Original branch 8-13 |
For branch 0 to branch 6, this table exhibits the branch spitting time, gene generation time and the generation interval of genes between adjacent branches
The mean value of time intervals of gene age under new dividing method is 64.3 million years, and time intervals in the right of Table 3 are fluctuating around this mean value. Thus this new spitting method is more suitable for us to study the evolutionary mechanisms of network.
Traditional evolutionary model
Supposing the number of nodes in the original network is N. The degree of node i is di. In traditional model, all newly added genes are described as duplication genes. Concretely, the flow chart of traditional model is shown in Fig. 6a. The detailed simulation process is described as follows [21, 22]. Step1: Gene duplication Parent gene is selected at random and duplicated. Duplication gene will reserve all connections of parent gene. The probability that the gene i(i=1,2,...,N) is chosen as a parent gene vp is . Step2: Edge deletion process Edges of duplication gene will be removed randomly with probability δ (δ is relatively large). Step3: Rewiring process Duplication gene will connect to the remaining genes with large degree, that is, the larger the degree of the remaining gene, the more likely it will be connected by a duplication gene. In other words, the duplication gene develops new links through a “rich-gets-richer" mechanism.
Assuming that there are n duplication genes added into the original network. The original network will evolve by adding these n duplication genes at the same time. The chosen of parent genes and reconnect genes will only base on the original network. Moreover, the δ in step2 is set as 0.8711 in our simulation (see “Results” section).
Our improved evolutionary model
According to the gene age divided by the above uniform repartition method, we obtain a family of gene coexpression networks. Firstly, we assume that the network 0 is consisted of genes which belong to branch 0. Then the genes belong to branch 1 are added into the network 0 to form the network 1. This process will continue until all genes are integrated into the network (Fig. 13). Finally, we obtain a family of evolutionary networks which is composed of network 0 to network 6. The seven networks (Table 4) reveal the evolutionary principle of gene coexpression network. By studying the seven networks step by step, we will derive the evolutionary process of human gene coexpression network. Furthermore, we will infer the evolutionary mechanism of duplication genes and de novo genes added into the network.
Fig. 13.

A part of the gene age-specific coexpression network 6. Genes with different age are labeled by different colors. The node in the network corresponds to a gene in human. Two genes are connected by an edge if they are co-expressed. In other words, their Pearson correlation coefficient is larger than 0.7
Table 4.
The origin time and termination time of networks
| Network | Components of the network | Number of genes | Original time (Myr) | Termination time (Myr) |
|---|---|---|---|---|
| 0 | New branch 0 | 6541 | -454.6 | -361.2 |
| 1 | New branch 0-1 | 7321 | -454.6 | -324.5 |
| 2 | New branch 0-2 | 7489 | -454.6 | -220.2 |
| 3 | New branch 0-3 | 7699 | -454.6 | -176.1 |
| 4 | New branch 0-4 | 7970 | -454.6 | -97.4 |
| 5 | New branch 0-5 | 7992 | -454.6 | -44.2 |
| 6 | New branch 0-6 | 8061 | -454.6 | 0 |
According to gene age, we infer a set of evolutionary networks which consist of network i(i=0,1,...,6). This table exhibits the origin time and corresponding terminate time for each network
In our proposed evolutionary model, the evolution of network is caused by new genes which contain duplication genes and de novo genes (Fig. 6b). For duplication genes, parent genes are chosen in the original network and then they will be duplicated to generate duplication genes. Duplication genes will reserve all connections of parent genes at the beginning. Later, a part of previous edges will be deleted and new connections will be added. Owing to the evolution of network is caused by the adding of duplication genes and de novo genes, we assume that the deletion and rewiring of edges are only related to newly added genes. For de novo genes, they will join in the network and establish connections with nodes in the original network. By analyzing the network 0-6, we can infer the evolutionary mechanisms that how are duplication genes and de novo genes added into the network. Finally, we can obtain an evolutionary model of human gene coexpression network.
Sensitivity analysis of evolutionary network
To analyze the sensitivity of the evolutionary network, we consider five parameter variables and calculate their sensitivity coefficients to the topological properties of the network. The five parameter variables are respectively the generation rate, the retention rate and the rewiring rate of duplication genes, the generation rate and the connection rate of de novo genes. The topological properties we have considered are respectively the average degree, transitivity, shortest pathlength, node betweenness and edge betweenness of the network. The sensitivity coefficient reflects the sensitivity of topological properties to parameter variables. Assuming that the topological feature of the network is A, and the parameter variable of this network is F. Then the formula for calculating the sensitivity coefficient is , where △A and △F are the changes in the values of the topological feature and the parameter variable, respectively. The larger the |S| is, the more sensitivity the topological feature A is to the uncertain parameter F. In our analysis of the sensitivity of evolutionary network, we take the evaluation index Ai(i=1,2,...,5) to be five topological properties, and the uncertain factor Fi(i=1,2,...,5) to be five parameter variables of our network.
Additional files
Table S1. This table contains gene origination mechanism for 8061 different genes. In this table, ‘Dl’, ‘D’, ‘Rl’ and ‘R’ all mean genes generated by duplication mechanism. ‘A’ and ‘Al’ mean genes generated by de novo mechanisms. There is only one gene we cannot find its origination and we regard it as a de novo gene. The reference data of gene origination mechanism is downloaded from reference [30] (http://gentree.ioz.ac.cn/index.php). (XLSX 239 kb)
R code for running evolutionary model 1. This R code is for running evolutionary model 1 when the number of new genes in each step is known. (R 11 kb)
The network 0 formed by genes from branch 0. This file is support for running evolutionary models 1 and 2. (CSV 15,567 kb)
The coreness of genes in the network 0. This file is support for running evolutionary models 1 and 2. (CSV 127 kb)
The network 1 formed by genes from branch 0 and branch 1. This file is support for running evolutionary models 1 and 2. (CSV 17,312 kb)
R code for running evolutionary model 2. This R code is for running evolutionary model 2 when the length of evolutionary time in each step is known. (R 12 kb)
The time intervals between each branch. This file is support for running evolutionary model 2. (CSV 1 kb)
Table S2. This table contains the original human RNA-seq data for 14341 different genes in 71 tissues. Element NA in this table means there is no expression value for this gene in the corresponding tissue. This table is downloaded from reference [28] (http://longlab.uchicago.edu/?q=SD_GB). (XLSX 5582 kb)
Table S3. This table contains the selected human RNA-seq data for 8237 different genes in 26 different tissues. (CSV 1085 kb)
Table S3. This table contains 8061 genes and 638624 links, and the Pearson correlation coefficients. (CSV 27,996 kb)
Table S4. This table contains gene age for 8061 different genes in original partitions. (XLSX 138 kb)
Acknowledgements
We are very grateful to editors and reviewers for their careful reading and valuable comments. We would like to thank Profs. Yicang Zhou and Yanni Xiao for their valuable discussion.
Abbreviations
- DNA-based duplication genes
Deoxyribonucleic acid-based duplication genes
- Myr
Million year
- RNA-based duplication genes
Ribonucleic acid-based duplication genes
- RNA-seq data
Ribonucleic acid-sequencing data
Authors’ contributions
JZ and YG analyzed and interpreted the data regarding the human RNA-seq data and gene age. YG and YL performed the numerical simulation, and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Funding
The study was supported by grants from the National Natural Science Foundation of China (11631012, 11971375, 11571272 and 11201368), grant from the National Science and Technology Major Project of China (2018ZX10721202), grant from the Natural Science Foundation of Shaanxi Province (2019JM-273), and grant from the China Postdoctoral Science Foundation (2014M560755). The funding body did not play any roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics approval and consent to participate
Not applicable. Because all of the data are downloaded from reference [28] (http://longlab.uchicago.edu/?q=SD_GB) and reference [30] (http://gentree.ioz.ac.cn/index.php).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yuexi Gu, Email: 260482792@qq.com.
Jian Zu, Email: jianzu@xjtu.edu.cn.
Yu Li, Email: 291247516@qq.com.
References
- 1.Hsiao YT, Lee WP. Inferring robust gene networks from expression data by a sensitivity-based incremental evolution method. BMC Bioinformatics. 2012;S8:13–7. doi: 10.1186/1471-2105-13-S7-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuo PD, Banzhaf W. Small world and scale-free network topologies in an artificial regulatory network model. In Pollack J, et al, editors, Artificial life IX: proceedings of the Ninth International Conference on the Simulation and Synthesis of Living Systems. MIT Press: 2004. pp. 404–409.
- 3.Chung F, Lu L, Dewey TG, et al. Duplication models for biological networks. J Comput Biol. 2003;10(5):677–87. doi: 10.1089/106652703322539024. [DOI] [PubMed] [Google Scholar]
- 4.Albert R, Jeong H, Barabási AL. Internet: Diameter of the world-wide web. Nature. 2000;401:130–1. doi: 10.1038/43601. [DOI] [Google Scholar]
- 5.Jeong H, Mason SP, Barabási AL, et al. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 6.Wagner A. The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol Biol Evol. 2001;18(7):1283–92. doi: 10.1093/oxfordjournals.molbev.a003913. [DOI] [PubMed] [Google Scholar]
- 7.Aiello W, Chung F, Lu L. A Random Graph Model for Massive Graphs. ACM. 2000. 10.1145/335305.335326.
- 8.Ravasz E, Barabási AL. Hierarchical organization in complex networks. Phys Rev E. 2003;67(2):026112. doi: 10.1103/PhysRevE.67.026112. [DOI] [PubMed] [Google Scholar]
- 9.Watts DJ, Strogatz SH. Collective dynamics of ’small-world’ networks. Nature. 1998;393(6684):440–2. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 10.Strogatz SH. Exploring complex networks. Nature. 2001;410(6825):268–76. doi: 10.1038/35065725. [DOI] [PubMed] [Google Scholar]
- 11.Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–13. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 12.Wagner A, Fell DA. The small world inside large metabolic networks. Proc R Soc London B: Biol Sci. 2001;268(1478):1803–10. doi: 10.1098/rspb.2001.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thieffry D, Huerta AM, Pérez-Rueda E, et al. From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in escherichia coli. Bioessays. 1998;20(5):433–40. doi: 10.1002/(SICI)1521-1878(199805)20:5<433::AID-BIES10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Barabási AL, Albert R. Emergence of scaling in random networks. Science. 1999;286(5439):509–12. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 15.Pupko T. Evolution after gene duplication. Trends Evol Biol. 2011;3(1):337. [Google Scholar]
- 16.Wagner A. Evolution of gene networks by gene duplications: a mathematical model and its implications on genome organization. Proc Natl Acad Sci. 1994;91(10):4387–91. doi: 10.1073/pnas.91.10.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 18.Consortium IHGS. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 19.Gu Z, Cavalcanti A, Chen FC, et al. Extent of gene duplication in the genomes of drosophila, nematode, and yeast. Mol Biol Evol. 2002;19(3):256–62. doi: 10.1093/oxfordjournals.molbev.a004079. [DOI] [PubMed] [Google Scholar]
- 20.Jeong H, Tombor B, Albert R, et al. The large-scale organization of metabolic networks. Nature. 2000;407(6804):651–4. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 21.Pastor-Satorras R, Smith E, Solé RV. Evolving protein interaction networks through gene duplication. J Theor Biol. 2003;222:199–210. doi: 10.1016/S0022-5193(03)00028-6. [DOI] [PubMed] [Google Scholar]
- 22.Bhan A, Galas DJ, Dewey TG. A duplication growth model of gene expression networks. Bioinformatics. 2002;18(11):1486–93. doi: 10.1093/bioinformatics/18.11.1486. [DOI] [PubMed] [Google Scholar]
- 23.Vazquez A, Maritan A, Flammini A, Maritan A, Vespignani A. Global protein function prediction from protein-protein interaction networks. Nat Biotechnol. 2003;21(6):697–700. doi: 10.1038/nbt825. [DOI] [PubMed] [Google Scholar]
- 24.Solé RV, Smith E, Pastor-Satorras R, et al. A model of large-scale proteome evolution. Adv Complex Syst. 2002;5(01):43–54. doi: 10.1142/S021952590200047X. [DOI] [Google Scholar]
- 25.Romano SA, Eguia MC. Characterization of degree frequency distribution in protein interaction networks. Phys Rev E. 2005;71(3):031901. doi: 10.1103/PhysRevE.71.031901. [DOI] [PubMed] [Google Scholar]
- 26.Ispolatov I, Krapivsky PL, Yuryev A. Duplication-divergence model of protein interaction network. Phys Rev E. 2005;71(6):061911. doi: 10.1103/PhysRevE.71.061911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ispolatov I, Krapivsky PL, Mazo I, et al. Cliques and duplication-divergence network growth. New J Phys. 2005;7(1):145. doi: 10.1088/1367-2630/7/1/145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Landback P, Gschwend AR, et al. New genes drive the evolution of gene interaction networks in the human and mouse genomes. Genome Biol. 2015;16(1):202. doi: 10.1186/s13059-015-0772-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan IK, Mario-Ramirez L, Wolf YI, et al. Conservation and coevolution in the scale-free human gene coexpression network. Mol Biol Evol. 2004;21(11):2058–70. doi: 10.1093/molbev/msh222. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YE, Vibranovski MD, Landback P, et al. Chromosomal redistribution of male-biased genes in mammalian evolution with two bursts of gene gain on the x chromosome. PLoS Biol. 2010;8(10):1000494. doi: 10.1371/journal.pbio.1000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bebek G, Berenbrink P, Cooper C, et al. The degree distribution of the generalized duplication model. Theor Comput Sci. 2006;369(1-3):239–49. doi: 10.1016/j.tcs.2006.08.045. [DOI] [Google Scholar]
- 32.Qian J, Luscombe NM, Gerstein M. Protein family and fold occurrence in genomes: power-law behaviour and evolutionary model. J Mol Biol. 2001;313(4):673–81. doi: 10.1006/jmbi.2001.5079. [DOI] [PubMed] [Google Scholar]
- 33.Vázquez A, Flammini A, Maritan A, et al. Modeling of protein interaction networks. Complexus. 2003;1(1):38–44. doi: 10.1159/000067642. [DOI] [Google Scholar]
- 34.Sanger F, Coulson AR. A rapid method for determining sequences in dna by primed synthesis with dna polymerase. J Mol Biol. 1975;94(3):441–8. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 35.Cornwell VMTL, M I, et al. Viper: Visualization pipeline for rna-seq, a snakemake workflow for efficient and complete rna-seq analysis. BMC Bioinformatics. 2018;19(1):135. doi: 10.1186/s12859-018-2139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, Gerstein M, Snyder M. Rna-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravasz E, Somera AL, Mongru DA, et al. Hierarchical organization of modularity in metabolic networks. Science. 2002;297(5586):1551–5. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 38.Freeman LC. A set of measures of centrality based on betweenness. Sociometry. 1977;40(1):35–41. doi: 10.2307/3033543. [DOI] [Google Scholar]
- 39.Boccaletti S, Latora V, Moreno Y, et al. Complex networks: Structure and dynamics. Phys Rep. 2006;424(4-5):175–308. doi: 10.1016/j.physrep.2005.10.009. [DOI] [Google Scholar]
- 40.Goh KI, Oh E, Kahng B, et al. Betweenness centrality correlation in social networks. Phys Rev E. 2003;67(1):017101. doi: 10.1103/PhysRevE.67.017101. [DOI] [PubMed] [Google Scholar]
- 41.Ma L, Ma C, Zhang HF, et al. Identifying influential spreaders in complex networks based on gravity formula. Physica A: Stat Mech Appl. 2016;451:205–12. doi: 10.1016/j.physa.2015.12.162. [DOI] [Google Scholar]
- 42.Carmi S, Havlin S, Kirkpatrick S, et al. A model of internet topology using k-shell decomposition. Proc Natl Acad Sci. 2007;104(27):11150–4. doi: 10.1073/pnas.0701175104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dorogovtsev SN, Goltsev AV, Mendes JFF. K-core organization of complex networks. Phys Rev Lett. 2006;96(4):040601. doi: 10.1103/PhysRevLett.96.040601. [DOI] [PubMed] [Google Scholar]
- 44.Lü L, Chen D, Ren XL, et al. Vital nodes identification in complex networks. Phys Rep. 2016;650:1–63. doi: 10.1016/j.physrep.2016.06.007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. This table contains gene origination mechanism for 8061 different genes. In this table, ‘Dl’, ‘D’, ‘Rl’ and ‘R’ all mean genes generated by duplication mechanism. ‘A’ and ‘Al’ mean genes generated by de novo mechanisms. There is only one gene we cannot find its origination and we regard it as a de novo gene. The reference data of gene origination mechanism is downloaded from reference [30] (http://gentree.ioz.ac.cn/index.php). (XLSX 239 kb)
R code for running evolutionary model 1. This R code is for running evolutionary model 1 when the number of new genes in each step is known. (R 11 kb)
The network 0 formed by genes from branch 0. This file is support for running evolutionary models 1 and 2. (CSV 15,567 kb)
The coreness of genes in the network 0. This file is support for running evolutionary models 1 and 2. (CSV 127 kb)
The network 1 formed by genes from branch 0 and branch 1. This file is support for running evolutionary models 1 and 2. (CSV 17,312 kb)
R code for running evolutionary model 2. This R code is for running evolutionary model 2 when the length of evolutionary time in each step is known. (R 12 kb)
The time intervals between each branch. This file is support for running evolutionary model 2. (CSV 1 kb)
Table S2. This table contains the original human RNA-seq data for 14341 different genes in 71 tissues. Element NA in this table means there is no expression value for this gene in the corresponding tissue. This table is downloaded from reference [28] (http://longlab.uchicago.edu/?q=SD_GB). (XLSX 5582 kb)
Table S3. This table contains the selected human RNA-seq data for 8237 different genes in 26 different tissues. (CSV 1085 kb)
Table S3. This table contains 8061 genes and 638624 links, and the Pearson correlation coefficients. (CSV 27,996 kb)
Table S4. This table contains gene age for 8061 different genes in original partitions. (XLSX 138 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.