

Abstract
Background
Geleophysic dysplasia (GPHYSD) is a disorder characterized by dysmorphic features, stiff joints and cardiac involvement due to defects of TGF‐β signaling. GPHYSD can be caused by mutations in FBN1, ADAMTLS2, and LTBP3 genes.
Methods and Results
Consistent with previous reports, we found intracellular inclusions of unknown material by electron microscopy (EM) in skin fibroblasts of two GPHYSD individuals carrying FBN1 mutations. Moreover, we found that the storage material is enclosed within lysosomes and is associated with the upregulation of several lysosomal genes. Treatment of GPHYSD fibroblasts carrying FBN1 mutations with the angiotensin II receptor type 1 inhibitor losartan that inhibits TGF‐β signaling did not reduce the storage but improved the extracellular deposition of fibrillin‐1 microfibrils.
Conclusion
Losartan is a promising candidate drug for treatment of GPHYSD due to FBN1 defects.
Keywords: extracellular matrix, geleophysic dysplasia, intracytoplasmic inclusions, losartan
1. INTRODUCTION
Geleophysic dysplasia (GPHYSD1, MIM231050; GPHYSD2, MIM614185; GPHYSD3, MIM617809) is a connective tissue disorder presenting with short stature, small hands and feet, cardiac valvular disease, hepatomegaly, joint contractures, and thickened skin (Rosser, Wilkinson, Hurst, McGaughran, & Donnai, 1995; Spranger, Gilbert, Tuffli, Rossiter, & Opitz, 1971; Vanace, Friedman, & Wagner, 1960). Although initially suspected to be a lysosomal storage disorder (Lipson, Kan, & Kozlowski, 1987; Wraith, Bankier, Chow, Danks, & Sardharwalla, 1990), GPHYSD was later recognized to be caused by mutations in genes encoding components of the extracellular matrix (ECM) including a disintegrin and metalloproteinase with thrombospondin motifs like 2 (ADAMTSL2, MIM612277) (GPHYSD1) (Le Goff et al., 2008), fibrillin 1 (FBN1, MIM134797) (GPHYSD2) (Le Goff et al., 2011), and latent transforming growth factor β (TGF‐β) binding protein 3 (LTBP3, MIM 602090) (GPHYSD3) (McInerney‐Leo et al., 2016). Transforming Growth Factor‐β (TGF‐β) is sequestered in an inactive and latent form by interactions with several ECM proteins and ordered polymers of fibrillin bind latent TGF‐β maintaining TFG‐β biologically inactive. Defects in GPHYSD genes perturb TGF‐β signaling and result in altered deposition of ECM (Le Goff et al., 2011, 2008). Mutations in FBN1 result in Marfan syndrome (MFS) (Robinson et al., 2006) but domain‐specific FBN1 mutations have been found in other connective tissue disorders including Weill‐Marchesani syndrome (Faivre et al., 2003), stiff skin syndrome (Loeys et al., 2010), isolated ectopia lentis (Lonnqvist et al., 1994), and acromicric dysplasia (Le Goff et al., 2011), in addition to GPHYSD. In GPHYSD patients, heterozygous FBN1 mutations are all located in exons 41 and 42 encoding for TGF‐β‐binding protein‐like domain 5 (TB5) (Le Goff et al., 2011).
In the present study, we report the ultrastructural observation of a storage phenotype in skin fibroblasts of GPHYSD patients carrying FBN1 mutations. Moreover, we found that the storage material is enclosed within lysosomes and is associated with upregulation of several lysosomal genes. Finally, we evaluated the efficacy of the angiotensin II receptor type 1 inhibitor losartan that inhibits TGF‐β signaling for correction of ECM deposition defect in fibroblasts of a GPHYSD patient carrying a FBN1 mutation.
2. CASE REPORTS
Clinical features of the subjects 1 and 3 from whom skin fibroblasts were obtained are summarized in Table 1. Subjects 1 and 2 are siblings and both presented with short stature and brachydactyly. Subject 1 is currently a 27‐year‐old female and she presented with short stature and pulmonic valve stenosis. She underwent a valvotomy at the age of 13 years. On physical exam at the age of 18 years, her height was 126.6 cm (below the 3rd centile) with an arm span of 116 cm. She had pleasant appearing facies, short arms and brachydactyly with a trigger left thumb. II/VI diastolic murmur was heard over the left sternal border. She delivered an affected son preterm at 33 3/7 weeks gestation. At 6 months, his corrected length was at the 10th centile but by the age of 18 months, his linear growth decelerated, and his height had fallen below the 3rd centile. His physical exam was remarkable for brachydactyly with significant shortening of the middle phalanx of II and V digits. Subject 2 is currently a 25‐year‐old male who also presented short stature and brachydactyly. At the age 10 years of his last evaluation, his height was 117.7 cm (below the 3rd centile) with an arm span 104 cm. He had short limbs, brachydactyly, and pleasant facies. Brother, sister and sister's son were cognitively normal. Both sibs were born to unaffected parents, thus suggesting gonadal mosaicism. Skin fibroblasts were not available for subject 2.
Table 1.
Summary of clinical, molecular, and ultrastructural findings
| Subject | |||
|---|---|---|---|
| 1 | 2a | 3b | |
| Age at last evaluation (years) | 10 | 13 | 1.8 |
| Gender | F | M | M |
| Ethnicity | Hispanic | Hispanic |
Hispanic/ African American |
| Clinical features | |||
| Short stature | + | + | + |
| Short neck | − | − | − |
| Laryngeal or tracheal stenosis | − | − | + |
| Chronic respiratory infections | − | − | − |
| Cardiac valve disease | + | − | + |
| Hepatomegaly | − | − | − |
| Motor delay | − | − | + |
| Skin stiffness | − | − | + |
| Small hands/feet | + | + | + |
| Joint contractures | − | − | + |
| Tiptoe gait | − | − | − |
| J‐shaped sella | + | + | + |
| Short small tubular bones | + | + | − |
| FBN1 variant |
c.5284G>A p.(Gly1762Ser) |
N.A. |
c.5117G>A p.(Cys1706Tyr) |
| EM inclusions | + | N.A. | + |
M, male; F, female; N.A., not available; EM, electron microscopy. FBN1 NCBI Reference Sequences were NC_000015.10 for the gene, NM_000138.4 for the mRNA, and NP_000129.3 for the protein.
Sibling of subject 1.
Previously described in Sule et al. (2013).
Clinical findings of subject 3 were previously reported [as subject 6 by Sule et al. (2013)]. Upon parental consent, skin biopsies were performed prior to genetic testing. Sanger sequencing of exons 41 and 42 of FBN1 identified heterozygous missense mutations which were previously reported (Le Goff et al., 2011) (Table 1).
3. MATERIALS AND METHODS
Genomic DNA was extracted from peripheral blood by standard procedures. PCR primers for FBN1 amplification were previously described (Le Goff et al., 2011, 2008, 2012). Sanger sequencing was performed by PRIMM. FBN1 NCBI Reference Sequences were NC_000015.10 for the gene, NM_000138.4 for the mRNA, and NP_000129.3 for the protein.
3.1. Cell culture studies
Fibroblast primary cultures were established from skin biopsy samples from GPHYSD, MS and MFS cases, and from healthy volunteers. Cells were cultured according to standard procedures and maintained in Dulbecco's Modified Eagle Medium (DMEM) (EuroClone) with 10% Fetal Bovine Serum (FBS) (Euroclone) and penicillin/streptomycin (Sigma‐Aldrich), in a humidified atmosphere containing 5% CO2 at 37°C. Cells were treated for 1 week (1 ng/ml) or 48 hr (10 ng/ml) with mature TGF‐β1 (Sigma‐Aldrich) or vehicle (4mM HCl‐0.1% human serum albumin). Losartan potassium salt (Sigma‐Aldrich) was dissolved into dimethyl sulfoxide (DMSO) (Sigma‐Aldrich) and used at the concentration of 200 μM for 14 days. At confluency, fibroblasts were trypsinized and resuspended in glutaraldehyde for electron microscopy (EM) analysis.
3.2. EM and Immuno‐EM
For immuno‐EM analysis of LAMP‐1 distribution in fibroblasts, cells were fixed with the mixture of 4% paraformaldehyde (PFA) and 0.05% glutaraldehyde for 10 min at room temperature, then washed with 4% PFA to remove the residual glutaraldehyde and fixed again with 4% PFA for 30 min at room temperature. Next, cells were incubated with the blocking/permeabilizing mixture (0.5% Bovine Serum Albumin, 0.1% saponin, 50 mM NH4Cl) for 30 min and subsequently with the primary monoclonal antibody against LAMP1 (Developmental Studies Hybridoma Bank; cat.# H4A30), diluted 1:500 in blocking/permeabilizing solution. On the following day, cells were washed and incubated with the secondary antibody, the anti‐rabbit Fab’ fragment coupled to 1.4‐nm gold particles (diluted 1:50 in blocking/ permeabilizing solution) for 2 hr at room temperature. Specimens were then postfixed as previously described (Polishchuk & Polishchuk, 2013). After dehydration, the specimens were embedded in epoxy resin and polymerized at 60°C for 72 hr. Thin 60 nm sections were cut at the Leica EM UC7 microtome. EM images were acquired from thin sections using a FEI Tecnai‐12 electron microscope equipped with a VELETTA CCD digital camera (FEI, Eindhoven, The Netherlands). To count the number of lysosomes within the cells, EM images were acquired at the same magnification. Thirty‐five cells (for each sample) randomly distributed on single thin section were taken in consideration. Lysosomes were manually counted on EM images.
3.3. Gene expression analyses
Total RNA was extracted from liver tissue in TRIzol reagent (Invitrogen) using RNeasy kit (Qiagen). RNA was reverse transcribed using a first‐strand complementary deoxyribonucleic acid kit with random primers according to the manufacturer's protocol (Applied Biosystems). The qRT‐PCR reactions were performed using SYBR Green Master Mix in a Roche Light Cycler 480 system (Roche). Primer sequences are available in Supplementary Table 1 in Data S1. PCR conditions were as follows: preheating, 5 min at 95°C; 40 cycles of 15 s at 95°C, 15 s at 60°C, and 25 s at 72°C. Quantification results were expressed in terms of cycle threshold (Ct). Ct values were averaged for each technical duplicate. For expression analysis, HPRT1 and B2M housekeeping genes were used as endogenous controls (reference markers) using LightCycler 480 software version 1.5. Differences between mean Ct values of tested genes and those of the reference gene were calculated as ΔCt gene = Ct gene − Ct reference. WT1 sample was used as calibrator and relative fold increase in expression levels was determined as E−ΔΔCt, E being primer efficiency.
3.4. Immunofluorescence stainings
FBN1 and COL1A1 immunofluorescence on fibroblasts were performed with anti‐FBN1 (Millipore; cat.# MAB1919) and anti‐COL1A1 (Millipore; cat.#AB758) antibodies as previously described (Piccolo et al., 2014). Confocal images were obtained using LSM710 confocal laser scanning microscope and ZEN2008 software (Carl Zeiss). Each experiment was performed in duplicate and at least five images per experiment were analyzed for each staining using ImageJ software.
3.5. Statistical analyses
Statistical significance was computed using the Student's two tail test or one sample two tail test. A p value < 0.05 was considered statistically significant.
4. RESULTS
Primary fibroblasts from skin biopsies (subjects 1 and 3) analyzed by EM showed intracellular inclusions with lamellar structure appearance, electron‐dense storage material, and large apparently empty vesicles (Figure 1a). Similar intracellular structures were also found in skin fibroblasts from three patients carrying SMAD4 mutations responsible for Myhre syndrome (MS) [reported by Piccolo et al, (Piccolo et al., 2014)], a condition with large clinical overlap to GPHYSD (Caputo et al., 2012; Le Goff et al., 2012) (Figure 1a and supplementary Figure 1 in Data S1). Immunogold with anti‐LAMP‐1 antibody on fibroblasts from subject 1 showed that the inclusions were decorated by LAMP‐1 signals, thus suggesting that they are enclosed within lysosomes (Figure 1b). Compared to wild type controls, fibroblasts from subject 1 also showed a significantly increased number of LAMP‐1–positive vesicles (Figure 1c) and upregulation of several lysosomal genes (Figure 1d). Incubation of wild type fibroblasts with TGF‐β in the medium resulting in the upregulation of TGF‐β target genes (COL1A1 and TIMP1) showed neither evidence of intracellular storage on EM analysis nor changes in lysosomal gene expression by qPCR (Supplementary Figure 2a‐b in Data S1), thus making unlikely that increased TGF‐β bioavailability is directly involved in the formation of intracellular inclusions.
Figure 1.
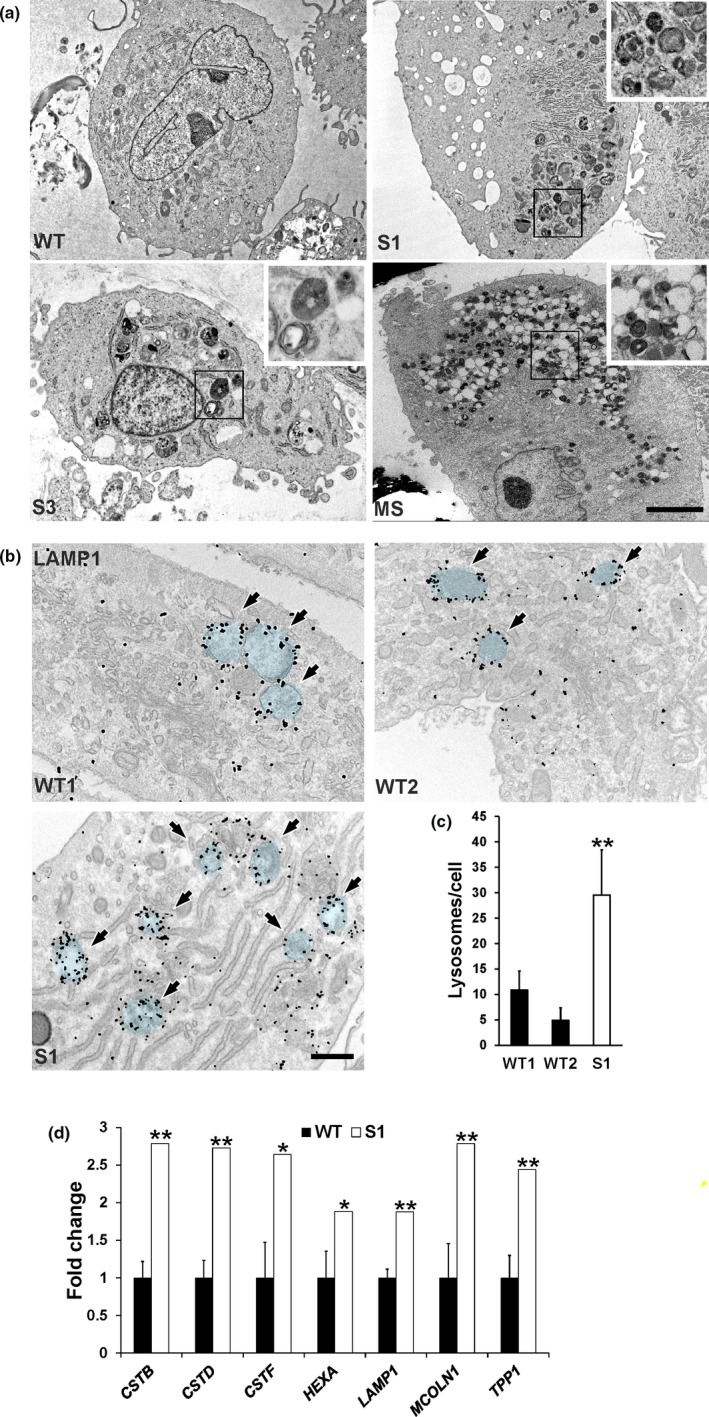
Lysosomal storage inclusions in GPHYSD fibroblasts. (a) Representative EM images of cultured skin fibroblasts from patients with GPHYSD [subject 1 (S1) and subject 3 (S3], Myhre syndrome (MS) carrying the p. (Arg496Cys) variant in SMAD4 gene showing intracytoplasmic multilamellar and electron‐dense inclusions in GPHYSD and MS fibroblasts. Cell from healthy subject (WT) is shown as control. Scale bar: 5 µm. (b) LAMP‐1 immunogold staining in subject 1 fibroblasts (S1) showing storage within LAMP‐1 decorated lysosomal vesicles highlighted in blue and indicated by arrows. Cells from two healthy subjects (WT1 and WT2) are shown as controls. Scale bar: 500 nm. (c) LAMP‐1–positive vesicle count in GPHYSD fibroblasts (subject 1, S1) compared to two WT controls (WT1 and WT2). Averages ± SEM are shown; **p < 0.005 versus WT1 and WT2; t test. (d) qPCR for lysosomal genes in GPHYSD fibroblasts from subject 1 compared to WT controls (n = 4). Averages ± SEM are shown; *p < 0.05, **p < 0.005; one sample t test
GPHYSD fibroblasts carrying FBN1 mutations have reduced and poorly organized ECM microfibrils (Le Goff et al., 2012). In a previous work, we showed that the angiotensin II type 1 receptor inhibitor losartan used to treat aortic aneurysm and prevent dissection in MFS (Brooke et al., 2008), improved microfibril deposition in MS fibroblasts (Piccolo et al., 2014). Therefore, we investigated whether losartan can improve ECM deposition also in GPHYSD carrying FBN1 mutations. Compared to vehicle‐treated controls, both GPHYSD and MFS fibroblasts incubated with losartan showed improved microfibril deposition as shown by FBN1 staining (Figure 2a,b). Losartan treatment had no effect on lysosomal inclusions evaluated by EM (Supplementary Figure 2c in Data S1).
Figure 2.
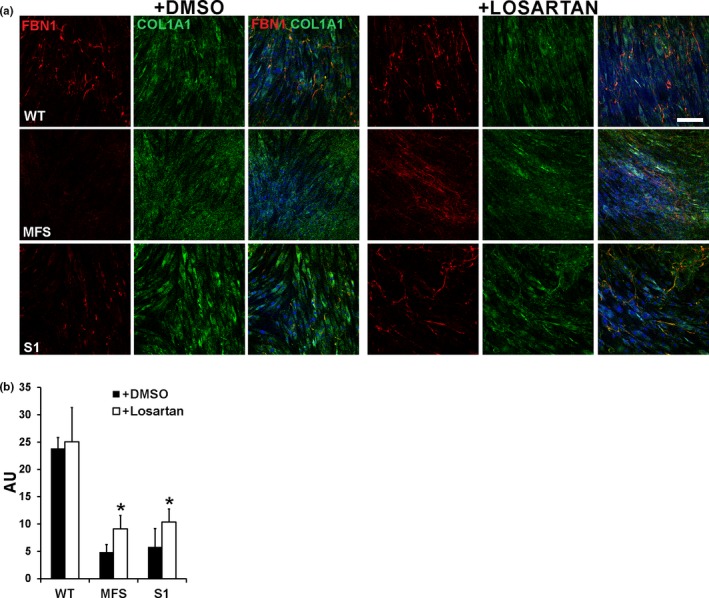
Losartan improves microfibril deposition defect in GPHYSD fibroblasts. Fibroblasts from GPHYSD and MFS patients and from healthy controls (WT) were treated for 14 days with vehicle (DMSO) or 200 μM losartan. (a) Staining for FBN1 (red) revealed improved microfibril deposition in losartan‐treated GPHYSD (subject 1, S1) and MFS fibroblasts carrying the p.Asp2104Ter variant in FBN1, compared to vehicle‐treated (DMSO) cells. COL1A1 (green) staining is shown as ECM deposition control and appears to be not affected by treatment. Nuclei were counterstained with DAPI (blue). Magnification: 20×. Scale bar: 100 μm. (b) Quantification of fluorescence intensity showing significant increase in FBN1 microfibrils deposition after losartan treatment in GPHYSD and MFS fibroblasts. Ratio between FBN1 fluorescence intensity and fluorescence area is expressed as arbitrary units (AU). Averages ± SEM are shown; *p < 0.05; t test
5. DISCUSSION
GPHYSD is a connective tissue disorder of an expanding group of diseases caused by defects of TGF‐β signaling (Doyle, Gerber, & Dietz, 2012). Because intracellular inclusion bodies were previously detected in cells of affected patients, GPHYSD was suspected to be a lysosomal storage disorder (Lipson et al., 1987; Rosser et al., 1995). Here, we confirmed that storage material accumulates within lysosomes in GPHYSD skin fibroblasts and is associated with increased expression of lysosomal genes. Interestingly, we also found that similar inclusions are detected in MS fibroblasts. Nevertheless, as previously shown (Hubmacher, Wang, Mecham, Reinhardt, & Apte, 2015), there is no direct relationship between increased TGF‐β bioavailability and generation of inclusions. Therefore, significance of such inclusions in the pathogenesis of the disease remains unclear.
In the present study, we confirmed that GPHYSD fibroblasts carrying FBN1 mutations exhibit few and shortened microfibrils that lack a detectable network organization (Le Goff et al., 2011) with a pattern similar to MS fibroblasts (Piccolo et al., 2014). Interestingly, losartan significantly improved microfibril deposition in GPHYSD fibroblasts, like in MFS and MS fibroblasts (Piccolo et al., 2014). Therefore, losartan has potential for treatment of the ECM defect in GPHYSD due to FBN1 mutations in addition to MS. However, the disease manifestations which can be improved by losartan need to be addressed in preclinical mouse studies and clinical trials. Nevertheless, being an approved and well‐tolerated drug, losartan holds premise for therapy of GPHYSD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by Fondazione Telethon Italy (TCBP37TELC and TCBMT3TELD) to N.B.‐P.
Piccolo P, Sabatino V, Mithbaokar P, et al. Skin fibroblasts of patients with geleophysic dysplasia due to FBN1 mutations have lysosomal inclusions and losartan improves their microfibril deposition defect. Mol Genet Genomic Med. 2019;7:e844 10.1002/mgg3.844
REFERENCES
- Brooke, B. S. , Habashi, J. P. , Judge, D. P. , Patel, N. , Loeys, B. , & Dietz, H. C. III (2008). Angiotensin II blockade and aortic‐root dilation in Marfan's syndrome. New England Journal of Medicine, 358(26), 2787–2795. 1056/NEJMoa0706585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo, V. , Cianetti, L. , Niceta, M. , Carta, C. , Ciolfi, A. , Bocchinfuso, G. , … Tartaglia, M. (2012). A restricted spectrum of mutations in the SMAD4 tumor‐suppressor gene underlies Myhre syndrome. American Journal of Human Genetics, 90(1), 161–169. 10.1016/j.ajhg.2011.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, J. J. , Gerber, E. E. , & Dietz, H. C. (2012). Matrix‐dependent perturbation of TGFbeta signaling and disease. FEBS Letters, 586(14), 2003–2015. 10.1016/j.febslet.2012.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre, L. , Gorlin, R. J. , Wirtz, M. K. , Godfrey, M. , Dagoneau, N. , Samples, J. R. , … Cormier‐Daire, V. (2003). In frame fibrillin‐1 gene deletion in autosomal dominant Weill‐Marchesani syndrome. Journal of Medical Genetics, 40(1), 34–36. 10.1136/jmg.40.1.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher, D. , Wang, L. W. , Mecham, R. P. , Reinhardt, D. P. , & Apte, S. S. (2015). Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia–a novel mouse model providing insights into geleophysic dysplasia. Disease Models & Mechanisms, 8(5), 487–499. 10.1242/dmm.017046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff, C. , Mahaut, C. , Abhyankar, A. , Le Goff, W. , Serre, V. , Afenjar, A. , … Cormier‐Daire, V. (2012). Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nature Genetics, 44(1), 85–88. 10.1038/ng.1016 [DOI] [PubMed] [Google Scholar]
- Le Goff, C. , Mahaut, C. , Wang, L. W. , Allali, S. , Abhyankar, A. , Jensen, S. , … Cormier‐Daire, V. (2011). Mutations in the TGFbeta binding‐protein‐like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. American Journal of Human Genetics, 89(1), 7–14. 10.1016/j.ajhg.2011.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff, C. , Morice‐Picard, F. , Dagoneau, N. , Wang, L. W. , Perrot, C. , Crow, Y. J. , … Cormier‐Daire, V. (2008). ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS‐like proteins in TGF‐beta bioavailability regulation. Nature Genetics, 40(9), 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson, A. H. , Kan, A. E. , Kozlowski, K. , Opitz, J. M. , & Bernstein, J. (1987). Geleophysic dysplasia–acromicric dysplasia with evidence of glycoprotein storage. American Journal of Medical Genetics. Supplement, 3, 181–189. 10.1002/ajmg.1320280522 [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Gerber, E. E. , Riegert‐Johnson, D. , Iqbal, S. , Whiteman, P. , McConnell, V. , … Dietz, H. C. (2010). Mutations in fibrillin‐1 cause congenital scleroderma: Stiff skin syndrome. Science Translational Medicine, 2(23), 23ra20 10.1126/scitranslmed.3000488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonnqvist, L. , Child, A. , Kainulainen, K. , Davidson, R. , Puhakka, L. , & Peltonen, L. (1994). A novel mutation of the fibrillin gene causing ectopia lentis. Genomics, 19(3), 573–576. 10.1006/geno.1994.1110 [DOI] [PubMed] [Google Scholar]
- McInerney‐Leo, A. M. , Le Goff, C. , Leo, P. J. , Kenna, T. J. , Keith, P. , Harris, J. E. , … Cormier‐Daire, V. (2016). Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. Journal of Medical Genetics, 53(7), 457–464. 10.1136/jmedgenet-2015-103647 [DOI] [PubMed] [Google Scholar]
- Piccolo, P. , Mithbaokar, P. , Sabatino, V. , Tolmie, J. , Melis, D. , Schiaffino, M. C. , … Brunetti‐Pierri, N. (2014). SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. European Journal of Human Genetics, 22(8), 988–994. 10.1038/ejhg.2013.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk, E. V. , & Polishchuk, R. S. (2013). Analysis of Golgi complex function using correlative light‐electron microscopy. Methods in Cell Biology, 118, 243–258. 10.1016/B978-0-12-417164-0.00015-X [DOI] [PubMed] [Google Scholar]
- Robinson, P. N. , Arteaga‐Solis, E. , Baldock, C. , Collod‐Beroud, G. , Booms, P. , De Paepe, A. , … Godfrey, M. (2006). The molecular genetics of Marfan syndrome and related disorders. Journal of Medical Genetics, 43(10), 769–787. 10.1136/jmg.2005.039669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser, E. M. , Wilkinson, A. R. , Hurst, J. A. , McGaughran, J. M. , & Donnai, D. (1995). Geleophysic dysplasia: A report of three affected boys–prenatal ultrasound does not detect recurrence. American Journal of Medical Genetics, 58(3), 217–221. 10.1002/ajmg.1320580304 [DOI] [PubMed] [Google Scholar]
- Spranger, J. W. , Gilbert, E. F. , Tuffli, G. A. , Rossiter, F. P. , & Opitz, J. M. (1971). Geleophysic dwarfism–a "focal" mucopolysaccharidosis? Lancet, 2(7715), 97–98. [DOI] [PubMed] [Google Scholar]
- Sule, G. , Campeau, P. M. , Zhang, V. W. , Nagamani, S. C. S. , Dawson, B. C. , Grover, M. , … Lee, B. H. (2013). Next‐generation sequencing for disorders of low and high bone mineral density. Osteoporosis International, 24(8), 2253–2259. 10.1007/s00198-013-2290-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanace, P. W. , Friedman, S. , & Wagner, B. M. (1960). Mitral stenosis in an atypical case of gargoylism: A case report with pathologic and histochemical studies of the cardiac tissues. Circulation, 21, 80–89. 10.1161/01.CIR.21.1.80 [DOI] [PubMed] [Google Scholar]
- Wraith, J. E. , Bankier, A. , Chow, C. W. , Danks, D. M. , & Sardharwalla, I. B. (1990). Geleophysic dysplasia. American Journal of Medical Genetics, 35(2), 153–156. 10.1002/ajmg.1320350202 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials