

Abstract
Introduction:
Receptor tyrosine kinases have been implicated in various vascular remodeling processes and cardiovascular disease. However, their role in the regulation of vascular tone is poorly understood. Herein, we evaluate the contribution of c-Kit signaling to vasoactive responses.
Methods:
The vascular reactivity of mesenteric arteries was assessed under isobaric conditions in c-Kit deficient (KitW/W-v) and littermate control mice (Kit+/+) using pressure myography. Protein levels of soluble guanylyl cyclase beta 1 (sGCβ1) were quantified by western blot. Mean arterial pressure was measured after high salt (8% NaCl) diet treatment using the tail-cuff method.
Results:
Smooth muscle cells (SMCs) from c-Kit deficient mice showed a 5-fold downregulation of sGCβ1 compared to controls. Endothelium-dependent relaxation of mesenteric arteries demonstrated a predominance of prostanoid vs. nitric oxide (NO) signaling in both animal groups. The dependence on prostanoid-induced dilation was higher in c-Kit mutant mice than in controls, as indicated by a significant impairment in vasorelaxation with indomethacin with respect to the latter. Endothelium-independent relaxation showed significant dysfunction of NO signaling in c-Kit deficient SMCs compared to controls. Mesenteric artery dilation was rescued by addition of a cGMP analog, but not with a NO donor, indicating a deficiency in cGMP production in c-Kit deficient SMCs. Finally, c-Kit deficient mice developed higher blood pressure on an 8% NaCl diet compared to their control littermates.
Conclusion:
c-Kit deficiency inhibits NO signaling in SMCs. The existence of this c-Kit/sGC signaling axis may be relevant for vascular reactivity and remodeling.
Keywords: arterial blood pressure, c-Kit, nitric oxide, prostacyclin, vascular reactivity, vasodilation
INTRODUCTION
Receptor tyrosine kinases (RTKs) are a class of cell surface receptors involved in signal transduction pathways. They play critical roles in a variety of vital cellular processes such as proliferation, differentiation, metabolism, and motility [1]. Dysfunctional RTKs are associated with a wide spectrum of cardiovascular problems such as arteriosclerosis, hypertension, and peripheral artery disease [2-5]. c-Kit, a widely known proto-oncogene and stem cell marker, is a member of the RTK family. It is expressed in many different cell types, including endothelial (ECs) and SMCs, where it participates in vascular remodeling [6,7].
Multiple antineoplastic drugs target RTKs, including c-Kit, to inhibit their angiogenic properties [8,9]. Hypertension is a common and serious adverse effect with RTK inhibitors, such that the Food and Drug Administration has recommendations in place to closely monitor blood pressure changes while patients are on therapy [10,11]. Despite the significance of this clinical problem, and because most RTK inhibitors target more than one kinase, there is a paucity of research on the contribution of each specific RTK to vascular reactivity.
The current information on the role of c-Kit signaling in vascular tone is poorly understood, in part because of the diversity of tissue sources and experimental conditions in which it has been obtained. c-Kit+ hematopoietic progenitors and mast cells are associated with the pathogenesis of pulmonary arterial hypertension [12]. Higher serum levels of a soluble form of c-Kit and its ligand stem cell factor (SCF) correlate with increased systolic blood pressure in humans [13,14], although it is not clear through which cellular mechanism. On the other hand, several studies support a role of the c-Kit receptor in vasorelaxation. c-Kit activation increases production of vasodilatory factors in human ECs [15]. In agreement with the latter, transcriptomic analyses of mouse c-Kit mutant and wild type SMCs uncovered a downregulation of nitric oxide (NO) signaling enzymes in the former [16]. c-Kit deficiency in rats was also associated with aldosteronism and increase in systemic blood pressure [17].
In this study, we used pressure myography to directly investigate the effects of c-Kit deficiency in vascular reactivity. Altogether, our findings indicate a role of vascular c-Kit in the regulation of vasoactive responses, and uncover a c-Kit/sGC signaling axis of possible relevance for vascular tone and remodeling.
MATERIALS AND METHODS
Animals
Experiments were done with 3-month-old c-Kit deficient mice (KitW/W-v) and littermate (Kit+/+) controls. KitW/W-v mice carry a c-Kit null mutation (W) and a hypomorphic allele (W-v). Compound heterozygotes exhibit reduced kinase activity and represent the most severe c-Kit deficient genotype that is not embryonic lethal [18]. KitW/W-v and Kit+/+ mice are obtained from the F1 generation of WB/ReJ KitW/J and C57BL/6J KitW-v/J crosses (Stock No. 000692 and 000049, Jackson Laboratories). Females and males were equally represented in all experimental groups. All experiments were conducted according to IACUC approvals and NIH guidelines.
Pressure Myography
The Pressure Myograph System model 110P (Danish Myo Technology, Denmark) was used in all the experiments. The mesenteric arcade was isolated and placed in physiological salt solution (PSS; 133.4 mM NaCl, 4.7 mM KCl, 14.9 mM NaHCO3, 2.1 mM CaCl2, 2.4 Mm MgSO4, 1.2 mM KH2PO4, 5.5 mM dextrose, and 26.3 μM CaNa2 EDTA). The surrounding fat tissue of the second-order mesenteric arteries was removed, after which the latter was mounted between two glass microcannulas (120 μm diameter) secured with surgical nylon suture. Mesenteric arteries were maintained aerated on the pressure myograph chamber in isotonic PSS with a special mix of 95% O2 and 5% CO2 at a constant temperature of 37°C.
Prior to running the experiment, we used the wake-up protocol to confirm the tissue quality of mesenteric arteries. This protocol consists of three pre-conditioning cycles: first with PSS containing 100 mM K+ (KPSS) to test the myogenic tone, next with KPSS followed by 10−5 M norepinephrine (NE; Sigma-Aldrich, St. Louis, MO) to record maximum contractility, and lastly with 10−5 M NE followed by 10−5 M acetylcholine (Ach; Sigma-Aldrich) to assess vasorelaxation. Briefly, vessels were gradually pressurized to 60 mmHg by increasing the pressure by 10 mmHg every 10 min. Arteries were washed twice with PSS over the next 10 min, and vascular function was tested in KPSS. Vessels were washed again with PSS over the next 10 min, followed by addition of, first, KPSS and, 5 min later, 10−5 M NE. At this point of maximum contraction, vessels were stretched longitudinally to optimize vasoreactivity [19]. To determine basal endothelial function, arteries were washed with PSS over 10 min, followed by pre-contraction with 10−5 M NE and 10−5 M Ach after 5 min. Finally, we allowed the vessels to equilibrate in PSS for 30 min before starting the experiment.
If the vessel responded appropriately with Ach (>10% increase in outside diameter with respect to the NE-induced diameter) during the warm-up protocol, we pre-contracted with 10−5 M NE and assessed endothelium-dependent vasodilation with progressively higher doses of Ach [10−12 to 10−5 M]. Next, vessels were washed with PSS, pre-incubated for 20 min with 10−5 M indomethacin (Sigma-Aldrich), pre-contracted with 10−5 M NE, and a second vasodilation curve was recorded with Ach [10−12 to 10−5 M]. Air bubbles were passed through the mesenteric arteries to remove the endothelium and test endothelium-independent vasodilation. A lack of vasodilatory response to 10−5 M Ach after 10−5 M NE-induced pre-contraction confirmed endothelial denudation. Next, vessels were pre-contracted with 10−5 M NE, and treated with progressively higher doses of sodium nitroprusside (SNP, 10−12 to 10−5 M; Sigma-Aldrich) or 8-bromo-cyclic GMP (10−11 to 10−5 M; Sigma-Aldrich). The pressure in the chamber was maintained at 60 mmHg during the entire experimentation protocol. Drug molarities refer to the final concentration in the chamber.
Smooth Muscle Cell (SMC) Isolation and Culture
Primary aortic SMCs were isolated from KitW/W-v and control Kit+/+ mice as described elsewhere [20]. Briefly, murine aortas were digested with collagenase type II, transferred to 10% fetal bovine serum in DMEM/F12, and cut into small pieces, to allow SMC migration out of the explants. Cells were cultured until 90% confluency. Cells were used within the first three passages to avoid fibroblast-like differentiation.
Western Blot
Murine SMCs were lysed in RIPA buffer (Santa Cruz Biotechnology, Santa Cruz, CA) supplemented with leupeptin (10 μg/ml), aprotinin (20 mU/ml), PMSF (10 μM), NaF (10 μM), and NaVO4 (10 μM). Cells lysates were loaded onto QIAshredder homogenizer columns (Qiagen, Hilden, Germany) and centrifuged at 9,000 g for 3 min. The Bio-Rad DC Protein Assay (Bio-Rad Laboratories, Hercules, CA) was used to quantify total proteins. Protein extracts were diluted 1:1 (vol/vol) in Laemmli buffer, 15 μg of protein were loaded on each lane of a 4–12% Tris-glycine gel (Invitrogen, Carlsbad, CA). After electrophoresis, proteins were transferred to a nitrocellulose membrane (GE Healthcare Biosciences, Piscataway, NJ), followed by incubation with either a rabbit monoclonal anti-sGCβ1 antibody (Abcam, Cambridge, UK) or an anti-β-actin antibody (Sigma-Aldrich, St Louis, MO). Membranes were then incubated with HRP-conjugated secondary antibodies, and developed using the WesternBreeze Chemiluminescent kit (Invitrogen).
Blood Pressure Measurement
Animals were subjected to high NaCl diet (8% NaCl; TD.92012, Teklad, Indianapolis, IN) ad libitum for three weeks. Systolic and diastolic pressures were determined using the CODA HT tail-cuff system with 4 activated channels (Kent Scientific Corporation, Torrington, CT). Through all measurements, mice were placed inside a plastic holder on a heated platform that ensured constant temperature. Mice were allowed to become accustomed to the measurement procedure a week before data recording.
Data Analysis
Statistical analysis was done using GraphPad Prism version 8.1.2 (San Diego, CA). The pressure myograph data was analyzed using two-way ANOVAs and the Sidak multiple comparison test. Normally distributed data are expressed as mean ± standard error of the mean (SEM) and compared using unpaired t-tests. Otherwise, data are expressed as median and interquartile range (IQR) and compared using the Mann-Whitney test. Differences were considered statistically significant at p<0.05.
RESULTS
Higher dependence on prostanoid-mediated dilation in c-Kit deficient arteries
Acetylcholine (Ach) is a well-known vasodilator that acts through several mechanisms, including activation of endothelial nitric oxide synthase (eNOS) and prostaglandin production in ECs [21,22]. Endothelium-dependent dilation was tested in c-Kit mutant and wild type mesenteric arteries in the presence or absence of eNOS and prostaglandin-endoperoxide synthase inhibitors, L-NAME and indomethacin, respectively (Fig. 1). There was no significant difference in Ach-induced relaxation between c-Kit deficient and wild type arteries in the absence of inhibitors (Fig. 1A-C). L-NAME did not cause any significant differences in the vasodilatory response in any of the experimental groups compared to Ach-only curves (Fig. 1A-B), nor between c-Kit mutant and wild type arteries (Fig. 1D).
Figure 1.

A-B) Endothelium-dependent acetylcholine (Ach) induced relaxation in wild type (A, Kit+/+) and c-Kit deficient (B, KitW/W-v) mesenteric arteries in the presence or absence of eNOS and prostaglandin-endoperoxide synthase inhibitors, L-NAME and indomethacin, respectively. C-E) Comparison of vasorelaxation curves from wild type and c-Kit deficient arteries under control conditions (C, no inhibitor), and after incubation with L-NAME (D) or indomethacin (E). Errors bars at each concentration point represent the mean±standard error of the mean (SEM).*P<0.05,**P<0.01,***P<0.001.
In contrast, indomethacin treatment caused significant reductions in vascular relaxation in both c-Kit deficient and wild type arteries compared to Ach-only curves (Fig. 1A-B). At the highest Ach concentration (10−5 M; p<0.01), there was a 25.6% decrease in relaxation in wild type animals with indomethacin compared to the Ach-only response (Fig. 1A). Interestingly, the impairment in relaxation by indomethacin was more pronounced in c-Kit deficient than in wild type arteries (Fig. 1E), with a 47.6% reduction in Ach-induced dilation at the maximum relaxation response compared to the Ach-only curve (Fig. 1B; p<0.001). The impairment in endothelium-dependent dilation in c-Kit mutant arteries after indomethacin suggests that these mutant mice rely more on prostanoid-mediated dilation compared to controls.
Defective nitric oxide (NO) mediated signaling in c-Kit deficient SMCs
Our group previously identified a remarkable downregulation of sGCβ1 RNA expression (GUCY1B3) in KitW/W-v SMCs compared to those from wild type animals [16]. This subunit is crucial for the activity of sGC in catalyzing the conversion of guanosine triphosphate (GTP) to the vasodilatory messenger cGMP upon NO binding [23]. In light of this evidence, we first determined whether the downregulation of sGCβ1 gene expression in KitW/W-v SMCs was also reflected at the protein level. Figure 2A shows the Western blot analysis of KitW/W-v and Kit+/+ SMCs confirming a 5-fold downregulation of sGCβ1 protein expression in c-Kit deficient SMCs.
Figure 2.

A) Western blot and quantification of sGCβ1 protein expression in primary aortic smooth muscle cells from c-Kit deficient (KitW/W-v) and wild type (Kit+/+) mice. sGCβ1 expression was normalized to protein expression of β-actin. Error bars represent the mean±standard error of the mean (SEM). *P<0.05 B-C) Endothelium-independent relaxation of wild type and c-Kit deficient mesenteric arteries in the presence of a nitric oxide donor (B, SNP) or a cell membrane-permeable cGMP analog (C, 8-bromo-cGMP). Errors bars at each concentration point represent the mean±standard error of the mean (SEM). *P<0.05,**P<0.01,***P<0.001, ****P<0.0001.
Next, we assessed endothelium-independent dilation in KitW/W-v and Kit+/+ mesenteric arteries using an NO donor (SNP) in the presence or absence of a cell-permeable cGMP analog (8-bromo-cGMP) after endothelium denudation (Fig. 2B-C). In agreement with the downregulation of sGCβ1, we observed a 21.9% reduction in SNP-induced vasorelaxation in c-Kit deficient arteries compared to wild type vessels (Fig. 2B), indicating a significant impartment in NO utilization by c-Kit mutant SMCs. To confirm these findings, we performed a rescue experiment using 8-bromo-cGMP. Similar 8-bromo-cGMP-induced vasorelaxation responses were obtained in c-Kit deficient and wild type arteries (Fig. 2C). This demonstrated that the impairment in endothelium-independent dilation in c-Kit mutant arteries occurs as a result of defective cGMP production.
c-Kit deficiency is associated with higher blood pressure after high salt diet
Arterial blood pressure was measured in c-Kit deficient and littermate controls before and after 3 weeks of high salt (8% NaCl) diet. The mean arterial pressure (MAP) was similar between experimental groups at baseline (median 125.3 [IQR 137.5-112.6] in KitW/W-v vs. 129.9 [133.7-117.1] in Kit+/+, p=0.94). In contrast, c-Kit mutant mice developed significantly higher MAP than wild type animals after challenge with high salt diet (Fig. 3).
Figure 3.
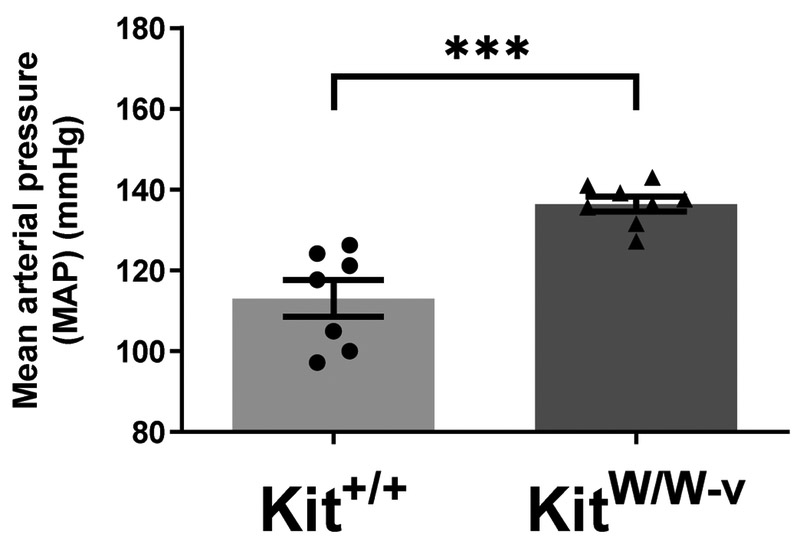
Mean arterial pressure of c-Kit deficient (KitW/W-v) and wild type (Kit+/+) mice after three weeks of high salt (8% NaCl) diet. Error bars represent the median±interquartile range (IQR).*P<0.05
DISCUSSION
High blood pressure is one of the main risk factors for cardiovascular diseases, which is the leading cause of mortality in the United States [24]. Hypertension is considered a class effect for RTKs inhibitors [10,11], but the molecular mechanisms are not fully understood. In this study, we demonstrated that 1) c-Kit deficient mesenteric arteries rely more on prostanoid-induced relaxation than wild type vessels, 2) c-Kit deficiency leads to dysfunction of NO-mediated relaxation in SMCs, at least in part, due to the sGC dysfunction and 3) c-Kit mutant mice develop higher blood pressure than littermate controls when challenged with a high salt diet.
The presence of c-Kit in vascular cells and the importance of this signaling pathway in different vascular processes have been previously reported [12,16,25], but the effects of this RTK in vascular reactivity were so far unknown. Our study provides mechanistic evidence of sGCβ1 downregulation in c-Kit deficient SMCs, and of dysfunctional NO-mediated responses in SMCs that were only rescued by addition of a cell-permeable cGMP analog. Previous studies have shown that c-Kit activation is associated with activation of the eNOS/NO pathway [26]. In ECs, the c-Kit ligand SCF stimulates downstream molecules in the c-Kit pathway such as Akt and ERK, while also inducing eNOS synthesis. Recent reports indicate that c-Kit signaling also stimulates the eNOS/NO pathway through increased synthesis and release of endothelin-3 (ET3) [27]. While these reports indicate that c-Kit activation in ECs is important for NO production, our findings demonstrate that, when c-Kit is defective, NO is not effectively utilized in SMCs, and consequently, vasorelaxation is impaired. It is well established that the NO pathway plays a critical role in vasodilation. Nitric oxide released from ECs binds to the beta subunit of sGC in SMCs, which converts GTP into cGMP. Dysfunction caused by reduced NO bioavailability or deficient sGC activity correlates with vascular diseases, such as systemic and pulmonary hypertension [28].
Interestingly, our study also shows that prostanoid-dependent vasodilation predominates over NO-induced relaxation in mesenteric arteries and more so in c-Kit deficient mice. The doses of L-NAME used in our experiments have produced significant differences in vasodilation in other studies [29], but failed to significantly reduce relaxation responses in this work. This may be related to the genetic background of our animals (which, to our knowledge, have never been used for vasoreactivity studies before) or to physiological characteristics of the mesenteric vessels. Previous reports indicate significant differences between mouse strains and vessels in the usage of vasodilatory pathways [2,22,30-34]. It is widely accepted that vasodilation of conduit vessels relies mostly on NO signaling, while resistance arteries including the mesenteric artery dilate through a combination of endothelium-derived relaxing factors such as NO, prostacyclin, and endothelium-derived hyperpolarizing factor (EDHF) [35-37]. Even so, there is significant diversity across resistance vascular beds with respect to the contribution of each vasodilatory pathway [38]. The higher dependence of c-Kit deficient arteries on prostanoid-induced dilation suggests a compensatory mechanism to dysfunctional NO signaling. Previous studies indicate that COX-derived vasodilators can compensate for a defective NO pathway in coronary and mesenteric arteries [35,39]. Nonetheless, compensatory mechanisms are also vascular bed-specific, so that they do not occur or not to the same extent in large arteries and other vessels [40].
c-Kit deficient animals also developed higher MAP than their littermate controls after 3 weeks of high salt diet. It has been reported that the NO pathway contributes to the body’s adaptation during a salt load diet, and that impairment in this pathway leads to salt-sensitivity and salt-induced hypertension [41,42]. The kidneys produce all three isoforms of NOS (endothelial [eNOS], inducible [iNOS], and neuronal [nNOS]), with eNOS being expressed in the afferent and efferent arterioles, vasa recta, proximal tubules, the thick ascending limbs, and collection ducts [40]. eNOS activity has been implicated in the regulation of natriuresis [40-42], such that impaired NO signaling in the SMCs of the renal arterioles of c-Kit deficient mice can significantly alter this process. There is evidence that c-Kit activation also regulates aldosterone synthesis in the adrenal gland [17]. Therefore, c-Kit deficiency can potentially influence the blood pressure at different levels of salt/water balance regulation. In conclusion, our study addresses the effect of c-Kit signaling in vascular reactivity. We uncovered a c-Kit/sGC pathway that leads to impaired NO signaling in c-Kit dysfunction, and suggests a major role of c-Kit in NO-mediated vasorelaxation.
Supplementary Material
HIGHLIGHTS.
Deficiency in c-Kit leads to impaired nitric oxide signaling in smooth muscle cells
c-Kit plays a role in nitric oxide-mediated vascular relaxation
c-Kit/sGC signaling axis contributes to vascular relaxation
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health grant R01HL109582 to R.I.V.-P., and K01HL145359 to R.M.L.-S
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Lemmon MA, Schlessinger J, Cell signaling by receptor tyrosine kinases, Cell 141 (2010) 1117–1134. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA, The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis, Atherosclerosis 186 (2006) 38–53. 10.1016/j.atherosclerosis.2005.06.038. [DOI] [PubMed] [Google Scholar]
- [3].Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, Offenstadt G, Leseche G, Cohen PL, Tedgui A, Mallat Z, Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis, Arterioscler. Thromb. Vasc. Biol 28 (2008) 1429–1431. 10.1161/ATVBAHA.108.169078. [DOI] [PubMed] [Google Scholar]
- [4].Zschabitz S, Grullich C, Lenvantinib: A Tyrosine Kinase Inhibitor of VEGFR 1-3, FGFR 1-4, PDGFRalpha, KIT and RET, Recent Results Cancer Res. 211 (2018) 187–198. 10.1007/978-3-319-91442-8_13. [DOI] [PubMed] [Google Scholar]
- [5].Matrougui K, Diabetes and microvascular pathophysiology: role of epidermal growth factor receptor tyrosine kinase, Diabetes Metab. Res. Rev 26 (2010) 13–16. 10.1002/dmrr.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Miyamoto T, Sasaguri Y, Sasaguri T, Azakami S, Yasukawa H, Kato S, Arima N, Sugama K, Morimatsu M, Expression of stem cell factor in human aortic endothelial and smooth muscle cells, Atherosclerosis 129 (1997) 207–213. [DOI] [PubMed] [Google Scholar]
- [7].Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, Okada M, Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells, J. Biol. Chem 279 (2004) 18600–18607. 10.1074/jbc.M311643200. [DOI] [PubMed] [Google Scholar]
- [8].Jeltsch M, Leppanen VM, Saharinen P, Alitalo K, Receptor Tyrosine Kinase-Mediated Angiogenesis, Cold Spring Harb. Perspect. Biol 5 (2013) 22 a009183. 10.1101/cshperspect.a009183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Regad T, Targeting RTK Signaling Pathways in Cancer, Cancers (Basel) 7 (2015) 1758–1784. 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Agarwal M, Thareja N, Benjamin M, Akhondi A, Mitchell GD, Tyrosine Kinase Inhibitor-Induced Hypertension, Curr. Oncol. Rep 20 (2018) 65 10.1007/s11912-018-0708-8. [DOI] [PubMed] [Google Scholar]
- [11].Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh ETH, Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 2, J. Am. Coll. Cardiol 70 (2017) 2552–2565. 10.1016/j.jacc.2017.09.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Farha S, Dweik R, Rahaghi F, Benza R, Hassoun P, Frantz R, Torres F, Quinn DA, Comhair S, Erzurum S, Asosingh K, Imatinib in pulmonary arterial hypertension: c-Kit inhibition, Pulm. Circ 4 (2014) 452–455. 10.1086/677359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhong HL, Lu XZ, Chen XM, Yang XH, Zhang HF, Zhou L, Wang L, Cao KJ, Huang J, Relationship between stem cell factor/c-kit expression in peripheral blood and blood pressure, J Hum Hypertens 24 (2010) 220–225. 10.1038/jhh.2009.62. [DOI] [PubMed] [Google Scholar]
- [14].Hoffmann J, Yin J, Kukucka M, Yin N, Saarikko I, Sterner-Kock A, Fujii H, Leong-Poi H, Kuppe H, Schermuly RT, Kuebler WM, Mast cells promote lung vascular remodelling in pulmonary hypertension (vol 37, pg 1400, 2011), Eur. Respir. J. 40 (2012) 515–517. 10.1183/09031936.50043310. [DOI] [PubMed] [Google Scholar]
- [15].Chen LL, Zhu J, Schumacher J, Wei C, Ramdas L, Prieto VG, Jimenez A, Velasco MA, Tripp SR, Andtbacka RHI, Gouw L, Rodgers GM, Zhang L, Chan BK, Cassidy PB, Benjamin RS, Leachman SA, Frazier ML, SCF-KIT signaling induces endothelin-3 synthesis and secretion: Thereby activates and regulates endothelin-B-receptor for generating temporally-and spatially-precise nitric oxide to modulate SCF- and or KIT-expressing cell functions, PLoS One 12 (2017) e0184154 10.1371/journal.pone.0184154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Song L, Martinez L, Zigmond ZM, Hernandez DR, Lassance-Soares RM, Selman G, Vazquez-Padron RI, c-Kit modifies the inflammatory status of smooth muscle cells, PeerJ 5 (2017) e3418 10.7717/peerj.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boyer HG, Wils J, Renouf S, Arabo A, Duparc C, Boutelet I, Lefebvre H, Louiset E, Dysregulation of Aldosterone Secretion in Mast Cell-Deficient Mice, Hypertension 70 (2017) 1256–1263. 10.1161/HYPERTENSIONAHA.117.09746. [DOI] [PubMed] [Google Scholar]
- [18].Bernstein A, Chabot B, Dubreuil P, Reith A, Nocka K, Majumder S, Ray P, Besmer P, The mouse W/c-kit locus, Ciba Found Symp 148 (1990) 158–166; discussion 166-172. [PubMed] [Google Scholar]
- [19].Coats P, Hillier C, Determination of an optimal axial-length tension for the study of isolated resistance arteries on a pressure myograph, Exp. Physiol 84 (1999) 1085–1094. [PubMed] [Google Scholar]
- [20].Ray JL, Leach R, Herbert JM, Benson M, Isolation of vascular smooth muscle cells from a single murine aorta, Methods Cell Sci 23 (2001) 185–188. [DOI] [PubMed] [Google Scholar]
- [21].Kellogg DL, Zhao JL, Coey U, Green JV, Acetylcholine-induced vasodilation is mediated by nitric oxide and prostaglandins in human skin, J. Appl. Physiol 98 (2005) 629–632. 10.1152/japplphysiol.00728.2004. [DOI] [PubMed] [Google Scholar]
- [22].Medow MS, Glover JL, Stewart JM, Nitric oxide and prostaglandin inhibition during acetylcholine-mediated cutaneous vasodilation in humans, Microcirculation 15 (2008) 569–579. 10.1080/10739680802091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Montfort WR, Wales JA, Weichsel A, Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor, Antioxid Redox Signal 26 (2017) 107–121. 10.1089/ars.2016.6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bromfield S, Muntner P, High blood pressure: the leading global burden of disease risk factor and the need for worldwide prevention programs, Curr. Hypertens. Rep 15 (2013) 134–136. 10.1007/s11906-013-0340-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hernandez DR, Artiles A, Duque JC, Martinez L, Pinto MT, Webster KA, Velazquez OC, Vazquez-Padron RI, Lassance-Soares RM, Loss of c-Kit function impairs arteriogenesis in a mouse model of hindlimb ischemia, Surgery 163 (2018) 877–882. 10.1016/j.surg.2017.10.052. [DOI] [PubMed] [Google Scholar]
- [26].Kim JY, Choi JS, Song SH, Im JE, Kim JM, Kim K, Kwon S, Shin HK, Joo CK, Lee BH, Suh W, Stem Cell Factor Is a Potent Endothelial Permeability Factor, Arteriosclerosis Thrombosis and Vascular Biology 34 (2014) 1459 10.1161/atvbaha.114.303575. [DOI] [PubMed] [Google Scholar]
- [27].Chen LL, Zhu J, Schumacher J, Wei CJ, Ramdas L, Prieto VG, Jimenez A, Velasco MA, Tripp SR, Andtbacka RHI, Gouw L, Rodgers GM, Zhang LS, Chan BK, Cassidy PB, Benjamin RS, Leachman SA, Frazier ML, SCF-KIT signaling induces endothelin-3 synthesis and secretion: Thereby activates and regulates endothelin-B-receptor for generating temporally-and spatially-precise nitric oxide to modulate SCF-and or KIT-expressing cell functions, PLoS One 12 (2017) 30 e0184154. 10.1371/journal.pone.0184154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R, Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension, Hypertension 20 (1992) 298–303. [DOI] [PubMed] [Google Scholar]
- [29].Manicam C, Staubitz JI, Pfeiffer N, Gericke A, Mechanisms of Endothelium-Dependent Vasodilation in the Ophthalmic Artery of Mice, Invest. Ophthalmol. Vis. Sci 55 (2014) 2. [Google Scholar]
- [30].Ross R, The pathogenesis of atherosclerosis: a perspective for the 1990s, Nature 362 (1993) 801–809. 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- [31].Muto A, Yi T, Harrison KD, Davalos A, Fancher TT, Ziegler KR, Feigel A, Kondo Y, Nishibe T, Sessa WC, Dardik A, Eph-B4 prevents venous adaptive remodeling in the adult arterial environment, J. Exp. Med 208 (2011) 561–575. 10.1084/jem.20101854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heath EI, Infante J, Lewis LD, Luu T, Stephenson J, Tan AR, Kasubhai S, LoRusso P, Ma B, Suttle AB, Kleha JF, Ball HA, Dar MM, A randomized, double-blind, placebo-controlled study to evaluate the effect of repeated oral doses of pazopanib on cardiac conduction in patients with solid tumors, Cancer Chemother. Pharmacol 71 (2013) 565–573. 10.1007/s00280-012-2030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Robinson ES, Khankin EV, Karumanchi SA, Humphreys BD, Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: mechanisms and potential use as a biomarker, Semin. Nephrol 30 (2010) 591–601. 10.1016/j.semnephrol.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ryan MJ, Didion SP, Davis DR, Faraci FM, Sigmund CD, Endothelial dysfunction and blood pressure variability in selected inbred mouse strains, Arterioscler. Thromb. Vasc. Biol 22 (2002) 42–48. [DOI] [PubMed] [Google Scholar]
- [35].Lagaud GJ, Skarsgard PL, Laher I, van Breemen C, Heterogeneity of endothelium-dependent vasodilation in pressurized cerebral and small mesenteric resistance arteries of the rat, J. Pharmacol. Exp. Ther 290 (1999) 832–839. [PubMed] [Google Scholar]
- [36].Jiang J, Zheng JP, Li Y, Gan Z, Jiang Y, Huang D, Li H, Liu Z, Ke Y, Differential contribution of endothelium-derived relaxing factors to vascular reactivity in conduit and resistance arteries from normotensive and hypertensive rats, Clin. Exp. Hypertens 38 (2016) 393–398. 10.3109/10641963.2016.1148155. [DOI] [PubMed] [Google Scholar]
- [37].Kang KT, Endothelium-derived Relaxing Factors of Small Resistance Arteries in Hypertension, Toxicol Res 30 (2014) 141–148. 10.5487/TR.2014.30.3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Durand MJ, Gutterman DD, Diversity in mechanisms of endothelium-dependent vasodilation in health and disease, Microcirculation 20 (2013) 239–247. 10.1111/micc.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lamping KG, Nuno DW, Shesely EG, Maeda N, Faraci FM, Vasodilator mechanisms in the coronary circulation of endothelial nitric oxide synthase-deficient mice, Am. J. Physiol. Heart Circ. Physiol 279 (2000) H1906–1912. 10.1152/ajpheart.2000.279.4.H1906. [DOI] [PubMed] [Google Scholar]
- [40].Ortiz PA, Garvin JL, Cardiovascular and renal control in NOS-deficient mouse models, Am. J. Physiol. Regul. Integr. Comp. Physiol 284 (2003) R628–638. 10.1152/ajpregu.00401.2002. [DOI] [PubMed] [Google Scholar]
- [41].Leonard AM, Chafe LL, Montani JP, Van Vliet BN, Increased salt-sensitivity in endothelial nitric oxide synthase-knockout mice, Am. J. Hypertens 19 (2006) 1264–1269. 10.1016/j.amjhyper.2006.05.025. [DOI] [PubMed] [Google Scholar]
- [42].Cubeddu LX, Alfieri AB, Hoffmann IS, Jimenez E, Roa CM, Cubeddu R, Palermo C, Baldonedo RM, Nitric oxide and salt sensitivity, Am. J. Hypertens 13 (2000) 973–979. 10.1016/s0895-7061(00)00283-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.