

Abstract
PEGylation is a biochemical modification process of bioactive molecules with polyethylene glycol (PEG), which lends several desirable properties to proteins/peptides, antibodies, and vesicles considered to be used for therapy or genetic modification of cells. However, PEGylation of proteins is a complex process and can be carried out using more than one strategy that depends on the nature of the protein and the desired application. Proteins of interest are covalently conjugated or non-covalently complexed with inert PEG strings. Purification of PEGylated protein is another critical step, which is mainly carried out based on electrostatic interactions or molecular sizes using chromatography. Several PEGylated drugs are being used for diseases like anemia, kidney disease, multiple sclerosis, hemophilia and cancers. With the advancement and increased specificity of the PEGylation process, the world of drug therapy, and specifically cancer therapy could benefit by utilizing this technique to create more stable and non-immunogenic therapies. In this article we describe the structure and functions of PEGylation and how this chemistry helps in drug discovery. Moreover, special emphasis has been given to CCN-family proteins that can be targeted or used as therapy to prevent or block cancer progression through PEGylation technology.
Keywords: PEGylation, Cancer, Polyethylene glycol, Nanoparticles, Immunogenicity
Introduction
Polyethylene glycol (PEG) is a non-immunogenic biological compound made of repeating ethylene glycol units (Fig. 1a) (Veronese 2001; Veronese et al. 2001). Covalent and non-covalent attachment of PEG to biological molecules such as proteins and enzymes, called PEGylation, has been shown to increase half-life, reduced immunogenicity, and improved solubility and stability of the later (Veronese 2001; Veronese et al. 2001). Each PEG molecule can combine with two or three water molecules, making the overall compound larger and more hydrophilic (Oelmeier et al. 2012). PEG molecules either branched or linear, have their own advantages when reacting with certain proteins. Branched PEG molecules tend to increase certain “stealth” properties of a conjugated biomolecule, thereby also increasing “in vivo” half-life. PEG conjugation has the ability to modify physiochemical properties and increase the retention of the therapeutic agents in the body, which are useful for designing newer drug therapies (Pfister and Morbidelli 2014) (Table 1).
Fig. 1.

(a). Poly ethylene glycol with the most frequently used end groups, diol on top, and methoxy PEG at the bottom panel. (b). First generation PEGylation using cyanuryl chloride as coupling agent
Table 1.
List of PEGylated compounds currently in use or approved for use
| Currently Approved Drug | Purpose and/or Effect | Reference |
|---|---|---|
| Krystexxa | Lowers uric acid levels in order to aid in removing gout crystals. | (Hershfield et al. 2014) |
| PEGASYS (peginterferon alpha 2b) | Used with other hepatitis C medicines to treat adults with chronic hepatitis C and certain liver problems. Can be used with ribavirin to treat both adults and children with chronic hepatitis C. | (Wang et al. 2000) |
| Adagen (pegademase) | Modified enzyme used for Enzyme Replacement Therapy (ERT). | (Booth and Gaspar 2009) |
| Oncaspar (pegaspargase) | Given to patients with acute lymphoblastic leukemia as part of a group of chemotherapy treatments. | (Abuchowski et al. 1984) |
| Somavert (pegvisomant) | A prescription medicine for acromegaly, a disease caused by the surplus of growth hormones in the body. The goal is to have a normal IGF-1 level in the blood. | (Thorner 1999; Thorner et al. 1999) |
| Neulasta (pegfilgrastim) | Administered to reduce the risk of infection after strong chemotherapy. | (Johnston et al. 2000) |
| Mircera (CERA; PEG-EPO) | Used to treat symptomatic anaemia associated with chronic kidney disease (CKD). | (Macdougall 2005; Macdougall and Roche 2005) |
| Cimzia (certolizumab) | An injected prescription medication that works to prevent inflammation that may result from an overactive immune system. | (Choy et al. 2002) |
| Macugen (pegaptanib) | Utilized for the treatment of neovascular age-related macular degeneration. | (Bayes et al. 2002) |
| Plegridy (peginterferon beta-1a) | Indicated for the treatment of patients with relapsing forms of multiple sclerosis. | (Pepinsky et al. 2001) |
Early work on PEGylated proteins and enzymes was conducted in Frank Davis’s laboratories during the late 1970’s. They laid down the founding materials for protein PEGylation as a targeting drug delivery system for researchers today. Davis’s work included linking a methoxy-PEG to the amino acids present in proteins using cyanuric chloride as the coupling agent (Fig. 1b). Their work demonstrated that PEGylated proteins had longer half-lives in the bloodstream and decreased immunogenicity (Dozier and Distefano 2015). Because of the newfound use of proteins in therapeutics, PEGylation has emerged as a well-established method in the field of targeted drug delivery systems.
The advantage of PEGylation is that it leads to retention and enhancement of favorable properties of protein therapeutics without loss of function. Chemical, molecular, and structural properties of PEGs, as well as their conformational behavior in aqueous solutions govern the pharmacological disposition of PEGs and PEGylated products in physiological compartments (Knop et al. 2010; Torchilin 2007). Key chemical properties that direct the capacity of PEG to modulate pharmacokinetic and pharmacodynamic profiles of small molecular drugs, proteins or nanoparticles are: (1) molecular weight (2) polydispersity (3) conformation, and (4) end group functionality. PEG molecules can vary in size and shape, thereby providing options to modulate pharmacological output for different drug therapies. This is especially useful when proteins are needed to reside in vivo for an extended period of time with non-significant degradation or loss of function (Su et al. 2017). PEG molecules are an important part for other site-specific targeted therapeutics such as with the use of nanoparticles (NP). Surface modification of the NP with PEGs of differing chain length, shape, density, and molecular weight allow for a more advanced drug delivery system for anti-cancer therapy with superior targeting capacity and biocompatibility (Dhiman et al. 2016; Mishra and Dey 2016). PEG molecules render NPs more biocompatible and efficacious by masking undesirable properties, such as surface cationic charge, or by enhancing water solubility. The high stability and low immunogenicity of PEGylated proteins result in sustained clinical response to drugs and allows for minimal dose and less frequent administration. PEGylation of liposomes improves stability, circulation time, and targeting ability, ultimately leading to enhanced permeation retention (EPR) effect. These effects improve the therapeutic outcome and reduces the toxicity of the encapsulated and/or conjugated drug (Milla et al. 2012).
PEGylation is the first successful technology to improve the pharmacokinetic profiles of therapeutic agents and has been applied in the clinic for more than 25 years. It is also the most established half-life extension technology in the clinic which has proven to be safe and FDA approved (Oh et al. 2015; Swierczewska et al. 2015, 2016). This review examines the functionalities of protein PEGylation, its application and significance in the area of targeted drug delivery, and potential drawbacks that might inhibit their widespread use in designing site-specific drug delivery systems.
Chemistry of PEG
The process of PEGylation starts by obtaining the PEG molecules from a monomeric ethylene oxide, using ring-opening polymerization. Polymerization with water can result in bifunctional or monofunctional PEG chains. Monofunctional PEG molecules or mono-methoxylated PEGs (mPEGs), obtained by methanol initiation, are typically preferred as the starting material for protein PEGylation (Pfister and Morbidelli 2014). The results from functionalizing PEG molecules is a mixture of molecules that are mono-dispersed with a range of molecular weights and therefore, typically requires additional purification steps (Pfister and Morbidelli 2014). Crude results from first-generation, non-specific binding are a combination of isomers and polymers of different sizes and molecular weights. There is usually a combination of mono-PEGylated, di-PEGylated and fully PEGylated conjugates, each of which have slightly different properties, especially in terms of hydrophilicity and charge distribution (Pfister and Morbidelli 2014). Since PEGylation is typically an additional step post-synthetic to protein, many methods have been tried to achieve the best results in the most economically viable route (Pfister and Morbidelli 2014).
Chemically describing, with a general structure of HO-(CH2CH2O) n-CH2CH2-OH, preparation of polyethylene glycol theoretically can be achieved through the reaction of ethylene oxide with water, ethylene glycol or ethylene glycol oligomers. The reaction could follow either anionic or cationic ring opening polymerization route, the former being preferred to realize a low polydispersity product (Fig. 1b). As the most frequently used route, poly (ethylene glycol) is synthesized through an anionic ring-opening polymerization of ethylene oxide initiated by nucleophilic attack of a hydroxide ion on the epoxide ring (Roberts et al. 2002). Polydispersity (the ratio of weight average to number average molecular weight of a polymer, Mw/Mn) of commercially available PEG is usually <1.1 (Zalipsky 1995). Monomethoxy PEG (mPEG) (CH3O-(CH2CH2O)n-CH2CH2-OH, which is heavily used for polypeptide modification, is synthesized by anionic ring opening polymerization initiated by methoxy ions. PEGs with different end groups can be realized by using different initiator/terminating reagents. Both homo (same functional groups on both ends of PEG chain) and hetero (different functional groups on both ends of PEG) forms have been synthesized. Different functional groups at the end of the PEG chain can also be introduced by the post-polymerization approach. The common functional groups that have already been synthesized and commercially available are listed in Fig. 2.
Fig. 2.

Common end groups that are available for PEGylation of proteins and different substrates
PEG’s unchallenged success in therapeutic formulation can be attributed to the following properties of this macromolecule: (a) broad-spectrum of solubility in both organic and aqueous media (b) minimum toxicity and immunogenicity compared to polymers of equivalent molecular weight (c) non-biodegradability (d) hydrophilicity, and (e) fast clearance. In addition, the stealth behavior of PEGs to physiological surveillance mechanisms such as immune or reticuloendothelial system (RES) has also triggered the use of PEG in variety of systemic and non-systemic preparations. Historically, and in most of the cases until now, attachment of small or macromolecular therapeutics are generally realized through covalent bonding with terminal primary OH groups of PEGS. The presence of a finite number of functions end groups on linear PEGs suppresses the tendencies of the molecule to crosslink, thereby improving dispersity and homogeneity of the intended conjugate, particularly if the target substrate has multiple numbers of complementary functional groups.
Molar mass of PEG controls the bio-distribution of PEG-conjugates
As any ring-opening polymerization, molecular weight of PEG is determined by controlling the ratio of monomer to initiator. Molar mass is one of the most critical factors that governs biocompatibility, stealth behavior and ability of PEG to modulate pharmacokinetic profiles of other conjugated protein molecules. Typically, PEG of molar mass 400 Da to 50 kDa has been used in different biomedical applications, with 1–5 kDa PEGs are often being used for conjugating antibodies and nanoparticles while 20–50 kDa PEGs are intended for use in conjugation with low molecular weight drugs or highly unstable products such as oligonucleotides and siRNA (Ref/US). With larger assemblies, addition of PEG decreases the opsonization and elimination by the reticuloendothelial system, while for smaller and unstable targets, PEGylation improves systemic half-life through minimization of renal clearance. Most consumer products available for healthcare, cosmetics and households contain PEGs of different molecular weights, reflective of the broad-spectrum biocompatibility of this product. For example, PEGs of 3–5 kDa have been approved by the FDA for use in laxative preparations (Ref/US) and has been considered as a GRAS (generally recognized as safe) product for use in pharmaceutical preparations. Molar mass of a molecule, along with the molecular architecture, determines the hydrodynamic volume it’s going to occupy in aqueous environment, which controls the rate of excretion of the polymer from circulation (Grayson and Godbey 2008; Luxenhofer et al. 2008).
Synthesis of PEG is accompanied by low polydispersity
Homogeneity of a synthesized polymer is indicated by a polydispersity index (PDI), which in turn is defined by the ratio of number average and weight average molecular weights of the product. A PDI value less than 1.1 indicates a homogenous distribution of the polymer in terms of the efficiency and reproducibility of the macromolecule synthesis (Pasut et al. 2009; Pasut and Veronese 2007; Veronese et al. 2005). Obtained through anionic polymerization of ethylene oxide, PEG exhibits PDI around 1.01, thus providing excellent uniformity in terms of pharmacokinetic properties (Knop et al. 2010).
Solubility of PEG
PEG exhibits moderate to high solubility in most of the common organic solvents, as well as in water. Hence, chemical modification of PEG at its end group is facile, as well as cleaning-up of the product through an aqueous purification method (such as dialysis of gel filtration chromatography) is relatively easy. Aqueous solubility is also key to the usage of PEG in biological applications. PEG is generally attached to biologically active molecules through its terminal primary hydroxyl (OH) groups. Hence, most of the OH-group chemistry, such as esterification, etherification, and carbonate linkage formation are employed for the preparation of PEG-conjugates. Although some of these pathways yield cleaner products, in many instances, it is difficult to separate the targeted, transformed product from the reaction mixture containing the unreacted polymer and the polymer that underwent a side reaction. Excellent differential solubility of PEG in various organic solvents and water enables easy separation of products to yield a clean, final conjugate. It is also notable that, while for small molecular drug conjugates, purification of the final product is extremely critical, protein-conjugated PEGs can be separated from the unreacted reaction components on the basis of molecular weight, charge or hydrophobicity. Hydrophilicity of PEG is also critical for maintaining and enhancing water solubility of non-polar drug molecules. Such solubility enhancement is essential not only for in vivo stability of drugs, but also for increasing storage stability. Such enhancement of physical and thermal stability is connected to so-termed ‘conformational cloud’ originated from the molecular flexibility of PEG (Knop et al. 2010).
Chemical diversification of PEG structure
A vast array of chemical diversification strategy of PEG has been tested, reported and commercialized. The main objective of such diversification is to render the polymer amenable to conjugation to small and macromolecular therapeutic agents, including drugs, oligo- and polypeptides, proteins (enzymes and antibody), oligonucleotides, and biomaterials surfaces. PEG has also been used as a cross-linker in case of which bifunctional derivatives of PEG has been synthesized. The functional end group requirement of PEG is largely directed by the complementary functional group of the substrate that will be coupled to PEG. For example, in order to conjugate with lysine residue that is present in most of the proteins, carboxylic acid terminated PEG is most frequently used. Other reactive amino acids present in proteins, which are typically conjugated with PEG include lysine, cysteine, histidine, arginine, aspartic acid, glutamic acid, serine, threonine and tyrosine (Roberts et al. 2002).
For lysine, alpha and epsilon amino groups are most often conjugated with PEG. Harsh chemical conjugation techniques are generally avoided. PEG derivatives that has been synthesized earlier for conjugating to amine groups of lysine (or other amine groups) are summarized in Fig. 1. These first-generation PEG modification strategies for amine groups of amino acids, as compiled by Harris et al., include: a) PEG dichlorotriazine b) PEG-tresylate c) PEG-succinimidyl carbonate d) PEG-benzotriazole carbonate e) PEG-p-nitrophenyl carbonate f) PEG-trichorophenyl carbonate g) PEG-carbonylimidazole and h) PEG-succinimidyl succinate. These PEG chemistries use acylation mechanisms to modify amine groups of proteins. In many cases, these reactions showed limitations in terms of impurities, low molecular weight products, unstable linkages, and lack of selectivity. Acylation of amino groups by PEG, mediated by urethane linkages has also been explored, where p-nitrophenyl carbonate, trichlorophenyl carbonate, and carbonylimidazole PEG has been used for polypeptide modification. PEG succinimidyl succinate (PEG-SS) is another first-generation PEG-reagent that has been used for protein PEGylation, but exhibited issues of hydrolysis and immunogenicity.
Reductive amination has found application in protein PEGylation with the advent of more targeted PEGylation reagents, such as PEG-aldehyde. However, this molecule has later been replaced by acetal derivatives of PEG-propionaldehyde or PEG-acetaldehyde, which is in-situ converted to aldehyde hydrate by acid hydrolysis. Acetal derivatives have longer storage stability than PEG-propionaldehyde or PEG-acetaldehyde.
Active esters of PEG carboxylic acid have been the most frequently utilized reagents for PEGylation chemistry (Fig. 3). Active ester covalently conjugates with amines to form amide bonds which are stable at physiological conditions.
Fig. 3.
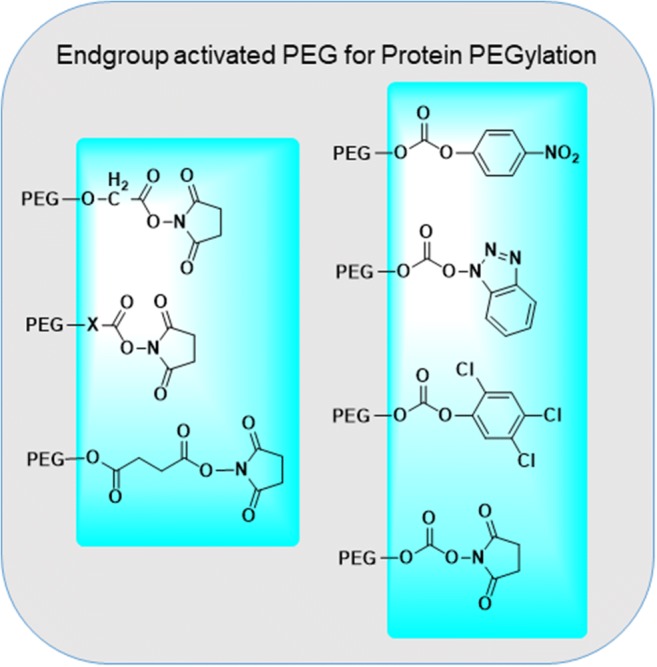
Activation of carboxylic end groups of PEGS for coupling to N-terminal of peptides. This coupling chemistry belong to the so-termed ‘first generation’ PEGylation strategy
Active esters of PEG can also be purified easily from unsubstituted or over-substituted compounds by ion-exchange chromatography. Most frequently encounters active esters of PEG carboxylic acids are N-hydroxysuccinimide (NHS) and carbodiimides. Carboxymethylated PEGs (CM-PEG) have been used as a substitute for SS-PEG to avoid premature dePEGylation of the PEGylated compounds. Succinimidyl ester of carboxylmethylated PEG is a very reactive intermediate (t1/2 of 0.75 min at pH 8 and at 25C). Distance between carboxylic acid group and PEG-backbone has a significant effect of hydrolysis of the PEGylated compound. Harris et al. synthesized propionic and butanoic acid derivatives fo PEG. It was found out that, SBA-PEG with two additional methylene units shows a longer hydrolysis half-life than SPA PEG. (23 min at pH 8 and 25 °C for PEG-SBA compared to 16.5 min for PEG-SPA under similar conditions). PEG-maleimides, vinyl sufone, iodoacetamide and orthopyridyl disulfide are representative PEG derivatives that can be used for the PEGylation of proteins via modifying the cysteine residue. Where PEG-vinyl sulfone reacts with thiol to form a stable thioether linkage, the reaction rate is slow in acidic conditions, but fast in slightly basic conditions (pH 7–8). On the other hand, PEG-maleimide (PEG-Mal) is reactive to thiol under acidic conditions. PEG-MA, as reported by Harris, is not stable in water and can undergo ring opening or addition of water across the double bond. Orthopyridyl disulfide PEG (PEG-OPSS) and PEG-iodoacetamide (PEG-IA) are also two reagents which found application in site-specific conjugation of PEGs to cysteine residue in protein (Roberts et al. 2002). While PEG-OPSS generates a disulfide bond in the product, which is susceptible to reduction, PEG-IA conjugation results in generation of iodine which may react with other amino acids. For carbohydrates, PEGylation chemistry essentially involves oxidation of the carbohydrate residue to aldehydes first. Hydrazide or amine derivatives of PEG are than reacted with these aldehyde groups to immobilize PEG on carbohydrates through hydrazone or Schiff base type linkers.
Reversible PEGylation of proteins is also possible by using a degradable linkage between the target protein and PEG. Intron® has been synthesized by Enzon to improve pharmacokinetic half-life of interferon alpha-2b (Kozlowski and Harris 2001). PEG was coupled to Nδ1 position of imidazole ring in histidine to form a carbamate linkage. Such carbamate linkages degrade over time, releasing free interferon. Pegasys®, which is the branched PEG40 kDa-interferon alpha-2a conjugate, however showed a better pharmacokinetic profile in vivo than the PEG-intron. Tag-free, cleavable PEGylation strategies have been reported on by different investigators (Yang et al. 2001). Zalipsky et al. used p- or o-disulfide of a benzyl urethane to design cleavable PEGylated proteins to recapitulate full bioactivity of the native protein upon PEG-cleavage (Zalipsky et al. 1999). Heterobifunctional PEG has also been synthesized which has found applications in drug conjugation, synthesizing targeted nanoparticles and in surface immobilization with bioactive substances.
Chemistry of PEGylation
PEGylation has evolved significantly since its first development from non-specific conjugations known as the ‘first generation PEGylation’. The variations of PEG conjugations including whether or not the PEG molecule is branched, the site of PEG attachment as well as the mass of the PEG molecule called for a “trial and error” method of PEGylation that brought to light many problems. Apart from achieving a solution of different sized PEG and protein conjugates, the resulting conjugates are also not uniform, resulting in many positional isomers. (Pfister and Morbidelli 2014; Su et al. 2017). This however, isn’t completely ineffective, as multiple drug therapies such as Pegasys, used to treat Hepatitis C, utilizes non-specific PEGylation (Pfister and Morbidelli 2014). Non-specific PEGylation typically uses amine conjugation. A subset of most frequently used PEGylation chemistry is presented in Fig. 4.
Fig. 4.

Common PEGylation of proteins using canonical amino acids
It has since evolved into a site-specific conjugation method known as ‘second generation PEGylation. The availability of more specific functions of PEG molecules, having the capability of reacting to particular moieties in the protein, should be accredited for the increase in PEGylation specificity (Bailon and Won 2009; Deiters et al. 2004; Jevsevar et al. 2010). There are multiple pathways for site-specific conjugation. N-terminal PEGylation, thiol and bridging PEGylation, histidine tags and enzymatic PEGylation (Fig. 5a) are a few of methods currently being used, based on where on the amino acid sequences the PEG molecules will attach (Pfister and Morbidelli 2014). The objective of site-specific PEGylation and main pathway is through reversible conjugation, or releasable prodrugs. (Dhiman et al. 2016). Reversible conjugation is even less inhibiting on drug activity than irreversible conjugation, used in first-generation PEGylation. Second-generation PEGylation looks to temporarily attach PEG molecules via cleavable linkages (Fig. 5b) (Dhiman et al. 2016). This way, drugs can be released according to a specified time schedule, in vivo via hydrolytic cleavage.
Fig. 5.

(a). Enzymatic ligation of PEG to glutamine residue of a protein, and (b). Attachment of cleavable PEG to a protein which is capable of releasing the active protein upon hydrolysis
The future of PEGylation looks toward third-generation PEGylation, which aims to achieve the highest potency and circulation half-life without compromising fast-acting, site specificity and lower dosages (Swierczewska et al. 2015, 2016). This is through noncovalent PEGylation based on electrostatic linkages (Pfister and Morbidelli 2014). One method that is being explored is through pre-targeting of PEG engagers. Used with effective results in targeting Epidermal Growth Factor Receptors (EGFRs) in Triple Negative Breast Cancer, PEG engager pre-targeting was used to minimize the regulatory challenges of lowered shielding and reduced uptake (Qian et al. 2013). This could be very useful for cancer therapeutics because it utilizes a targeting molecule and separate linker molecule. Based on the linker molecule, the pro-drug can enter tumor cells via endocytosis which can be receptor-mediated or not, based on the type of cancer cell and pro-drug (Dhiman et al. 2016). Site-specific ligation of PEG to amino acids is an exclusive strategy (Nischan and Hackenberger 2014) that can result in PEGylation of proteins bearing either cysteine, or non-canonical amino acids (Kim et al. 2013; Lang and Chin 2014). Michael type thiol-maleimide reactions (Fig. 6a) and azide-alkyne click cycloaddition (Deiters et al. 2004) (Fig. 6b, top panel) have gained extensive traction in realizing such PEGylation strategies. To avoid introduction and removal challenges of copper ions, metal free chemoselective reactions have been introduced for site-specific ligation of PEGs to azide containing proteins. Known as ‘Strain promoted azide alkyne cycloaddition (SPAAC), developed in the laboratories of Bertozzi, Boons and in van Delfts laboratories separately, this process relies on the ring strain of cyclooctyne derivative of PEG (Fig. 6b, bottom panel).
Fig. 6.

(a). Thiol reactive PEGs for site-specific PEGylation of proteins containing cysteine residue. (b). CuAAC PEGylation of human superoxide dismutase1, and (B) SPAAC PEGylation of lipase CalB
As an illustrative example of such site specific ligations, Candida antartica lipase B (CalB) was expressed in an auxotrophic strain of E.Coli. The enzyme contained five (05) azido homo alanines, one of which was selectively exposed by solvent. A dibenzocyclooctyne PEG derivative of 2 kDa molecular weight, was able to connect itself to the azido-CalB in only 3 h in PBS buffer at room tempeature (Codelli et al. 2008; Ning et al. 2008; van Geel et al. 2012). This type of chemoselective reaction will open an exclusive opportunity to PEGylate wide variety of therapeutic proteins which contains the non-canonical amino acids located in non-functional domain.
Additionally, the capacity of PEG to neutralize the cytotoxicity of a therapy is critical for improving its potency in treating diseases. As an instance, mitochondrial oxidative damage can be treated by the protecting the mitochondria with mitochondriotropic antioxidants. However, this therapeutic approach has raised speculation regarding its cytotoxicity (Fernandes et al. 2018). PEGylation was used to moderate the toxicity of AntiOxCIN6 which is a mitochondriotropic antioxidant with cytotoxic properties particularly to hepatocarcinoma (HepG2) (Fernandes et al. 2018). This modification resulted in maintenance of the antioxidant abilities related to CAF in the CAF - PEG - TPP conjugate (known as CPTPP) and that the PEGylation reverted the loss of the ability to chelate iron associated with just AntiOxCIN6 (Fernandes et al. 2018). In addition, it was observed that CPTPP was not toxic to human HepG2 cells whereas both AntiOxCIN6 and CAF, when alone, acted in a harmful way to the same cell lines. The successful modulation of the AntiOxCIN6 cytotoxicity along with the maintained oxygen consumption of mitochondria and efficacy of the antioxidant during the PEGylation demonstrated the ability of PEG to act as a method that not only increases the half-life of a drug and retains its efficacy, but also one that can reduce the cytotoxicity of a treatment.
Functional significance of PEGylation
Protein therapeutics and protein engineering are one of the newest and most promising biologic drug therapy routes. Due to their high specificity and rapid onset, they are preferred to synthetic therapeutics. (Bailon and Won 2009; Pfister and Morbidelli 2014) Currently, these biologic drugs cannot be used to their full potential because of short half-lives, protein degradation and other interfering pharmacokinetic (PK) properties of these therapies (Swierczewska et al. 2015; Veronese et al. 2005). With protein PEGylation, these biomolecules can have an increased half-life in the body and be protected from rapid renal filtration via the kidneys (Bailon and Won 2009). PEGylation can bestow a number of noteworthy and distinct pharmacological advantages over the unmodified form of the biomolecule such as, improved drug solubility, reduced dosage frequency, toxicity and rate of kidney clearance, an extended circulating life, increased drug stability, enhanced protection from proteolytic degradation, decreased immunogenicity and antigenicity, and minimal loss of biological activity (Chen et al. 2015). This can all be done without a loss of function, making it an attractive for further drug therapies, and especially cancer therapies.
One of the most common anti-cancer therapeutics is the use of monoclonal antibodies 1(Dhiman et al. 2016). These antibodies attach to specific sites on tumor cells that can activate apoptosis, or cell death, or block cell growth pathways. However, since they don’t typically have the Fc region that binds to cell surface receptors and is linked to antibody solubility and stability (Swierczewska et al. 2015), they’re in vivo half life is short. Through PEGylation, antibody glomerular filtration and immunogenicity has been reduced, all the while, maintaining binding affinity for the receptors (Dhiman et al. 2016). The attachment of PEG to angiogenesis inhibitor (CDP791) increased its efficacy and has been shown in clinical studies of colorectal, ovarian and renal cancers (Dhiman et al. 2016).
PEGylation has been utilized in drug therapies that have been FDA- approved and had a proven track record of success. The first marketed drug that used PEGylation appeared in 1990, called Adagen, used to treat adenosine deaminase (ADA) deficiencies in severe combined immunodeficiency disease (SCID) (Pfister and Morbidelli 2014). Since then, over 10 PEGylated drug therapies have been FDA-approved and on the market, while more than 20 are currently undergoing clinical trials (Swierczewska et al. 2015). These longer-acting solutions have made for a less frequent need to apply the drug and a lower dosage as well.
Why PEGylation is important for protein therapy
Over the last thirty years, protein therapy has emerged as a major pharmaceutical method for treating diseases. When conjugated with PEG (resulting in PEGylation), proteins are granted new capabilities that improve their clinical potential. For example, PEGylation delays proteolytic degradation and glomerular filtration through the kidneys in vivo (Bailon and Won 2009). In addition, the most successful protein conjugates have been with PEG (Carter 2011). This is due in part to the increased half-life of the serum as a result of increased hydrodynamic volume. An obstacle that is occasionally faced when dealing with protein therapy is the immunogenicity, or the ability to provoke an immune response, of the protein. The immunogenicity of a protein can make a drug less effective, so PEGylation overcomes this problem by reducing the immunogenicity of proteins (although not all PEGylated proteins do this) (Jevsevar et al. 2010; Pasut et al. 2008). PEGylation has the capability of altering the physicochemical properties in the parent protein, including electrostatic and hydrophobic properties (Nucci et al. 1991). PEGylation allows for prolonged circulation time of conjugated therapeutics in the body through a decreased rate of kidney clearance and reduction of proteolysis and opsonization. The high hydration capacity in PEG molecules helps increase the hydrodynamic radius of the conjugate approximately 5–10 folds higher than if it would be from the molecular weight alone. The increased radius then allows for lesser chances of glomerular filtration and aids its solubility (Pisal et al. 2010). PEGylation has been chosen as the method of choice in extending protein half-life after many years of intense studies. It has been selected due to its extraordinary flexibility, hydrophilicity, variably size, and low toxicity. As of now, there are over 10 different kinds of PEGylated products approved by the FDA, with many others in developmental stages (Dozier and Distefano 2015).
Pitfalls of PEGylation
PEGylation has come a long way since it first arrived in the market 30 years ago. It has undergone procedural changes from first-generation PEGylation to second-generation PEGylation and now, there are attempts to increase the efficacy with third-generation PEGylation. Although becoming one of the most widely used drug delivery technologies, PEGylation has had structural challenges. The size and position of PEG molecules on the conjugates strongly affects its properties. PEG dispersity index, degree of PEGylation, and PEGylation site specificity are some of the key problems with PEGylation that need attention.
Cyclic dimer of ethylene oxide, 1,4-dioxane, is the major side product associated with the synthesis of PEG. International Agency for Research on Cancer (IARC) categorizes dioxane as being possibly carcinogenic in humans based on data generated from animal models. 1,4-dioxane is generally stripped off from the final product under low pressure. Side products also include residual ethylene oxide and formaldehyde, both of which are classified by IARC in group 1 (carcinogen in humans). Hence, using pharmaceutical grade PEG for biomedical applications is always recommended (Knop et al. 2010). In earlier synthetic processes, PEG synthesis was also accompanied by the presence of PEG diols, which in some cases exceeded 15% of the composition of mPEG. Presence of PEG diols has been addressed by Harris et al., by using benzyloxy-PEG with diol impurity, followed by exhaustive methylation and deprotection of benzyl group. In this technique, inert dimethyl ether derivative of PEGs can be removed after polypeptide attachment. Ion-exchange chromatography has also been employed to remove PEG-diols after converting PEG to PEG carboxylic acid (Roberts et al. 2002).
There have been multiple studies to confront these issues. One study reported that PEGylation sites could be found through the comparison of native and PEGylated proteins, but the method was not sufficient in locating PEGylation sites to a certain residue (Qian et al. 2013). In addition, although the PEG chain can be identified in smaller peptides, the PEG site may be impossible to detect with larger peptides. The problem lies in the difficulty of isolating and purifying parts of the enzyme digestion and also in the obstruction of the specific cleavage by proteolytic enzymes (Veronese 2001). Another obstacle researchers have faced is the polydispersity of PEG, which ranges in value from 1.01 for low molecular weight (3–5 kDa) up to 1.2 for high molecular weight (20 kDa) (Veronese 2001). This is an undesirable characteristic due to its similarity in dispersity to the PEG conjugates.
In addition to these problems, it has also been found that the same process that prevents proteolytic enzymes from advancing towards the PEGylated protein can also refuse a substrate from the active site of the protein (Veronese 2001). This scenario is primarily observed in enzymes with greater molecular weight (polysaccharides, peptides, proteins) and greatly reduces the advantages of PEG conjugation. In order to counter this complication, researchers have developed a series of methods such as using an active-site protecting agent or an inhibitor. Although the problem was reduced through the use of an active-site protecting agent, the possibility of PEGylation still occurring in the area around the protected site renders the results insufficient. In response to this result, a process, based on the use of an inhibitor linked with an insoluble resin (agarose) was devised (Veronese 2001). This method, when held at proper pH and ionic fortitude, protects both the active site and the area surrounding it. The enzyme, after the removal of the inhibitor, continued to reflect biological activity towards substrates such as albumin and blood clots (with urokinase).
PEGylated therapies have also exhibited multiple side-effects on patients. PEGylated drugs, being small, can enter the vasculature easily, not only of the tumor, but also normal body tissues. HFS (Hand and foot syndrome), mucositis, and rash were common side-effects observed.
PEGylation of CCN-proteins for future cancer therapies
Cells are uniquely organized their daily life to survive, differentiate and grow. Cells must take inventory of their surrounding supplies (signaling molecules, growth factors etc) for the above functions and make the decision of cells’s next step would be. Interestingly, Secretory proteins of CCN family are capable of performing all of the above, and malfunctions of these proteins result carcinogenic growth. For example, over expressions of CCN1(CYR61) and CCN2 (CTGF) play oncogenic roles while CCN5 (WISP-2) upregulation plays a tumor suppressor role (Banerjee et al. 2008, 2016; Dhar et al. 2008; Haque et al. 2011a, b; Holloway et al. 2005; Jiang et al. 2004; Johnson et al. 2014; Leask 2011, 2013; O'Kelly and Koeffler 2005). Following discoveries of these proteins, scientists further found up and downstream signaling molecules that are directly or indirectly associated with tumorigenesis, cancer stemness and drug resistance. Therefore. we believe that antibody therapy of CCN1 or CCN2 and making the CCN5 protein a therapeutic drug through PEGylation (Fig. 7) would be an ideal strategy for cancer therapy.
Fig. 7.
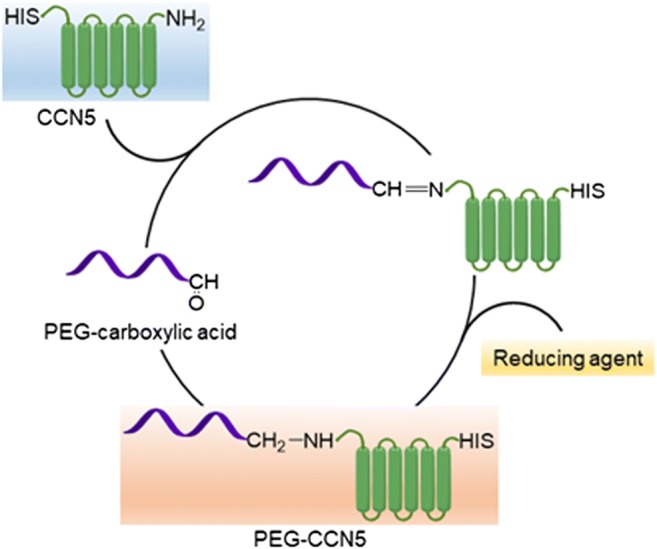
Reductive amination of CCN5 using mPEG propionaldehyde of different molecular weight resulted in PEGylated CCN5, which extended the biological half-life of the protein
Conclusions
PEGylation, as a process, has evolved rapidly through research over the past three decades. Its diverse functionality grants it a prestigious standing in the world of site-specific targeting therapeutics. Currently, there are around 10 FDA approved PEGylated drugs on the market that treat a variety of diseases ranging from Hepatitis C to Multiple Sclerosis. The enhancements that these drugs grant proteins, such as extended half-life, reduced/modulated protein cytotoxicity, maintained protein efficacy, and binding affinity to cell receptors are just the current advantages, and future research looks to delve deeper into the benefits PEGylation can provide. The third generation of PEGylation will attempt to provide the greatest potency and circulating half-life without compromising site-specificity and low dosage. If completed successfully, with the help of further research PEGylation has the potential to become the primary method for protein therapy.
Acknowledgements
We thank the members of Kansas City VA Research Office and Midwest Biomedical Research Foundation Administrative and clerical supports.
Abbreviations
- PEG
Polyethylene glycol
- NP
Nanoparticle
- MW
Molecular weight
Author’s contribution
Conception and design: S. K. Banerjee, S. Banerjee, M. Quadir, and V. Gupta, A. Ghosh; Writing and review of the manuscript: V. Gupta, M. Quadir, S. Bhavanasi, G. Ghosh, T. Siahaan, and S.K. Banerjee; Administrative, technical or material support: M. Quadir and S. K. Banerjee, and Study supervision: S. K. Banerjee.
Funding
The work is supported by Merit review grant from Department of Veterans Affairs (Sushanta K. Banerjee, 5I01BX001989–04 and Snigdha Banerjee, I01BX001002–05), KUMC Lied Basic Science Grant Program (SKB), and Grace Hortense Greenley Trust, directed by The Research Foundation in memory of Eva Lee Caldwell (SKB). This work is partially supported by NIH grant P20 GM109024 from the National Institute of General Medical Science (NIGMS) (MQ), NSF under Grant No. 092354 (MQ), NIH Grant Number 2P20 RR015566 from the National Center for Research Resources (MQ), NIH grant 1R01 GM 114080 (NIGMS) and NSF Grant No. IIA-1355466 from North Dakota Established Program to Stimulate Comperative Research (EPSCoR) through the Center for Sustanable Materials Science (MQ).
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Compliance with ethical standards
Ethics approval and consent to participate
Compliance with ethical standard of VA Medical Center.
Consent for publication
All the authors of this manuscript have agreed to publish this article.
Competing interests
No potential conflicts of interest were disclosed.
Footnotes
Sneha Bhavanasi, Kevin Singh, Gaurav Ghosh and Kritin Vasamreddy Summer students
Vijayalaxmi Gupta and Sneha Bhavanasi contributed equally to this work.
Contributor Information
Mohiuddin Quadir, Email: mohiuddin.quadir@ndsu.edu.
Snigdha Banerjee, Email: sbanerjee@kumc.edu.
Sushanta K. Banerjee, Phone: (816) 861-4700, Email: sbanerjee@kumc.edu, Email: cancerresearchunit@icloud.com
References
- Abuchowski A, Kazo GM, Verhoest CR, Jr, et al. Cancer therapy with chemically modified enzymes. I. Antitumor properties of polyethylene glycol-asparaginase conjugates. Cancer Biochem Biophys. 1984;7:175–186. [PubMed] [Google Scholar]
- Bailon P, Won CY. PEG-modified biopharmaceuticals. Expert Opin Drug Deliv. 2009;6:1–16. doi: 10.1517/17425240802650568. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Dhar G, Haque I, et al. CCN5/WISP-2 expression in breast adenocarcinoma is associated with less frequent progression of the disease and suppresses the invasive phenotypes of tumor cells. Cancer Res. 2008;68:7606–7612. doi: 10.1158/0008-5472.CAN-08-1461. [DOI] [PubMed] [Google Scholar]
- Banerjee SK, Maity G, Haque I, et al. Human pancreatic cancer progression: an anarchy among CCN-siblings. Journal of cell communication and signaling. 2016;10:207–216. doi: 10.1007/s12079-016-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2002;24:703–729. [PubMed] [Google Scholar]
- Booth C, Gaspar HB. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID) Biologics. 2009;3:349–358. [PMC free article] [PubMed] [Google Scholar]
- Carter PJ. Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res. 2011;317:1261–1269. doi: 10.1016/j.yexcr.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Chen X, Hu C, Zhang W, et al. Metformin inhibits the proliferation, metastasis, and cancer stem-like sphere formation in osteosarcoma MG63 cells in vitro. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:9873–9883. doi: 10.1007/s13277-015-3751-1. [DOI] [PubMed] [Google Scholar]
- Choy EH, Hazleman B, Smith M, et al. Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: a phase II double-blinded, randomized, dose-escalating trial. Rheumatology. 2002;41:1133–1137. doi: 10.1093/rheumatology/41.10.1133. [DOI] [PubMed] [Google Scholar]
- Codelli JA, Baskin JM, Agard NJ, et al. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiters A, Cropp T, Summerer D, et al. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg Med Chem Lett. 2004;14:5743–5745. doi: 10.1016/j.bmcl.2004.09.059. [DOI] [PubMed] [Google Scholar]
- Dhar G, Banerjee S, Dhar K, et al. Gain of oncogenic function of p53 mutants induces invasive phenotypes in human breast cancer cells by silencing CCN5/WISP-2. Cancer Res. 2008;68:4580–4587. doi: 10.1158/0008-5472.CAN-08-0316. [DOI] [PubMed] [Google Scholar]
- Dhiman S, Mishra N, Sharma S. Development of PEGylated solid lipid nanoparticles of pentoxifylline for their beneficial pharmacological potential in pathological cardiac hypertrophy. Artif Cells Nanomed Biotechnol. 2016;44:1901–1908. doi: 10.3109/21691401.2015.1111234. [DOI] [PubMed] [Google Scholar]
- Dozier JK, Distefano MD. Site-specific PEGylation of therapeutic proteins. Int J Mol Sci. 2015;16:25831–25864. doi: 10.3390/ijms161025831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes C, Benfeito S, Amorim R, et al. Desrisking the cytotoxicity of a Mitochondriotropic antioxidant based on Caffeic acid by a PEGylated strategy. Bioconjug Chem. 2018;29:2723–2733. doi: 10.1021/acs.bioconjchem.8b00383. [DOI] [PubMed] [Google Scholar]
- Grayson SM, Godbey WT. The role of macromolecular architecture in passively targeted polymeric carriers for drug and gene delivery. J Drug Target. 2008;16:329–356. doi: 10.1080/10611860801969616. [DOI] [PubMed] [Google Scholar]
- Haque I, Banerjee S, Mehta S, et al. Cysteine-rich 61-connective tissue growth factor-nephroblastoma-overexpressed 5 (CCN5)/Wnt-1-induced signaling protein-2 (WISP-2) regulates microRNA-10b via hypoxia-inducible factor-1alpha-TWIST signaling networks in human breast cancer cells. JBiolChem. 2011;286:43475–43485. doi: 10.1074/jbc.M111.284158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque I, Mehta S, Majumder M, et al. Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol Cancer. 2011;10:8. doi: 10.1186/1476-4598-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershfield MS, Ganson NJ, Kelly SJ, et al. Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res Ther. 2014;16:R63. doi: 10.1186/ar4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway SE, Beck AW, Girard L, et al. Increased expression of Cyr61 (CCN1) identified in peritoneal metastases from human pancreatic cancer. J Am Coll Surg. 2005;200:371–377. doi: 10.1016/j.jamcollsurg.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Jevsevar S, Kunstelj M, Porekar VG. PEGylation of therapeutic proteins. Biotechnol J. 2010;5:113–128. doi: 10.1002/biot.200900218. [DOI] [PubMed] [Google Scholar]
- Jiang WG, Watkins G, Fodstad O, et al. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr Relat Cancer. 2004;11:781–791. doi: 10.1677/erc.1.00825. [DOI] [PubMed] [Google Scholar]
- Johnson SK, Stewart JP, Bam R, et al. CYR61/CCN1 overexpression in the myeloma microenvironment is associated with superior survival and reduced bone disease. Blood. 2014;124:2051–2060. doi: 10.1182/blood-2014-02-555813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston E, Crawford J, Blackwell S, et al. Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol. 2000;18:2522–2528. doi: 10.1200/JCO.2000.18.13.2522. [DOI] [PubMed] [Google Scholar]
- Kim C, Axup J, Schultz P. Protein conjugation with genetically encoded unnatural amino acids. Curr Opin Chem Biol. 2013;17:412–419. doi: 10.1016/j.cbpa.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop K, Hoogenboom R, Fischer D, et al. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- Kozlowski A, Harris JM. Improvements in protein PEGylation: pegylated interferons for treatment of hepatitis C. Journal of controlled release : official journal of the Controlled Release Society. 2001;72:217–224. doi: 10.1016/S0168-3659(01)00277-2. [DOI] [PubMed] [Google Scholar]
- Lang K, Chin J. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev. 2014;114:4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Leask A. CCN1: a novel target for pancreatic cancer. Journal of cell communication and signaling. 2011;5:123–124. doi: 10.1007/s12079-011-0127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A. Sonic advance: CCN1 regulates sonic hedgehog in pancreatic cancer. Journal of cell communication and signaling. 2013;7:61–62. doi: 10.1007/s12079-012-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenhofer R, Bezen M, Jotdan R. Kinetic investigations on the polymerization of 2-Oxazolines using Pluritriflate Initators. Macromolecular Rapid Communication. 2008;29:1509–1513. doi: 10.1002/marc.200800293. [DOI] [Google Scholar]
- Macdougall IC. CERA (continuous erythropoietin receptor activator): a new erythropoiesis-stimulating agent for the treatment of anemia. Curr Hematol Rep. 2005;4:436–440. [PubMed] [Google Scholar]
- Macdougall IC, Roche A. Administration of intravenous iron sucrose as a 2-minute push to CKD patients: a prospective evaluation of 2,297 injections. Am J Kidney Dis. 2005;46:283–289. doi: 10.1053/j.ajkd.2005.04.032. [DOI] [PubMed] [Google Scholar]
- Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13:105–119. doi: 10.2174/138920012798356934. [DOI] [PubMed] [Google Scholar]
- Mishra, P.N., B.; Dey, R.K. (2016). PEGylation in anti-cancer therapy: an overview. Asian Journal of Pharmaceutical Sciences 11, 337–348
- Ning X, Guo J, Wolfert M, et al. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angewandte Chemie-International Edition. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nischan N, Hackenberger CP. Site-specific PEGylation of proteins: recent developments. J Org Chem. 2014;79:10727–10733. doi: 10.1021/jo502136n. [DOI] [PubMed] [Google Scholar]
- Nucci R, Raia CA, Vaccaro C, et al. Allosteric modifier and substrate binding of donkey deoxycytidylate aminohydrolase (EC 3.5.4.12) Arch Biochem Biophys. 1991;289:19–25. doi: 10.1016/0003-9861(91)90436-M. [DOI] [PubMed] [Google Scholar]
- O'Kelly JK, Koeffler HP. The role of CCN1 in tumorigenesis and cancer progression. In: Perbal BT, editor. In CCN proteins: anew family of cell growth and differentiation regulators. London: Imperial College Press; 2005. pp. 273–291. [Google Scholar]
- Oelmeier SA, Dismer F, Hubbuch J. Molecular dynamics simulations on aqueous two-phase systems - single PEG-molecules in solution. BMC Biophys. 2012;5:14. doi: 10.1186/2046-1682-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Swierczewska M, Kim TH, et al. Delivery of tumor-homing TRAIL sensitizer with long-acting TRAIL as a therapy for TRAIL-resistant tumors. Journal of controlled release : official journal of the Controlled Release Society. 2015;220:671–681. doi: 10.1016/j.jconrel.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasut G, Canal F, Dalla Via L, et al. Antitumoral activity of PEG-gemcitabine prodrugs targeted by folic acid. Journal of controlled release : official journal of the Controlled Release Society. 2008;127:239–248. doi: 10.1016/j.jconrel.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Pasut G, Greco F, Mero A, et al. Polymer-drug conjugates for combination anticancer therapy: investigating the mechanism of action. J Med Chem. 2009;52:6499–6502. doi: 10.1021/jm900804m. [DOI] [PubMed] [Google Scholar]
- Pasut G, Veronese F. Polymer-drug conjugation, recent achievements and general strategies. Prog Polym Sci. 2007;32:933–961. doi: 10.1016/j.progpolymsci.2007.05.008. [DOI] [Google Scholar]
- Pepinsky RB, LePage DJ, Gill A, et al. Improved pharmacokinetic properties of a polyethylene glycol-modified form of interferon-beta-1a with preserved in vitro bioactivity. J Pharmacol Exp Ther. 2001;297:1059–1066. [PubMed] [Google Scholar]
- Pfister D, Morbidelli M. Process for protein PEGylation. Journal of controlled release : official journal of the Controlled Release Society. 2014;180:134–149. doi: 10.1016/j.jconrel.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J Pharm Sci. 2010;99:2557–2575. doi: 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Dong H, Tian H, et al. Characterization of a site-specific PEGylated analog of exendin-4 and determination of the PEGylation site. Int J Pharm. 2013;454:553–558. doi: 10.1016/j.ijpharm.2013.06.059. [DOI] [PubMed] [Google Scholar]
- Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459–476. doi: 10.1016/S0169-409X(02)00022-4. [DOI] [PubMed] [Google Scholar]
- Su YC, Burnouf PA, Chuang KH, et al. Conditional internalization of PEGylated nanomedicines by PEG engagers for triple negative breast cancer therapy. Nat Commun. 2017;8:15507. doi: 10.1038/ncomms15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swierczewska M, Han HS, Kim K, et al. Polysaccharide-based nanoparticles for theranostic nanomedicine. Adv Drug Deliv Rev. 2016;99:70–84. doi: 10.1016/j.addr.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swierczewska M, Lee KC, Lee S. What is the future of PEGylated therapies? Expert Opin Emerg Drugs. 2015;20:531–536. doi: 10.1517/14728214.2015.1113254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorner MO. The discovery of growth hormone-releasing hormone. J Clin Endocrinol Metab. 1999;84:4671–4676. doi: 10.1210/jcem.84.12.6210. [DOI] [PubMed] [Google Scholar]
- Thorner MO, Strasburger CJ, Wu Z, et al. Growth hormone (GH) receptor blockade with a PEG-modified GH (B2036-PEG) lowers serum insulin-like growth factor-I but does not acutely stimulate serum GH. J Clin Endocrinol Metab. 1999;84:2098–2103. doi: 10.1210/jcem.84.6.5732. [DOI] [PubMed] [Google Scholar]
- Torchilin VP. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007;9:E128–E147. doi: 10.1208/aapsj0902015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geel R, Pruijn G, van Delft F, et al. Preventing thiol-Yne addition improves the specificity of strain-promoted Azide-alkyne cycloaddition. Bioconjug Chem. 2012;23:392–398. doi: 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- Veronese FM. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials. 2001;22:405–417. doi: 10.1016/S0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- Veronese FM, Sacca B, Polverino de Laureto P, et al. New PEGs for peptide and protein modification, suitable for identification of the PEGylation site. Bioconjug Chem. 2001;12:62–70. doi: 10.1021/bc000061m. [DOI] [PubMed] [Google Scholar]
- Veronese FM, Schiavon O, Pasut G, et al. PEG-doxorubicin conjugates: influence of polymer structure on drug release, in vitro cytotoxicity, biodistribution, and antitumor activity. Bioconjug Chem. 2005;16:775–784. doi: 10.1021/bc040241m. [DOI] [PubMed] [Google Scholar]
- Wang YS, Youngster S, Bausch J, et al. Identification of the major positional isomer of pegylated interferon alpha-2b. Biochemistry. 2000;39:10634–10640. doi: 10.1021/bi000617t. [DOI] [PubMed] [Google Scholar]
- Yang JB, Duan ZJ, Yao W, et al. Synergistic transcriptional activation of human acyl-coenzyme a: cholesterol acyltransterase-1 gene by interferon-gamma and all-trans-retinoic acid THP-1 cells. J Biol Chem. 2001;276:20989–20998. doi: 10.1074/jbc.M011488200. [DOI] [PubMed] [Google Scholar]
- Zalipsky S. Functionalized poly(ethylene glycol) for preparation of biologically relevant conjugates. Bioconjug Chem. 1995;6:150–165. doi: 10.1021/bc00032a002. [DOI] [PubMed] [Google Scholar]
- Zalipsky S, Qazen M, Walker JA, 2nd, et al. New detachable poly(ethylene glycol) conjugates: cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjug Chem. 1999;10:703–707. doi: 10.1021/bc990031n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.