

Abstract
Aims/hypothesis
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are two peptides that function to promote insulin secretion. Dipeptidyl peptidase-4 (DPP-4) inhibitors increase the bioavailability of both GLP-1 and GIP but the dogma continues to be that it is the increase in GLP-1 that contributes to the improved glucose homeostasis. We have previously demonstrated that pancreatic rather than intestinal GLP-1 is necessary for improvements in glucose homeostasis in mice. Therefore, we hypothesise that a combination of pancreatic GLP-1 and GIP is necessary for the full effect of DPP-4 inhibitors on glucose homeostasis.
Methods
We have genetically engineered mouse lines in which the preproglucagon gene (Gcg) is absent in the entire body (GcgRAΔNull) or is expressed exclusively in the intestine (GcgRAΔVilCre) or pancreas and duodenum (GcgRAΔPDX1Cre). These mice were used to examine oral glucose tolerance and GLP-1 and GIP responses to a DPP-4 inhibitor alone, or in combination with incretin receptor antagonists.
Results
Administration of the DPP-4 inhibitor, linagliptin, improved glucose tolerance in GcgRAΔnull mice and control littermates and in GcgRAΔVilCre and GcgRAΔPDX1Cre mice. The potent GLP-1 receptor antagonist, exendin-[9–39] (Ex9), blunted improvements in glucose tolerance in linagliptin-treated control mice and in GcgRAΔPDX1Cre mice. Ex9 had no effect on glucose tolerance in linagliptin-treated GcgRAΔNull or in GcgRAΔVilCre mice. In addition to GLP-1, linagliptin also increased postprandial plasma levels of GIP to a similar degree in all genotypes. When linagliptin was co-administered with a GIP-antagonising antibody, the impact of linagliptin was partially blunted in wild-type mice and was fully blocked in GcgRAΔNull mice.
Conclusions/interpretation
Taken together, these data suggest that increases in pancreatic GLP-1 and GIP are necessary for the full effect of DPP-4 inhibitors on glucose tolerance.
Keywords: DPP-4 inhibitor, GIP, GLP-1, Glucose homeostasis, Incretin
Introduction
Glucagon-like peptide-1 (GLP-1), encoded as a fragment of proglucagon in the preproglucagon gene (Gcg), is expressed in the brain, pancreatic alpha cells and enteroendocrine cells within the intestine. GLP-1 is thought to act in the central nervous system to regulate feeding behaviour and on pancreatic beta cell GLP-1 receptors (GLP-1Rs) to stimulate insulin secretion. Two potent therapeutic strategies based on the GLP-1 system have been developed for the treatment of type 2 diabetes [1]. The first includes injectable long-acting GLP-1R agonists and the second increases the bioavailability of endogenous GLP-1 by pharmacologically inhibiting dipeptidyl peptidase-4 (DPP-4), the enzyme that removes the N-terminal amino acid and consequently inactivates GLP-1 [2].
While DPP-4 is important for regulating circulating active GLP-1 levels by limiting the half-life to 1–2 min [3], it also acts on other peptides involved in metabolic regulation including glucose-dependent insulinotropic polypeptide (GIP) [4]. A previous study in mice suggests that increasing the bioavailability of both GIP and GLP-1 contributes to the ability of DPP-4 inhibitors to improve glucose tolerance [5]. However, the lack of efficacy of GIP agonists in stimulating insulin secretion and promoting glucose disposal in individuals with type 2 diabetes [6, 7] raises doubts about whether both GLP-1 and GIP mediate the therapeutic effects of DPP-4 inhibitors in humans [8]. Thus, the standard explanation as to how DPP-4 inhibitors exert their therapeutic effects is their promotion of GLP-1 signalling. However, recent clinical data suggest that elevation in plasma GLP-1 does not account for all the glucoregulatory effects of DPP-4 inhibitors [2].
Using Cre/loxP technology, we have generated a novel genetic mouse model that allows for the reactivation of endogenous Gcg with tissue specificity. Using this model we have found that proglucagon products produced in pancreatic alpha cells, rather than intestinal L cells, are the principle mediator of glucose tolerance [9]. Thus, the aim of this study was to determine whether pancreas-derived or intestine-derived GLP-1 is the predominant source of the plasma GLP-1 that increases with DPP-4 inhibitors and that, in combination with an increase in GIP, drives the improvements in glucose regulation.
Methods
Animals
We developed a mouse model with a loxP flanked transcriptional blocking cassette inserted between exons 2 and 3 of the Gcg gene, as described previously [9]. These mice were extensively phenotyped in a previous publication [9]. The resulting mouse is null for Gcg expression (GcgRAΔNull) and therefore does not produce any proglucagon peptides (including GLP-1 and glucagon) [9]. In some studies GcgRAΔNull mice and wild-type littermates were used as controls. In other studies we crossed the GcgRAΔNull mice to lines expressing Cre recombinase with either the villin-1 promoter (VilCre; Stock # 004586, Jackson Laboratories, Bar Harbor, ME, USA) or pancreatic duodenal homeobox-1 (PDXICre; Stock # 014647, Jackson Laboratories) promoter generated mouse lines with endogenous Gcg reactivated specifically in intestinal epithelial L cells (GcgRAΔVilCre) and pancreatic alpha cells and within the duodenum (GcgRAΔPDX1Cre), respectively [9]. Since the intestinal reactivation, which also includes the duodenum, and the pancreatic/duodenal reactivation have very different responses, then the duodenum is not a major contributor to this process, and the GcgRAΔPDX1Cre mouse is considered to be a pancreatic reactivation model for the purposes of this study. All reactivated mice were compared with littermate mice expressing Cre alone (VilCre or PDXICre, respectively). All mice were male on a mixed background, were at least 8 weeks old and were individually housed and maintained on a 12 h light-dark cycle (lights off at 17:00 hours) at 25°C and 50–60% humidity with ad libitum access to water and standard chow diet. All procedures for animal use were approved by the University of Michigan and University of Cincinnati Institutional Animal Care and Use Committee.
Effect of acute DPP-4 inhibition on glucose tolerance
Mice were fasted for 4–5 h prior to oral glucose tolerance tests (OGTT). Baseline glucose levels were taken before two doses of linagliptin (3 or 30 mg/kg) were administered by gavage 30 min prior to an oral glucose load (100 μl of 20%, wt/vol. in water, dextrose). Because both doses were effective at lowering glucose, we chose an intermediate dose (10 mg/kg) of linagliptin for the remaining studies. In all experiments 0.5% natrosol (wt/vol. in water; 9004–62-0; Millipore Sigma, St Louis, MO, USA) was used as the vehicle for linagliptin. In separate studies, baseline glucose was measured and then mice were given an i.p. injection of 50 μg/100 μl of exendin-[9–39] (Ex9) (Bachem Bioscience, Torrance, CA, USA) or saline (NaCl 154 mmol/l) 45 min before an oral glucose load (200 μl of 20% wt/vol. in water, glucose) and 15 min before oral linagliptin (10 mg/kg). Blood was sampled from the tail vein at baseline and at −15, 15, 30, 45, 60 and 120 min after glucose. To study the role of GIP in the response to linagliptin, mice were administered a GIP receptor-antagonising antibody (GIPrAb; obtained from Boehringer Ingelheim, Ingelheim am Rhein, Germany). The structure of this compound and the protocol of administration was as reported previously [10]. Briefly, mice were administered the GIPrAb (30 μg/kg), fasted for 24 h, then linagliptin (10 mg/kg) or vehicle was administered prior to an oral glucose (2 g/kg) tolerance test. Blood was sampled from the tail vein at baseline and at 0, 10, 20, 40, 60 and 120 min post glucose gavage.
When appropriate, mice were utilised in a cross-over design whereby each animal was exposed to vehicle or treatment conditions in distinct experiments with each experiment separated by at least one week. All mice were randomised to groups, and experimenters were blind to groups during blood glucose measurements. However, for all experiments each animal was studied under a specific drug condition once (i.e. data are from distinct samples). Animals were excluded from an analysis if the handler noted a bad injection of drug and/or glucose or if they lost weight and became ill between experiments.
Effect of chronic DPP-4 inhibition on glucose tolerance
In a separate cohort, mice were fed a high-fat diet (19 kJ [4.54 kcal]/g, 41% fat; Research Diets, New Brunswick, NJ, USA) for 16 weeks. The DPP-4 inhibitor vildagliptin was given by i.p. injection at a dose (150 μg/200 μl Millipore Sigma, St Louis, MO, USA) equipotent (data not shown) to 10 mg/kg linagliptin, daily for 11 days. A control group was administered with saline. Body weight and food intake was measured daily and on day 11 body composition was assessed and an OGTT was performed.
Assays
Mice were administered either linagliptin (10 mg/kg) or vehicle (0.5% natrosol) via oral gavage 30 min before oral glucose (200 μl, 50%, wt/vol. in water, glucose) and 45 min prior to blood collection. Blood was collected in heparinised syringes and placed in a tube with a mixture of DPP-4 inhibitor (Millipore, Burlington, MA, USA), heparin, EDTA and aprotonin. Plasma was assayed for active GLP-1 to validate genotyping using sandwich ELISA kits (K150JWC-1; Mesoscale Discovery, Rockville, MD, USA). Plasma was assayed for active mouse GIP using an ELISA (27702; IBL America, Minneapolis, MN, USA). Plasma insulin was analysed using a standard ELISA kit (90080; Crystal Chem, Elk Grove Village, IL, USA). DPP-4 activity was measured using a specific peptide substrate with a terminal coumarin derivative (H-Ala-Pro-7-amido-4-trifluoromethylcoumarin; Bachem, Bubendorf, Switzerland), allowing quantification in a fluorescence microplate reader (Wallac Victor 1420 Multilabel Counter; Perkin Elmer, Waltham, MA, USA) upon cleavage by DPP-4 [11].
Gene expression
Mouse whole pancreas and epithelial scrapes from duodenum and ileum were immediately homogenised in Trizol (Thermo Fisher Scientific, Waltham, MA, USA) using a Tissuelyser II (Qiagen, Hilden, Germany). Total RNA was extracted using the TaqMan Reverse Transcription kit (Thermo Fisher Scientific). The quality and concentration of the isolated total RNA were determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific). cDNA was isolated (iScript cDNA synthesis kit; BioRad, Hercules, CA, USA) and real-time quantitative PCR (qPCR) was performed using a TaqMan 7900 Sequence Detection System with TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA). mRNA expression was evaluated using the TaqMan gene probe mouse Gcg (Mm01269054_m1) and normalised to the ribosomal RPL32 gene (Rn00820748_g1).
Statistical analysis
Data are presented as mean ± SD (bar graphs) or mean ± SEM (line graphs), and statistical significance was set at p<0.05 for all analyses. The data were analysed using mixed-model ANOVAs to evaluate significant main effects of drug, genotype or time (where appropriate) and significant interactions among these independent variables. Where appropriate a Tukey’s post hoc analysis was performed to determine where the significant differences lie. Because of the complex statistical design (drug, genotype, time) and potent effect of both the drug and genotype, sometimes there was no interaction with time (main effect of drug and/or genotype). These main effects are indicated in the text and figure legends. Statistical interactions are represented by symbols within figures
Results
Gcg peptides are not necessary for glucose-lowering effect of DPP-4 inhibitors
Similar to previous findings, GcgRAΔNull and GcgRAΔVilCre mice weighed significantly more than control mice (electronic supplementary material [ESM] Table 1). Basal levels of glucose, total GLP-1 and total GIP are reported in ESM Table 1. In response to the DPP-4 inhibitor linagliptin, plasma DPP-4 activity was significantly reduced in VilCre control, GcgRAΔVilCre and GcgRAΔNull mice (Fig. 1a). Plasma levels of active GLP-1 were significantly increased by linagliptin in VilCre control and GcgRAΔVilCre mice and, as expected, GLP-1 was not detectable in the plasma of GcgRAANuU mice regardless of treatment (Fig. 1b). Plasma active GIP was significantly increased to a similar degree in VilCre control, GcgRAAVilCre and GcgRAΔNull mice after linagliptin administration (Fig. 1c). However, we did not observe an increase in plasma active GIP in GcgRAΔNull mice compared with VilCre control mice, as previously reported in a similar Gcg-deficient mouse model [12].
Fig. 1.
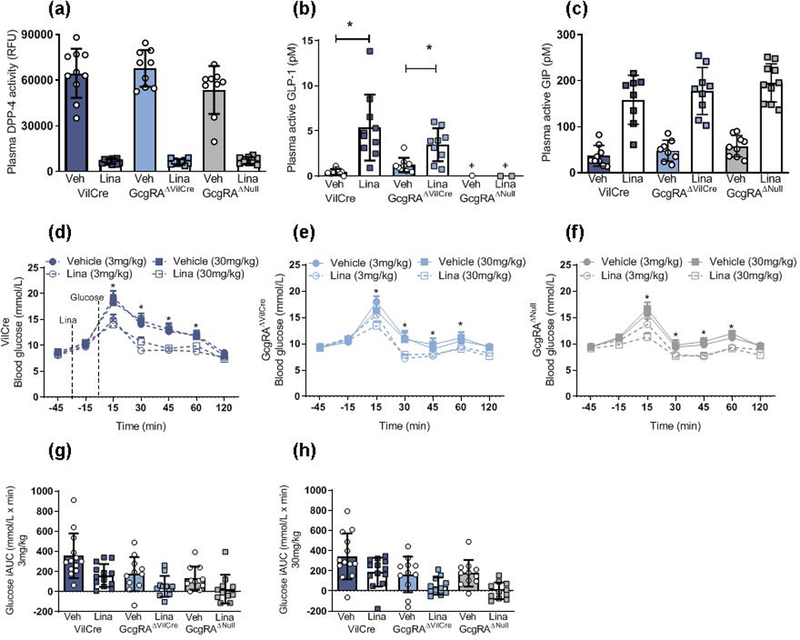
Linagliptin improves glucose tolerance in GcgRAΔNull mice. (a) Linagliptin reduced plasma DPP-4 activity in VilCre control, GcgRAΔNull and GcgRAΔVilCre mice (p<0.0001, two-way ANOVA, main effect of linagliptin vs vehicle across genotypes; n=9–10). (b) Linagliptin significantly increased plasma active GLP-1 levels in VilCre control and GcgRAΔVilCre mice but plasma active GLP-1 was not detected in GcgRAΔNull mice (***p<0.001, two-way ANOVA, linagliptin vs vehicle; †p<0.05 GcgRAΔNull vehicle and linagliptin vs all other groups, genotype × linagliptin interaction; n=9–10). (c) Linagliptin increased plasma active GIP levels across all genotypes (p<0.0001, two-way ANOVA, main effect of linagliptin vs vehicle across genotypes; n=9–10). (d–f) Linagliptin at 3 mg/kg and 3G mg/kg significantly improved oral glucose tolerance in VilCre control mice (d; n=13), GcgRAΔVilCre mice (e; n=11) and GcgRAΔNull mice (f; n=11) (*p<0.05; three-way ANOVA; time × drug interaction for both 3 mg and 3G mg linagliptin doses vs respective vehicle across genotypes). (g, h) The integrated AUC for glucose responses to 3 mg/kg linagliptin (g) and 3G mg/kg linagliptin (h) graphs shown in d–f (p<0.001, two-way ANOVA, main effect of linagliptin vs vehicle across genotypes; p<0.001, main effect of genotype across drug doses). iAUC, integrated AUC; Lina, linagliptin; RFU, relative fluorescence units; Veh, vehicle
As we have found previously [9], GcgRAΔNull and GcgRAΔVilCre mice displayed improved glucose tolerance. Our previous work concluded that this was a consequence of glucagon deficiency rather than improved insulin sensitivity or secretion, as determined by a hyperinsulinaemic-euglycaemic clamp and an IVGTT [9]. Here, we found that linagliptin significantly improved glucose tolerance at doses of 3 mg/kg and 30 mg/kg in VilCre control, GcgRAΔVilCre and GcgRAΔNull mice at 15, 30, 45 and 60 min after an oral glucose gavage (Fig. 1d–h).
Tissue-specific source of GLP-1 after DPP-4 inhibition
DPP-4 inhibitors are generally held to improve glucose homeostasis by extending the bioavailability of circulating GLP-1 derived from distal enteroendocrine L cells. To determine whether the specific tissue source of GLP-1 influences DPP-4 inhibitor effectiveness we gave a potent GLP-1R antagonist, Ex9, to GcgRAΔVilCre mice. Linagliptin (10 mg/kg) lowered peak glucose concentration, which occurred at 15 min in all mice, while Ex9 impaired basal glucose and tolerance to an oral glucose load in VilCre control mice but had no effect in GcgRAΔVilCre or GcgRAΔNull mice (Fig. 2a–d). Given that Ex9 is a GLP-1 antagonist and thus requires intact ligand, we expected that it would have no impact on GcgRAΔNull mice that do not produce GLP-1 (Fig. 2c). However, despite normalised postprandial plasma GLP-1 levels in response to linagliptin in the GcgRAΔVilCre mice (Fig. 1b), Ex9 alone or with linagliptin did not impair glucose tolerance in these mice (Fig. 2b,d). Ex9 and linagliptin administered together to VilCre control mice but not GcgRAΔVilCre or GcgRAΔNull mice produced elevated basal glucose (−15 min) and all mice had a significantly lower glucose excursion compared with Ex9 treatment alone at 15–60 min after the glucose load. These findings are consistent with our previous findings [9] but do not fit with the conventional model, whereby GLP-1 secreted into the circulation from the gut mediates the effects of DPP-4 inhibitor action [13].
Fig. 2.
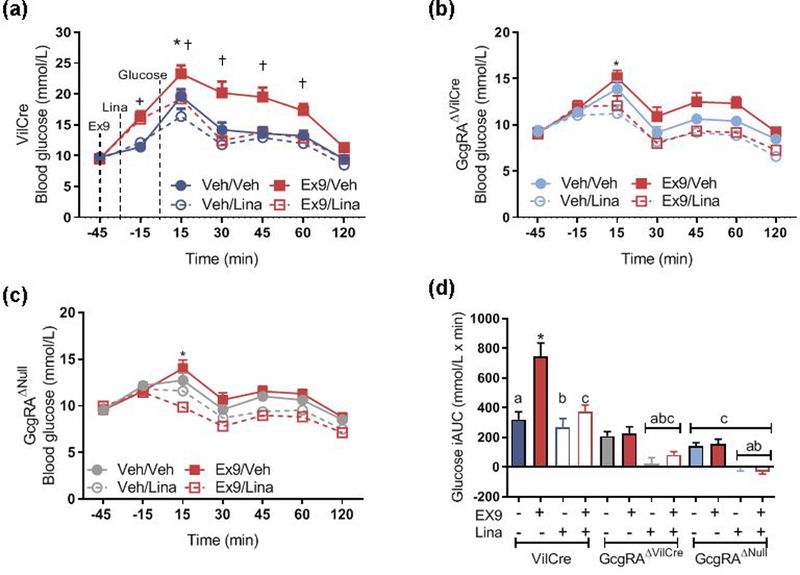
Ex9 blunts the improvement in glucose tolerance after linagliptin in control mice. (a–c) Ex9 increased glucose tolerance in VilCre (n=15/drug group) control mice (a) but had no effect in littermate GcgRAΔVilCre (b; n=13 in Veh/Veh and Ex9/Lina groups; n=14 in Ex9/Veh and Veh/Lina groups) or GcgRAΔNull mice (c; n=15/drug group) during an OGTT (†p<0.05, Ex9/Veh in VilCre vs GcgRAΔNull and GcgRAΔVilCre, genotype × time × Ex9 interaction; *p<0.05, Veh/Veh vs Veh/Lina across genotypes, time × linagliptin × Ex9 interaction; n=13–16). Graphs (a–c) were statistically analysed in a repeated measure four-way ANOVA with a Tukey post hoc test but are presented separately for clarity. (d) Integrated AUC for the graphs shown in a–c. *p<0.05 vs all other bars. Bars with the same symbols are significantly different compared with VilCre control mice (p<0.05, Ex9 × linagliptin × genotype interaction; n=13–16. Data are analysed with a three-way ANOVA with a Tukey post hoc test. iAUC, integrated AUC; Lina, linagliptin; Veh, vehicle
To determine whether GLP-1 produced in pancreatic islets is a target for DPP-4 inhibitors, Gcg was reactivated in alpha cells of GcgRAΔNull mice by crossing Gcg-null mice to PDX1Cre mice to generate GcgRAΔPDX1Cre mice. These mice have similar body weights to the PDX1Cre control mice alone and to a pooled group of PDX1Cre and VilCre control mice (ESM Table 1). Basal levels of glucose, total GLP-1 and total GIP in GcgRAΔPDX1Cre mice are shown in ESM Table 1. These mice have normalised expression of Gcg in the pancreas and duodenum but no Gcg expression within the ileum (Fig. 3a). Plasma active GLP-1 levels were increased in both the PDX1Cre control mice and GcgRAΔPDX1Cre mice after linagliptin administration (Fig. 3b), although PDX1Cre mice had overall higher levels of active GLP-1. This was not surprising as we would not predict the pancreas to be a major source of circulatory GLP-1 as it likely acts locally in a paracrine manner. In addition, we saw a similar increase in plasma active GIP levels in response to linagliptin in PDX1Cre control mice and GcgRAΔPDX1Cre mice (Fig. 3c). Linagliptin significantly improved glucose tolerance in GcgRAΔPDX1Cre mice to a similar extent as in PDX1Cre control mice (Fig. 3d,e).
Fig. 3.
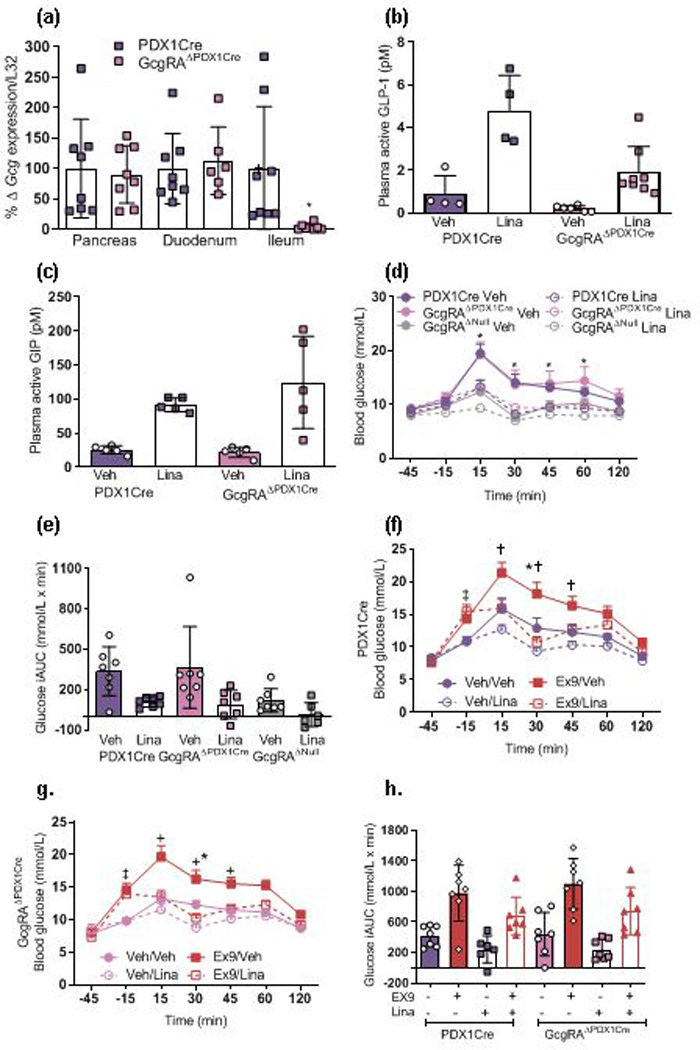
Ex9 blocks DPP-4 inhibitor action in GcgRAΔPDX1Cre mice. (a) Tissue harvested 30 min after oral linagliptin (10 mg/kg) and 15 min after oral glucose revealed lowered Gcg gene expression in the ileum (n=8) but not pancreas (n=8) and duodenum (n=6) of GcgRAΔPDX1Cre vs PDX1Cre control mice (*p<0.05, one-tailed t test within each tissue). (b) Active plasma GLP-1 levels increased after linagliptin treatment in GcgRAΔPDX1Cre mice and to a greater extent in PDX1Cre control mice (p<0.0001, two-way ANOVA; main effect of linagliptin vs vehicle across genotypes; p<0.001, main effect of genotype; n=4–8). (c) Linagliptin significantly increased plasma active GIP levels 15 min after a glucose load in both PDX1Cre control and GcgRAΔPDX1Cre mice (p<0.0001, two-way ANOVA; main effect of linagliptin vs vehicle across genotypes; n=5). (d) Regardless of genotype, linagliptin-treated mice displayed reduced glucose levels at 15, 30, 45 and 60 min after an oral glucose load (**p<0.01, three-way ANOVA; time x linagliptin interaction for all genotypes; n=6–7). (e) iAUC for glucose excursion shown in (d) (p<0.001, two-way ANOVA; main effect of linagliptin vs vehicle across genotypes; p<0.05, main effect of genotype; n=6–7). (f, g) Linagliptin-treated (Veh/Lina) PDX1Cre and GcgRAΔPDX1Cre littermate mice displayed lowered blood glucose levels 30 min after an oral glucose gavage compared with vehicle-treated (Veh/Veh) mice (***p<0.001, time × Ex9 × linagliptin interaction; n=7). Ex9-treated mice (Ex9/Veh) had a greater glucose excursion compared with all other treatment groups 15, 30 and 45 min after glucose (†††p<0.001, four-way ANOVA, time × Ex9 × linagliptin interaction). Mice administered with combined Ex9 and linagliptin (Ex9/Lina) had greater glucose levels compared with Veh/Veh and Veh/Lina groups 15 min before an oral glucose load, but not at any other time point (‡‡‡p<0.001, four-way ANOVA, time × Ex9 × linagliptin interaction). Although presented separately, a repeated measure four-way ANOVA was used to determine the impact of genotype, Ex9, linagliptin and time on glucose tolerance. (h) In both PDX1Cre and GcgRAΔPDX1Cre mice, Ex9 increased (p<0.001, main effect of Ex9 across genotypes), while linagliptin reduced (p<0.0001, three-way ANOVA, main effect of linagliptin across genotypes; n=6–7), blood glucose integrated AUC levels vs Veh. iAUC, integrated AUC; Lina, linagliptin; Veh, vehicle
We then assessed the insulin response to linagliptin (10 mg/kg) after oral glucose administration in all mice. No differences in baseline insulin levels were detected across genotypes (ESM Fig. 1a). Linagliptin did not have any additional impact on the insulin response 5 or 15 min after a glucose load (ESM Fig. 1b,c).
Unlike mice with only intestinal Gcg production, Ex9 impaired oral glucose tolerance in GcgRAΔPDX1Cre and control PDX1Cre mice (Fig. 3f–h) and was able to lessen the glucose-lowering effect of linagliptin in both PDX1Cre control and GcgRAΔPDX1Cre mice. These data suggest that Gcg expression and GLP-1 synthesis within the pancreas is necessary for the glucoregulatory role of DPP-4 inhibitors.
The role of GIP in response to DPP-4 inhibition
To test the specific contribution made by GIP to linagliptin-induced improvements in glucose tolerance, a GIP antagonising antibody (GIPrAB) [10] was administered to littermate wild-type and GcgRAΔNull mice. Blockade of GIP receptors impaired glucose tolerance in both wild-type and GcgRAΔNull mice (Fig. 4a,b). In wild-type mice that were co-administered linagliptin and GIPrAB, linagliptin was able to prevent the impairment of glucose tolerance by the GIPrAB (Fig. 4a and ESM Fig. 2a). However, the GIPrAB retained its ability to impair glucose tolerance in GcgRAΔNul1 mice when the antibody was co-administered with linagliptin (Fig. 4b and ESM Fig. 2a). These data highlight the dual action of DPP-4 inhibition, which targets both GLP-1 and GIP to improve glucose homeostasis. Previous studies have suggest that GIP signalling is muted during obesity and/or type 2 diabetes [7, 14]. Based on this observation, DPP-4 inhibitors may be less effective in high-fat-diet-fed GcgRAΔNull and GcgRAΔVilCre mice that depend on GIP to mediate DPP-4 inhibitory effects. We administered vildagliptin, another DPP-4 inhibitor, to mice after 16 weeks of high-fat diet. Vildagliptin did not affect fat mass or food intake over 11 days of treatment (data not shown). Despite the prolonged exposure to high-fat diet, VilCre control, GcgRAΔNul1 and GcgRAΔVilCre mice all showed improved oral glucose tolerance in response to vildagliptin (Fig. 4c and ESM Fig. 2b).
Fig. 4.
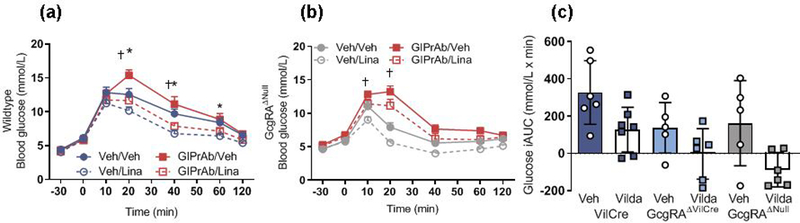
Response to GIPrAB after linagliptin in GcgRAΔNull mice. (a, b) GIPrAB impaired oral glucose tolerance and blunted the linagliptin-induced decrease in glucose in littermate wild-type mice (a), but in GcgRAΔNull mice, which have no circulating GLP-1, the GIPrAB retained the full antagonistic effect whether administered with vehicle or linagliptin (b) (††p<0.01, four-way ANOVA, time × GIPrAB × genotype interaction, Veh/Veh vs GIPrAB/Veh; *p<0.05, time × linagliptin × genotype interaction, Veh/Veh vs Veh/Lina. Wild-type: Veh/Veh, n=11; Veh/Lina, n=12; GIPrAB/Veh, n=16; GIPrAB/Lina, n=14; GcgRAΔNull: Veh/Veh, n=9; Veh/Lina, n=10; GIPrAB/Veh, n=15; GIPrAB/Lina, n=15). (c) Eleven consecutive days of vildagliptin treatment improved glucose tolerance in high-fat-diet-induced obese VilCre control mice, GcgRAΔNull mice and GcgRAΔVilCre mice (p<0.001, two-way ANOVA; main effect of vildagliptin vs vehicle across genotypes; n=5–7). A genotype effect was noted under vehicle conditions such that GcgRAΔNull and GcgRAΔVilCre mice had improved glucose tolerance compared with littermate VilCre control mice (p<0.01, main effect of genotype). iAUC, integrated AUC; Lina, linagliptin; Veh, vehicle; Vilda, vildagliptin
Discussion
DPP-4 inhibitors are effective and widely used therapeutics for type 2 diabetes. The conventionally accepted mechanism of action underlying the efficacy of DPP-4 inhibitors is that they improve glucose homeostasis through increased gut-derived plasma GLP-1 concentrations. However, preclinical work and recent clinical studies suggest that this view may be incomplete and ignores potentially important contributions from GIP. We have recently generated a transgenic mouse line that allows for tissue-selective expression of the endogenous Gcg gene [9]. Using this model, we determined that pancreatic but not intestinal GLP-1 was the critical peptide pool necessary for regulation of normal glucose tolerance. In addition, studying the impact of DPP-4 inhibitors in combination with GLP-1 and GIP receptor antagonists in our Gcg-null animals allowed us to determine the relative roles of these two incretin peptides. Consistent with previous work [15, 16], we found that in the absence of GLP-1 production, GIP is sufficient to maintain improved glucose tolerance after DPP-4 inhibitor administration. Moreover, our findings indicate that the endocrine pancreas is also an important source of GLP-1 targeted by DPP-4 inhibitors to confer better glucose tolerance.
The proteolytic actions of DPP-4 target approximately 40 currently known substrates, with additional substrates continually being discovered [17]. Despite this growing list, many of these compounds are primarily in vitro substrates with little evidence supporting the in vivo regulation of their activity by DPP-4 [17]. Currently, the best evidence supports GLP-1 and GIP as important targets of DPP-4’s action in animals and humans that can be tied to physiological outcomes. Indeed, an early formulation of a DPP-4 inhibitor improved oral glucose tolerance in GLP-1R knockout mice and in GIP receptor knockout mice [15]. However, double knockout of both GLP-1 and GIP receptors did not provide any significant improvement in oral glucose tolerance following DPP-4 inhibitor administration [15, 16]. Two recent studies that included individuals with and without type 2 diabetes found that GLP-1 contributed ~50% of the glucose-lowering and insulin-stimulatory effects of DPP-4 inhibitors [2, 18]. Using both genetic and pharmacological inhibition, our work is consistent with these data in that linagliptin retained the ability to improve glucose tolerance in animals devoid of all Gcg peptides.
Recent work has also identified the source of DPP-4 critical to GLP-1 and GIP degradation as being endothelial cells, rather than enteric epithelial or hematopoietic cells [19]. Although doses of DPP-4 inhibitors that reduce enzyme activity in the intestinal lumen without prominent systemic effects (gut-selective) improve glucose tolerance and increase both GLP-1 and GIP, they are not as potent as doses that decrease plasma DPP-4 activity as well [19, 20].
Interestingly, gut-selective doses of sitagliptin retained the ability to improve glucose tolerance in mice with genetic ablation of DPP-4 from endothelial cells, albeit only in animals fed a chow diet and not those receiving a high-fat diet. The studies described here examined the acute effect of linagliptin in improving glucose tolerance in chow-fed mice and found that pancreatic GLP-1 is an important source for the inhibitory effects of DPP-4.
Our previous work in the GcgRAΔNull and GcgRAΔVilCre mouse models demonstrated that the minimal glucose excursion after an oral glucose load was not due to improved insulin sensitivity or increased insulin secretion [9]. We hypothesise that the absence of glucagon leads to improved oral glucose tolerance similar to that seen in glucagon receptor knockout mice, which also have minimal glucose excursions after an oral glucose load. Despite this, linagliptin still improved glucose tolerance in GcgRAΔNull mice, yet we found no additivity of linagliptin and glucose on insulin levels. It is possible that we would need to assess a full time course to fully appreciate the impact of linagliptin on insulin levels in these mice. It is also possible that DPP-4 inhibitors have effects that go beyond insulin secretion. Indeed, previous work has determined that vildagliptin improves glucose tolerance secondary to increased insulin secretion and proliferation of beta cells [21]. Overall, the key point from these data is that the ability of linagliptin to regulate glucose tolerance depends upon both GIP signalling and the tissue-specific source (i.e. pancreas) of available GLP-1.
We cannot exclude the possibility that GLP-1 or GIP receptor number or sensitivity could influence responses to ligands or antagonists of those receptors. However, in experiments where the concentration of Ex9 is doubled (data not shown), we still did not see a significant Ex9-induced impairment of glucose tolerance in GcgRAΔNul1 or GcgRAΔVilCre mice. As expected, linagliptin did not increase active GLP-1 levels to control levels in the GcgRAΔPDX1Cre mice as the pancreas is not a major contributor to circulating GLP-1. In line with previous reports [22–24], we believe pancreas-produced GLP-1 acts in a paracrine manner. In an important series of experiments, recent findings from a study using perifused islets demonstrated that both GLP-1 and glucagon secreted from alpha cells can act on beta cell GLP-1Rs to regulate insulin secretion [25]. Thus, alpha cell preproglucagon products are clearly important in regulating local insulin levels (i.e. paracrine action). While DPP-4 is expressed in mouse and pig pancreatic endocrine cells [26, 27] within the pancreatic duct, and in pancreatic alpha cells in humans [28, 29], glucagon is not one of its major substrates. Thus, DPP-4 inhibitors could easily act on pancreatic islets to specifically protect the local pool of GLP-1 produced by alpha cells. Interestingly, there has been speculation that DPP-4 plays a role in mediating the balance of glucagon and GLP-1 from pancreatic alpha cells [28]. Indeed, previous work has found that linagliptin protects human isolated islets from glucose-, lipid-and cytokine-induced toxicity [30]. Additionally, islets isolated from individuals with type 2 diabetes exhibit decreased DPP-4 activity in order to preserve GLP-1 secretion in culture, further implicating a functional role of endogenous islet GLP-1 in diabetes [29].
In order to transfer preclinical knowledge about tissue-specific sources of Gcg peptides to improve upon current pharmacotherapies for type 2 diabetes, it is important to understand what stimuli provoke paracrine communication within the pancreas. Preclinical models may be used to help determine which physiological conditions regulate one pool of GLP-1 from another. Conditions such as metabolic stress, acute or chronic inflammation or nutritional availability all might impact the regulation of GLP-1 from intestinal or pancreatic sources [13]. Indeed, it has been suggested that pancreatic Gcg-encoded peptides are essential components in adaptation to metabolic stress and that a lack of communication within the endocrine pancreas contributes to variations in glucose tolerance [25]. Further, human islet architecture has a more favourable organisation for paracrine signalling when compared with mouse islets [22, 31]. Although we cannot rule out the possibility of species differences in GLP-1 biology, we feel that understanding the nuances of the potential importance of pancreatic Gcg-encoded peptides to islet function will be informative to future studies in humans.
Previous studies in Zucker rats suggest that GIP signalling is muted during obesity [14]. However, we found that 11 days of vildagliptin treatment was still able to improve glucose tolerance in diet-induced obese GcgRAΔNull and GcgRAΔVilCre mice. In general, when hyperglycaemia is corrected in individuals with type 2 diabetes using a variety of interventions, including sulfonylureas and insulin, GIP sensitivity can be restored [32, 33]. Similar effects have been observed in obese rats following DPP-4 inhibitor treatment [14]. While it is unlikely that GLP-1 and/or GIP contribute to the pathology of type 2 diabetes mellitus, pharmacotherapeutics targeting both GIP receptors and GLP-1Rs in individuals with type 2 diabetes may offer further benefits than GLP-1R agonism alone [34]. Thus, it is possible, given the better than normal glucose tolerance inherent in the GcgRAΔNull and GcgRAΔVilCre mouse lines, that GIP signalling was preserved, therefore, enabling vildagliptin to improve glucose tolerance.
In summary, our data demonstrate that via GIP signalling linagliptin is able to retain its ability to improve glucose tolerance in mice devoid of Gcg-encoded peptides, including GLP-1 and oxyntomodulin (two targets of DPP-4 degradation). Further, using a combination of genetic and pharmacological tools, our data suggest that islet-derived rather than intestine-derived GLP-1 also contributes to the therapeutic efficacy of DPP-4 inhibitors.
Supplementary Material
Research in context.
What is already known about this subject?
Glucagon-like peptide-1 (GLP-1) is an enteric peptide that promotes insulin secretion
Prolonging the bioavailability of GLP-1 by inhibiting a primary inactivating enzyme, dipeptidyl peptidase-4 (DPP-4), improves glucose homeostasis
What is the key question?
Does the source of GLP-1 (either pancreatic or intestinal) influence the effectiveness of DPP-4 inhibitors?
What are the new findings?
The GLP-1 receptor antagonist exendin-[9–39] blunted improvements in glucose tolerance in linagliptin-treated control mice and in mice expressing Gcg only in the pancreas (GcgRAΔPDX1Cre), but had no effect on Gcg whole-body knockout mice or mice expressing Gcg only in the intestine (GcgRAΔVilCre); this indicates that GLP-1 produced within the endocrine cells of the pancreas is necessary for the full response to DPP-4 inhibitors
Co-administration of a GIP-antagonising antibody blunted the impact of linagliptin in wild-type mice and fully blocked its effect in Gcg whole-body knockout mice, indicating that, in the absence of GLP-1, GIP is sufficient to induce the full glycaemic response to DPP-4 inhibitors
How might this impact on clinical practice in the foreseeable future?
If these findings are also valid in humans, then understanding how to increase the bioavailability of pancreatic GLP-1 could improve the impact of drugs that target GLP-1 on glucose homeostasis
Acknowledgements
We would like to thank J. Magrisso (Department of Surgery, University of Michigan, USA) for his technical assistance.
Funding
This work is supported in part by NIH Awards DK082480 (DAS) and DK093848 (RJS). The investigators also received research support from Boehringer Ingelheim (DAS, RJS), Ethicon Endo-Surgery (DAS, RJS), Sanofi (RJS) and Novo Nordisk A/S (DAS, RJS).
Abbreviations
- DPP-4
Dipeptidyl peptidase-4
- Ex9
Exendin-[9–39]
- GcgRAΔNull
Gcg null mouse model
- GcgRAΔPDX1Cre
Mouse model with Gcg reactivated in the pancreas
- GcgRAΔVilCre
Mouse model with Gcg reactivated in the intestine
- GIP
Glucose-dependent insulinotropic polypeptide
- GIPrAB
GIP receptor-antagonising antibody
- GLP-1
Glucagon-like peptide-1
- GLP-1R
GLP-1 receptor
- PDX1
Pancreatic duodenal homeobox-1
- PDX1Cre
Wild-type littermate of GcgRAΔPDX1Cre
- VilCre
Wild-type littermate of GcgRAΔVilCre
Footnotes
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author by reasonable request.
Duality of interest
RJS has received research support from Ethicon Endo-Surgery, Novo Nordisk, Sanofi and Janssen. RJS has served on scientific advisory boards for Ethicon Endo-Surgery, Daiichi Sankyo, Janssen, Novartis, Nestle, Takeda, Boehringer Ingelheim, Sanofi and Novo Nordisk and is a paid speaker for Ethicon Endo-Surgery. DAS has received research support from Ethicon Endo-Surgery, Novo Nordisk and Boehringer Ingelheim.All other authors declare no duality of interest associated with this manuscript.
References
- 1.Fujioka K, O’Neil PM, Davies M, et al. (2016) Early weight loss with liraglutide 3.0 mg predicts 1-year weight loss and is associated with improvements in clinical markers. Obesity 24:2278–2288. 10.1002/oby.21629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nauck MA, Kind J, Kothe LD LD, et al. (2016) Quantification of the contribution of GLP-1 to mediating insulinotropic effects of DPP-4 inhibition with vildagliptin in healthy subjects and patients with type 2 diabetes using exendin [9–39] as a GLP-1 receptor antagonist. Diabetes 65:2440–2447. 10.2337/db16-0107 [DOI] [PubMed] [Google Scholar]
- 3.Meier JJ, Nauck MA, Kranz D, et al. (2004) Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes 53:654–62 [DOI] [PubMed] [Google Scholar]
- 4.Vilsbøll T, Agersø H, Lauritsen T, et al. (2006) The elimination rates of intact GIP as well as its primary metabolite, GIP 3–42, are similar in type 2 diabetic patients and healthy subjects. Regul Pept 137:168–172. https://doi.org/10.1016Zj.regpep.2006.07.007 [DOI] [PubMed] [Google Scholar]
- 5.Ali S, Lamont BJ, Charron MJ, Drucker DJ (2011) Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest 121:1917–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vilsbøll T, Krarup T, Madsbad S, Holst JJ (2002) Defective amplification of the late phase insulin response to glucose by GIP in obese type II diabetic patients. Diabetologia 45:1111–1119. 10.1007/s00125-002-0878-6 [DOI] [PubMed] [Google Scholar]
- 7.Nauck MA, Heimesaat MM, Orskov C, et al. (1993) Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91:301–307. 10.1172/JCI116186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seino Y, Fukushima M, Yabe D (2010) GIP and GLP-1, the two incretin hormones: similarities and differences. J Diabetes Invest. 1: 8–23. 10.1111/j.2040-1124.2010.00022.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambers AP, Sorrell JE, Haller A, et al. (2017) The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab 25:927–934. 10.1016/j.cmet.2017.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravn P, Madhurantakam C, Kunze S, et al. (2013) Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J Biol Chem 288:19760–19772. 10.1074/jbc.M112.426288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas L, Eckhardt M, Langkopf E, et al. (2008) (R)-8-(3-Amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther 325:175–182. 10.1124/jpet.107.135723 [DOI] [PubMed] [Google Scholar]
- 12.Fukami A, Seino Y, Ozaki N, et al. (2013) Ectopic expression of GIP in pancreatic β-cells maintains enhanced insulin secretion in mice with complete absence of proglucagon-derived peptides. Diabetes 62:510–518. 10.2337/db12-0294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yabe D, Seino Y, Seino Y (2018) Incretin concept revised: the origin of the insulinotropic function of glucagon-like peptide-1: the gut, the islets or both? J Diabetes Investig 9:21–24. 10.1111/jdi.12718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piteau S, Olver A, Kim S-J, et al. (2007) Reversal of islet GIP receptor down-regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat. Biochem Biophys Res Commun 362:1007–1012. 10.1016/j.bbrc.2007.08.115 [DOI] [PubMed] [Google Scholar]
- 15.Hansotia T, Baggio LL, Delmeire D, et al. (2004) Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes 53:1326–1335 [DOI] [PubMed] [Google Scholar]
- 16.Flock G, Baggio LL, Longuet C, Drucker DJ (2007) Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes 56:3006–3013. 10.2337/db07-0697 [DOI] [PubMed] [Google Scholar]
- 17.Mulvihill EE, Drucker DJ (2014) Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev 35:992–1019. 10.1210/er.2014-1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aulinger BA, Bedorf A, Kutscherauer G, et al. (2014) Defining the role of GLP-1 in the enteroinsulinar axis in type 2 diabetes using DPP-4 inhibition and GLP-1 receptor blockade. Diabetes 63:1079–1092. 10.2337/db13-1455 [DOI] [PubMed] [Google Scholar]
- 19.Mulvihill EE, Varin EM, Gladanac B, et al. (2017) Cellular sites and mechanisms linking reduction of dipeptidyl peptidase-4 activity to control of incretin hormone action and glucose homeostasis. Cell Metab 25:152–165. 10.1016/j.cmet.2016.10.007 [DOI] [PubMed] [Google Scholar]
- 20.Waget A, Cabou C, Masseboeuf M, et al. (2011) Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology 152:3018–3029. 10.1210/en.2011-0286 [DOI] [PubMed] [Google Scholar]
- 21.Kawamori D, Shirakawa J, Liew CW, et al. (2017) GLP-1 signalling compensates for impaired insulin signalling in regulating beta cell proliferation in PIRKO mice. Diabetologia 60:1442–1453. 10.1007/s00125-017-4303-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchetti P, Lupi R, Bugliani M, et al. (2012) A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 55:3262–3272. 10.1007/s00125-012-2716-9 [DOI] [PubMed] [Google Scholar]
- 23.Traub S, Meier DT, Schulze F, et al. (2017) Pancreatic a cell-derived glucagon-related peptides are required for P cell adaptation and glucose homeostasis. Cell Rep 18:3192–3203. https://doi.org/10.1016Zj.celrep.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 24.Lin H V, Wang J, Wang J, et al. (2018) GPR142 prompts glucagon-like peptide-1 release from islets to improve β cell function. Mol Metab 11:205–211. 10.1016/j.molmet.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capozzi ME, Svendsen B, Encisco SE, et al. (2019) P Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 4: e126742. 10.1172/jci.insight.126742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorrell C, Grompe MT, Pan FC, et al. (2011) Isolation of mouse pancreatic alpha, beta, duct and acinar populations with cell surface markers. Mol Cell Endocrinol 339:144–150. 10.1016/j.mce.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulsen MD, Hansen GH, Dabelsteen E, et al. (1993) Dipeptidyl peptidase IV is sorted to the secretory granules in pancreatic islet A-cells. J Histochem Cytochem 41:81–88. 10.1177/41.L8093256 [DOI] [PubMed] [Google Scholar]
- 28.Augstein P, Naselli G, Loudovaris T, et al. (2015) Localization of dipeptidyl peptidase-4 (CD26) to human pancreatic ducts and islet alpha cells. Diabetes Res Clin Pract 110:291–300. 10.1016/j.diabres.2015.10.010 [DOI] [PubMed] [Google Scholar]
- 29.Omar BA, Liehua L, Yamada Y, et al. (2014) Dipeptidyl peptidase 4 (DPP-4) is expressed in mouse and human islets and its activity is decreased in human islets from individuals with type 2 diabetes. Diabetologia 57:1876–1883. 10.1007/s00125-014-3299-4 [DOI] [PubMed] [Google Scholar]
- 30.Shah P, Ardestani A, Dharmadhikari G, et al. (2013) The DPP-4 inhibitor linagliptin restores β-cell function and survival in human isolated islets through GLP-1 stabilization. J Clin Endocrinol Metab 98:E1163–E1172. 10.1210/jc.2013-1029 [DOI] [PubMed] [Google Scholar]
- 31.Whalley NM, Pritchard LE, Smith DM, White A (2011) Processing of proglucagon to GLP-1 in pancreatic α-cells: is this a paracrine mechanism enabling GLP-1 to act on β-cells? J Endocrinol 211:99–106. 10.1530/J0E-11-0094 [DOI] [PubMed] [Google Scholar]
- 32.Aaboe K, Knop FK, Vilsboll T, et al. (2009) KATP channel closure ameliorates the impaired insulinotropic effect of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. J Clin Endocrinol Metab 94:603–608. 10.1210/jc.2008-1731 [DOI] [PubMed] [Google Scholar]
- 33.Højberg P V, Vilsbøll T, Rabøl R, et al. (2009) Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52:199–207. 10.1007/s00125-008-1195-5 [DOI] [PubMed] [Google Scholar]
- 34.Frias JP, Nauck MA, Van J, et al. (2018) Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392:2180–2193. 10.1016/S0140-6736(18)32260-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.