

Abstract
Background
Hereditary hemorrhagic telangiectasia (HHT) is a vascular dysplasia disorder characterized by epistaxis, mucocutaneous telangiectasias and arteriovenous malformations in internal organs. Recurrent epistaxis is the primary complaint in 90%‐96% of HHT patients and the other symptoms come with age. The aim of this study was to analyze HHT‐associated gene variant spectrum in Chinese HHT patients and to assess whether genetic testing could contribute to the early diagnosis.
Methodology/Principal
Thirty one HHT families including 62 individuals were recruited. Variants in the coding regions of four genes involved in HHT were amplified and analyzed using Sanger sequencing and multiplex ligation‐dependent probe amplification (MLPA).
Results
Twenty unique variants, including 8 novel variants were found in 24 of the 31 (77.4%) kindred. Diagnosis is confirmed for 7 possible individuals from 6 kindred. Thirteen ACVRL1 variants were detected from 17 isolated HHT families. Variants in ACVRL1 from 8/17 (47.1%) families were located in exon8. Seven ENG variants were found in 7 unrelated families throughout the coding region.
Conclusion
We conclude that ACVRL1 gene variant is 2.4 times more prevalent than that in ENG in Chinese individuals with HHT, and exon8 of the ACVRL1 gene may be a hotspot region. Genetic testing could contribute to early diagnosis for HHT.
Keywords: ACVRL1, ENG, epistaxis, hereditary hemorrhagic telangiectasia, variants
1. INTRODUCTION
Hereditary hemorrhagic telangiectasia, also known as Rendu‐Osler‐Weber syndrome, is a rare autosomal dominant genetic disorder, which affects 1 in 5–8,000 individuals (Faughnan et al., 2011; Govani & Shovlin, 2009; Lesca et al., 2007; Shovlin, 2010). Characteristic features of HHT include recurrent epistaxis, the presence of mucocutaneous telangiectasias, arteriovenous malformations (AVMs) in internal organs, and family history of HHT (Guttmacher, Marchuk, & White, 1995). The clinical diagnosis of HHT is based on the Curacao criteria (Shovlin et al., 2000), which propose that three or more of the four characteristic features described above define a definite diagnosis, where as two of these features suggest a “possible” diagnosis and one or none of these features indicate unlikely HHT. The penetrance for HHT is age‐dependent. Epistaxis is the first and the primary manifestation in 90%–96% of HHT patients(Guttmacher et al., 1995). Thus, the diagnosis for children and sporadic patients with recurrent epistaxis only is hard to decide.
At least four genes, including Endoglin (ENG, OMIM: 131195)(McAllister et al., 1994) resulting in HHT1 (OMIM: 187300), Activin A Receptor Type II‐like 1 (ACVRL1, OMIM: 601284) resulting in HHT2 (OMIM: 600376) (Johnson et al., 1996), SMAD family member 4(SMAD4, OMIM: 600993) resulting in HHT syndrome associated with juvenile polyposis (JP‐HHT, OMIM: 175050) (Gallione et al., 2006) and Bone morphogenetic proteins 9(BMP9, OMIM: 605120) resulting in a vascular anomaly syndrome (HHT5, OMIM: 615506) (Wooderchak‐Donahue et al., 2013), are thought to be responsible for about 90% HHT patients diagnosed by the clinical features. The remaining ∼10% of HHT patients have an unidentified genetic cause, which may be resulted from intronic variants in the known genes or caused by a novel gene (McDonald et al., 2011; Wooderchak‐Donahue et al., 2018). The aforementioned genes were all part of the transforming growth factor (TGFβ) signal pathway and integral to angiogenesis. Pathogenic variants in any of these genes may disrupt the balance between pro‐ and antiangiogenic signals for normal vascular development, resulting in HHT.
Previous studies have indicated that the disorder was caused predominantly by variants in either ENG (McAllister et al., 1994; McDonald et al., 1994; Shovlin et al., 1994) or ACVRL1 (Johnson et al., 1995; Vincent et al., 1995) genes. More than 500 variants have been reported in the two genes. Many of the variants were specific for each family, however, recurrent or founder variant has been reported in some populations, suggesting that the variant spectrum for HHT families may vary in different populations. Indeed, it has been shown that American, North European and Japanese families have fewer ACVRL1 variants than ENG variants (Komiyama, Ishiguro, Yamada, Morisaki, & Morisaki, 2014; McDonald et al., 2011). Presently there is only one report in the literature on the clinical and genetic characteristics of Chinese HHT patients (Chen et al., 2013). Thus, the aim of our study was to expand on this database on the variant spectrum of Chinese patients with HHT, and to assess whether genetic testing could set the diagnosis for Chinese patients with HHT.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The study was approved by the Ethics Committee of Beijing TongRen Hospital and performed in accordance with the guidelines of the World Medical Association’s Declaration of Helsinki. Written informed consent was obtained from all subjects or from next of kin, and carers or guardians of minors/children.
2.2. Cohort
A total of 62 individuals, including 36 females and 26 males, from 31 unrelated families with one or more members suffering from HHT were recruited from the outpatient clinic of Otolaryngology, Head and Neck Surgery Department at Beijing TongRen Hospital, who come from the different provinces in China. All the patients were of Han Chinese origin and aged between 4 years old to 73 years old; with a mean age of 42.9 ± 15.7 years. Clinical diagnosis of HHT was made according to the Curacao criteria (Shovlin et al., 2000). A cohort of 100 individuals without recurrent epistaxis, telangiectasias and the family history of HHT were also recruited as normal controls. Subjects were excluded if they or their first degree family members had any inherited vascular diseases.
2.3. DNA extraction
DNA was extracted from the peripheral blood leukocytes using the DNA Isolation Kit (Roche, Indianapolis, USA).
2.3.1. Single nucleotide variants and indel analysis
The protein coding sequences together with intron/exon boundaries of the four related genes (ENG, NM_000118.3; ACVRL1, NM_000020.2; SMAD4, NM_005359.5; BMP9,NM_016204.2) were amplified using polymerase chain reaction (PCR) for all DNA samples. The purified PCR products were directly sequenced using BigDye Terminator v.3.1 Cycle sequencing Kit (Applied Biosystems, Foster City, USA) and analyzed on ABI 3,730 DNA Analyzer (Applied Biosystems, Foster City, USA). PCR and sequencing primer pairs were designed using online Primer 3.0 software (Koressaar & Remm, 2007) (Table S1). The coding region and the flanking sequences (about 50 bases around the coding region) of the four genes were captured. Nucleotide alterations were identified by sequence alignment with the NCBI Reference Sequence (Build137). When a novel missense variant was identified, the paralog and ortholog sequences were compared using the CLUSTAL O (1.2.4) Multiple Sequence Alignment Program (Bayrak‐Toydemir, Mao, Lewin, & McDonald, 2004). The functional impact on the protein as an amino acid substitution was assessed using SIFT software (Bayrak‐Toydemir et al., 2006).
2.4. Deletion/duplication detection
Large deletions and duplications in the ACVRL1 and ENG genes of individuals who tested negative via PCR amplification and sequencing were detected using the SALSA MLPAkit (P093‐B1 HHT/PPH1, MRC‐Holland, the Netherlands), according to the manufacturer’s instructions. MLPA peak plots were analyzed using the Coffalyser. Net software (MRC‐Holland) to normalize and calculate the dosage ratios. Limit dosage ratios of ≤0.7 and ≥1.35 were set for deletion and duplication, respectively.
Additionally, when a variant likely to be pathogenic was identified in a proband, the variant was screened in other family members to assess whether the variant was co‐segregated with the patients and normal individuals. Furthermore, one hundred unrelated normal individuals were analyzed for each novel variant detected.
2.5. Evaluation of variants
The classification for variants uses the joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Variants have been classified as pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign and benign (Richards et al., 2015). The calculation and analysis for the probability of observed cosegregation was according to the method recommended by Jarvik & Browning (2016).
3. RESULTS
Overall, 62 individuals from 31 HHT families were recruited. Among them, five individuals were sporadic with no family history of HHT and the other 57 individuals came from 26 families with other affected members (Figure S1).
Table 1 shows the characteristics of all the participants in the study. Epistaxis was the most frequent clinical feature in our cohort and all the individuals had the manifestation. Overall, 32 patients were diagnosed as definite HHT patients and 11 as possible HHT patients, with the HHT onset age ranging from 3 to 50 years old. Four subjects were classified as “carriers” based on the presence of pathogenic gene variant and the missing symptoms, which may be explained by their rather young age (the age were described in Table 1).
Table 1.
Clinical features and variant analyses results
| Family ID | Individual ID | Gender | Age | Onset age | Epistaxis | MT a | AVMs b | Family history | Diagnosis | Gene | Exon | Nucleotide change | Amino acid change | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F1_III:1 | Male | 55 | 17 | Yes | No | GIT | Yes | HHT | ACVRL1 | EXON8 | c.1231C>T | p.Arg411Trp | Pathogenic |
| F1 | F1_III:2 | Male | 47 | 18 | Yes | Na | Na | Yes | Possible HHT | ACVRL1 | EXON8 | c.1231C>T | p.Arg411Trp | Pathogenic |
| F1 | F1_III:3 | Female | 40 | 17 | Yes | Na | Na | Yes | Possible HHT | ACVRL1 | EXON8 | c.1231C>T | p.Arg411Trp | Pathogenic |
| F1 | F1_IV:1 | Male | 28 | / | No | No | Na | Yes | Carrier | ACVRL1 | EXON8 | c.1231C>T | p.Arg411Trp | Pathogenic |
| F2 | F2_II:1 | Female | 58 | 20 | Yes | Yes | PAVMs | Yes | HHT | ACVRL1 | EXON3 | c.200G>A | p.Arg67Gln | Pathogenic |
| F2 | F2_III:1 | Male | 31 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F3 | F3_II:1 | Female | 72 | 18 | Yes | Yes | HAVMs | Yes | HHT | ENG | EXON5 | c.593del | p.Pro198Argfs*24 | Pathogenic |
| F3 | F3_III:1 | Male | 48 | 20 | Yes | Yes | Na | Yes | HHT | ENG | EXON5 | c.593del | p.Pro198Argfs*24 | Pathogenic |
| F4 | F4_II:1 | Male | 32 | 26 | Yes | Yes | HAVMs | Yes | HHT | ACVRL1 | INTRON4 | c.526−3C>G | / | VUS |
| F5 | F5_I:1 | Male | 73 | 23 | Yes | Na | HAVMs,PAVMs | Yes | HHT | ENG | EXON7 | c.841A>G | p.Ile281Val | VUS |
| F6 | F6_II:2 | Male | 42 | 20 | Yes | No | Na | Yes | Possible HHT | ACVRL1 | EXON10 | c.1436G>C | p.Arg479Pro | VUS |
| F6 | F6_II:1 | Male | 44 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F7 | F7_II:1 | Female | 61 | 25 | Yes | Yes | GIT | Yes | HHT | / | / | / | / | |
| F7 | F7_III:1 | Male | 43 | 15 | Yes | Yes | Na | Yes | HHT | / | / | / | / | |
| F7 | F7_III:2 | Female | 50 | 13 | Yes | Yes | Na | Yes | HHT | / | / | / | / | |
| F7 | F7_IV:2 | Female | 22 | 13 | Yes | Yes | Na | Yes | HHT | / | / | / | / | |
| F7 | F7_III:3 | Male | 30 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F7 | F7_II:2 | Male | 58 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F7 | F7_IV:1 | Male | 32 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F7 | F7_III:4 | Female | 37 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F7 | F7_II:3 | Female | 58 | / | No | No | Na | Yes | Normal | / | / | / | / | |
| F8 | F8_III:1 | Female | 35 | 3 | Yes | Yes | Na | Yes | HHT | ENG | EXON7 | c.840del | p.Ile281Serfs*78 | Pathogenic |
| F8 | F8_IV:1 | Male | 10 | 3 | Yes | No | Na | Yes | Possible HHT | ENG | EXON7 | c.840del | p.Ile281Serfs*78 | Pathogenic |
| F8 | F8_II:1 | Male | 60 | 18 | Yes | Yes | HAVMs,GIT | Yes | HHT | ENG | EXON7 | c.840del | p.Ile281Serfs*78 | Pathogenic |
| F9 | F9_III:2 | Male | 41 | 17 | Yes | Yes | Na | Yes | HHT | ENG | EXON14 | c.1878+7C>T | / | Likely pathologenic |
| F9 | F9_III:1 | Female | 43 | / | No | No | Na | Yes | Normal | / | / | / | / | / |
| F9 | F9_III:4 | Male | 40 | 12 | Yes | Yes | Na | Yes | HHT | ENG | EXON14 | c.1878+7C>T | / | Likely pathologenic |
| F9 | F9_IV:1 | Female | 10 | 4 | Yes | No | Na | Yes | Possible HHT | ENG | EXON14 | c.1878+7C>T | / | Likely pathologenic |
| F9 | F9_II:1 | Female | 67 | 20 | Yes | Yes | GIT | Yes | HHT | ENG | EXON14 | c.1878+7C>T | / | Likely pathologenic |
| F9 | F9_III:3 | Female | 39 | / | No | No | Na | No | Normal | / | / | / | / | / |
| F11 | F11_II:1 | Male | 50 | 10 | Yes | Yes | Na | Yes | HHT | / | / | / | / | / |
| F12 | F12_III:1 | Male | 53 | 20 | Yes | Yes | HAVMs | Yes | HHT | ACVRL1 | EXON8 | c.1232G>A | p.Arg411Gln | Pathogenic |
| F13 | F13_II:1 | Female | 37 | 5 | Yes | Na | HAVMs | Yes | HHT | / | / | / | / | / |
| F14 | F14_II:1 | Female | 53 | 18 | Yes | Yes | PAVMs | Yes | HHT | ENG | EXON4 | c.496del | p.Gln166Argfs*56 | Pathogenic |
| F14 | F14_III:1 | Female | 28 | / | No | Na | Na | Yes | Carrier | ENG | EXON4 | c.496del | p.Gln166Argfs*56 | Pathogenic |
| F14 | F14_II:2 | Female | 50 | / | No | Na | Na | Yes | Normal | / | / | / | / | / |
| F15 | F15_II:1 | Female | 45 | 17 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON7 | c.853C>T | p.Leu285Phe | VUS |
| F15 | F15_III:1 | Female | 10 | / | No | No | Na | Yes | Normal | / | / | / | / | / |
| F16 | F16_II:1 | Male | 57 | 30 | Yes | Yes | HAVMs | Yes | HHT | ACVRL1 | EXON8 | c.1120C>T | p.Arg374Trp | Pathogenic |
| F16 | F16_III:1 | Male | 23 | / | No | No | Na | Yes | Carrier | ACVRL1 | EXON8 | c.1120C>T | p.Arg374Trp | Pathogenic |
| F18 | F18_II:1 | Female | 33 | 3 | Yes | Na | Na | Yes | Possible HHT | / | / | / | / | / |
| F18 | F18_III:1 | Female | 11 | / | No | No | Na | Yes | Normal | / | / | / | / | / |
| F19 | F19_II:1 | Male | 57 | 50 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON7 | c.1042G>A | p.Asp348Asn | VUS |
| F20 | F20_III:1 | Male | 36 | 28 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON8 | c.1207C>G | p.Leu403Val | VUS |
| F21 | F21_III:1 | Female | 58 | 15 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON8 | c.1135G>A | p.Glu379Lys | Pathogenic |
| F21 | F21_III:2 | Female | 50 | 14 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON8 | c.1135G>A | p.Glu379Lys | Pathogenic |
| F21 | F21_IV:1 | Female | 32 | / | No | No | Na | Yes | Normal | / | / | / | / | / |
| F22 | F22_II:1 | Female | 53 | 45 | Yes | Yes | HAVMs | Yes | HHT | / | / | / | / | / |
| F23 | F23_II:1 | Female | 41 | 3 | Yes | Yes | Na | Yes | HHT | / | / | / | / | / |
| F24 | F24_II:1 | Female | 42 | 14 | Yes | Yes | HAVMs | Yes | HHT | ENG | EXON6 | c.772del | p.Tyr258Thrfs*101 | Pathogenic |
| F25 | F25_I:1 | Male | 45 | 13 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON8 | c.1232G>A | p.Arg411Gln | Pathogenic |
| F25 | F25_II:1 | Female | 4 | / | No | No | Na | Yes | Carrier | ACVRL1 | EXON8 | c.1232G>A | p.Arg411Gln | Pathogenic |
| F26 | F26_III:1 | Male | 45 | 14 | Yes | Yes | Na | Yes | HHT | ACVRL1 | EXON3 | c.200G>A | p.Arg67Gln | Pathogenic |
| F27 | F27_II:1 | Female | 59 | 20 | Yes | No | Na | Yes | Possible HHT | ACVRL1 | EXON5 | c.576del | p.Leu193Trpfs*65 | Pathogenic |
| F27 | F27_III:1 | Female | 36 | / | No | No | Na | No | Normal | / | / | / | / | / |
| F27 | F27_III:2 | Female | 33 | / | No | No | Na | No | Normal | / | / | / | / | / |
| F28 | F28_III:1 | Male | 43 | 13 | Yes | No | Na | Yes | Possible HHT | ENG | INTRON3 | c.360+1G>A | / | Pathogenic |
| S1 | S1 | Female | 52 | 45 | Yes | Yes | GIT | No | HHT | ACVRL1 | EXON5 |
c.552_559delins
TCTGCTCAGGTGCAGTCT |
p.Gly185Leufs*43 | Pathogenic |
| S2 | S2 | Female | 73 | 15 | Yes | Yes | GIT | No | HHT | ACVRL1 | EXON8 | c.1232G>A | p.Arg411Gln | Pathogenic |
| S4 | S4 | Female | 63 | 20 | Yes | Yes | Na | No | Possible HHT | ACVRL1 | EXON8 | c.1120C>T | p.Arg374Trp | Pathogenic |
| S6 | S6 | Female | 31 | 3 | Yes | Yes | Na | No | Possible HHT | ACVRL1 | EXON3 | c.106T>C | p.Cys36Arg | VUS |
| S8 | S8 | Female | 54 | 14 | Yes | No | PAVMs | No | Possible HHT | / | / | / | / | / |
Abbreviations: Na, not available; Yes, the manifestation is present; No, the manifestation is absent; GIT, Gastrointestinal telangiectases; PAVMs, pulmonary arteriovenous malformations; HAVMs, hepatic arteriovenous malformations; Variants in bold red letters are novel.
Mucocutaneous telangiectasias.
Arteriovenous malformations.
A total of 20 variants were identified in 24 of 31 kindred (sequences shown in Figure S2), with 24 definite HHT cases from19 kindred, 9 possible HHT individuals from 8 kindred and 4 carriers from 4 kindred, which were responsible for 77.4% (24/31) of all HHT families. No variant was detected in 7 families (7/31, 22.6%), including 5 families with 8 definite HHT patients and 2 families with 2 possible HHT patients (Table 1).
All the 20 variants were single nucleotide variants (SNVs) or small indels located in ACVRL1 and ENG gene. We didn’t find any pathogenic variant in the SMAD4 or BMP9 gene. No gross alteration was found in the MLPA analysis for ACVRL1 and ENG.
A total of 13 variants in ACVRL1 gene were detected from 17 isolated HHT families (17/24 families, 70.8%). The ACVRL1 variant of c.200G>A in exon3, the c.1120C>T and c.1232G>A in exon8 were recurrent in unrelated families. Overall, variants found in 8/17 (47.1%) of all families with ACVRL1 variants were located in exon8 (including 5 unique variants) (Figure 1). The distribution of other ACVRL1 variants was illustrated on Figure 1. Similarly, seven variants in ENG gene were identified in 7 HHT families (7/24 families, 29.2%). The distribution of the seven ENG variants was showed on Figure 1.
Figure 1.
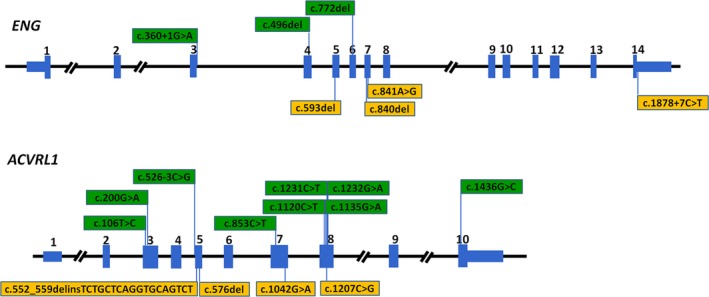
Variants found in the HHT families and their sketches on the ENG and ACVRL1 genes. The novel variant was shown as orange bars, and the reported variants are shown as green bars
A total of eight novel variants, which have not been reported previously, were found in this study. Four of these were detected in the ENG gene (c.593del, c.840del and c.1878+7C>T and c.841A>G), and the other four novel variants were located in the ACVRL1 gene (c.576del, c.1207C>G, and c.552_559delinsTCTGCTCAGGTGCAGTCT and c.1042G>A). We further analyzed the pathogenic potential for the different type of novel variants in the following section.
Four of the novel variants were out‐of‐frame indels, which may be pathogenic as haploinsufficiency of ENG or ACVRL1 was an underlying cause of HHT (Pece‐Barbara, Cymerman, Vera, Marchuk, & Letarte, 1999). These variants have never been found in public databases (dbSNPs, 1000Genome and ESP) neither the previous investigations. Patient’s phenotype is highly specific for HHT. Variant of c.552_559delinsTCTGCTCAGGTGCAGTCT in ACVRL1 was found in a sporadic individual. In regards to c.593del, c.840del in ENG and the c.576del in ACVRL1, variants testing for the family members found cosegregation with disease in more than one family member. Variant of c.593del was found in a four generation family (family ID: F3, Figure S1). Both the proband and her son, who were clinically diagnosed as definite HHT, were heterozygous with the variant. Samples of the other family members were not obtained. The second out‐of‐frame variant (c.840del) was detected in a four generation family with three patients. The proband, her father and her son were heterozygous with variant of c.840del (family ID: F8, Figure S1). The proband and her father were clinically diagnosed as HHT with more than three features. And the proband’s son, a possible HHT individual, was confirmed by the genetic testing. The novel c.576del was found in a four generation family (family ID: F27, Figure S1). In this family, a HHT patient and two normal individuals were recruited. The HHT patient was heterozygous with the c.576del variant and the two normal individuals were wildtype. And the genotype for all obtained members in F27 was co‐segregated with the manifestation of the HHT. All the above findings indicated that these novel variants were likely to be pathogenic.
The fifth novel variant (c.1878+7C>T,) was a substitution in the 3′UTR region of the ENG. It was found in a four generation family (family ID: F9, Figure S1). In this family, 6 DNA samples from 4 patients and 2 normal individuals were obtained and tested. All 4 patients were heterozygous with this variant of c.1878+7C>T, and the 2 normal individuals were wild type for this site. The variant was rare and not found in the previous reports and the public databases (dbSNPs, 1000Genome and ESP), thus, we assumed the variant only entered the pedigree once. The probability of observed cosegregation was calculated. The untyped relative (II:2) who must had passed the variant was assumed to be heterozygous. Considering definitely affected individuals (Ⅱ:3, Ⅲ:3, Ⅲ:5 and Ⅳ:3), we observed four meioses, so the affected individuals contributed a factor of (1/2)4 to the value of probability of observed cosegregation. The normal individual (Ⅲ:1) from the family contributes a factor of (1−(1/2)) = 1/2. Thus, for family F9, the probability of observed cosegregation was 1/32, which is a strong evidence for the pathogenicity of c.1878+7C>T in the ACVRL1 (Jarvik & Browning, 2016).
The other three novel missense variants (c.841A>G in ENG, c.1207C>G and c.1042G>A in ACVRL1) were absent in the 100 normal individuals. Variants of c.1207C>G and c.1042G>A were not found in the public databases (dbSNPs, 1000Genome and ESP) and the previous studies. This variant of c.841A>G was reported in the EXAC (The Exome Aggregation Consortium) database (Cymerman, Vera, Karabegovic, Abdalla, & Letarte, 2003). The Minor allele frequency of this variant was 1.7e‐5 in the dbSNPs database. SIFT was applied to predict whether the missense variants affect protein function. It was predicted to be “damaging”. Alignment for amino acid sequences from different species found that the novel missense variants were located in the conserved region (Figures S3 and S4).
4. DISCUSSION
HHT presents clinically as a variety of symptoms including recurrent epistaxis, mucocutaneous telangiectasias, and visceral AVMs in lung, liver, gastrointestinal tract, brain or spinal cord. In the present study, most of the patients diagnosed as definite HHT patients demonstrated epistaxis, mucocutaneous telangiectasia and family history. Nose bleeds were observed in all definite individuals, but also the 11 possible HHT individuals.
Our study has demonstrated that 77.4% of the kindred (24/31, 24 families including 24 definite HHT cases, 9 possible HHT cases, 4 carriers) had ACVRL1 or ENG variant, which is in accordance with the findings of previous reports (Bayrak‐Toydemir et al., 2006; Chen et al., 2013; Heimdal et al., 2016). However, the variant carrier percent in HHT from the ARUP institute is higher than that of our study, as the non‐coding region variants are screened, which may contribute to about 1% of HHT patients (Wooderchak‐Donahue et al., 2018). What’s more, none variant was found in SMAD4 and BMP9 gene, which may account for about 1%–2% of HHT patients. In the present study, a likely or clearly pathogenic variant was detected in 7 possible HHT cases and 4 individuals without any symptoms except for positive family history, which enabled us to set the diagnosis of HHT for the recurrent epistaxis patients and find the high risk individuals early. These findings suggest that the Curacao Criteria should be revised to take into consideration the results of genetic testing, which could confirm the recurrent nose bleeds and find the high risk individuals early (Torring, Brusgaard, Ousager, Andersen, & Kjeldsen, 2014).
Previous studies have indicated that there is a considerable variation in the ENG/ACVRL1 variant ratio in different populations. While one study in French HHT patients has demonstrated the ENG/ACVRL1 variant ratio range from 0.37 to 0.51 (Lesca et al., 2007), a ENG/ACVRL1 variant ratio of 0.72 (13/18) has been found in Canadian patients (Abdalla et al., 2005), a ratio of 1.22 in American (USA) patients (Bayrak‐Toydemir et al., 2006) and a ratio of 2.0 in Danish patients (Brusgaard et al., 2004). In the current study, 17 families (17/24, 70.8%) had variants in ACVRL1 gene and 7 families (7/24, 29.2%) in the ENG, providing an ENG/ACVRL1 variant ratio of 0.41 (7/17), which is comparable to the findings in the French patients. In comparison, another study of patients from 14 Chinese families has indicated a ratio is 0.25 (2/8) (Chen et al., 2013). It is possible that the wide variation noted in the ENG/ACVRL1 variant ratios in different populations may be a consequence of differences in patient numbers and study methods employed in the different studies. However despite these differences, the findings from the two studies of Chinese HHT patients indicate that ACVRL1 variants, which were 2.4–4 times greater than ENG variants, are the predominant cause of HHT in the Chinese patients.
In ACVRL1, the c.1120C>T (p.Arg374Trp) and c.1232G>A (p.Arg411Gln) variants on exon8 were seen twice and three times in apparently unrelated families. These two variants have been reported in several families in previous studies (Abdalla, Cymerman, Johnson, Deber, & Letarte, 2003; Bayrak‐Toydemir et al., 2006; Berg et al., 1997; Harrison et al., 2003; Johnson et al., 1996; Kjeldsen et al., 2001; Lesca et al., 2004; Trembath et al., 2001), suggesting that these codons may be the hotspot or founder region of the ACVRL1 gene, which may need further studies in a larger sample size to proved. In this study, variants in 8/17(47.1%) families were located in exon8 of ACVRL1 gene. Indeed, a study by Chen and colleagues (Chen et al., 2013) investigating 14 Chinese HHT families also reported 8 unique ACVRL1 variants, of which 3/8 (37.5%) variants (c. 1121G>A, c.1124A>G and c.1195T>C) were located in exon8. These findings suggest that exon8 of the ACVRL1 gene may be a hotspot region, which may be useful in the effective genetic testing for HHT. In contrast, seven of the ENG variants were widely distributed throughout the gene, none of which was observed in multiple families.
In this study, a total of eight novel variants were found and the pathogenicity was evaluated. Four of the novel out‐of‐frame indels were proved to be pathogenic for HHT. The variant of c.1878+7C>T in ENG was found in a four generation family. The cosegregation with HHT in this family was a strong evidence for the pathogenicity of c.1878+7C>T. Besides that, the variant was rare and absent in the public databases. These results strongly supported that it was to be “likely pathogenic”, although all variants in the two terminal exons except a large deletion (two exons) were currently listed as benign or pending classification in ARUP ([Link]; Jarvik & Browning, 2016; Richards et al., 2015). The three missense variants were absent or rare in the public database. Multiple lines of computational evidence support a deleterious effect on the gene. However, the functional study for the pathogenicity of these variants is required in the future studies. So, the three missense variants were classified as “variant of unknown significance (VUS)” according to standards and guidelines of the American College of Medical Genetics and Genomics (Richards et al., 2015).
Indeed, a total of 12 variants have ever been reported in the previous studies, including two out‐of‐frame indels (c.496del([Link]; Lesca et al., 2004) and c.772del(Fernandez et al., 2006; Olivieri et al., 2007)), two assumed splice‐site variants (c.526‐3C>G(Torring et al., 2014) and c.360+1G>A(Cymerman et al., 2003, 2000; Pece et al., 1997)) and eight missense variants (c.1231C>T(Abdalla, Geisthoff, et al., 2003; Trembath et al., 2001; Zhang et al., 2004), c.200G>A(Bayrak‐Toydemir et al., 2004; Berg et al., 1997; Olivieri et al., 2007; Schulte et al., 2005), c.1232G>A(Abdalla, Geisthoff, et al., 2003; Bayrak‐Toydemir et al., 2004; Berg et al., 1997; Johnson et al., 1996), c.1120C>T(Abdalla, Cymerman, et al., 2003; Berg et al., 1997; Kjeldsen et al., 2001), c.1135G>A(Bayrak‐Toydemir et al., 2004; Brusgaard et al., 2004; Lesca et al., 2004), c.1436G>C(Bayrak‐Toydemir et al., 2006; Lesca et al., 2006), c.853C>T(Bayrak‐Toydemir et al., 2004; Lesca et al., 2004; J. McDonald et al., 2011) and c.106T>C([Link])). The two out‐of frame indels located in ENG have been proved to be pathogenic. Variant of c.360+1G>A located in the invariant splice‐site sequence GU (+1+2) or AG (−1 –2), and expected to cause aberrant splicing. Five of the eight missense variants (c.1231C>T, c.200G>A, c.1232G>A, c.1120C>T and c.1135G>A) have been reported to be pathogenic in the previous studies (Bossler, Richards, George, Godmilow, & Ganguly, 2006). The other three (c.1436G>C, c.853C>T and c.106T>C) were found to be all located in the conserved region aligning with amino acid sequences from different species (Figure S3), suggesting potential pathogenic role in HHT. However, it may need more information to support this.
In conclusion, our study has demonstrated that 24 of the 31 (77.4%) kindred carry the variant from ACVRL1 or ENG gene. And no variant to be likely pathogenic in SMAD4 and BMP9 was found. HHT patients with ACVRL1 variants are 2.4–4 times more than those with ENG variants in Chinese. About 47.1% of the ACVRL1 variants are located in exon8, despite a wide distribution throughout the gene. These findings suggest that exon8 of the ACVRL1 gene may be a hotspot region for HHT in Chinese patients. And c.1120C>T (p.Arg374Trp) and c.1232G>A (p.Arg411Gln) in ACVRL1 may be the commonest variants. Further studies with a larger sample size and functional analysis for the variants are needed to confirm this.
CONFLICT OF INTEREST
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supporting information
ACKNOWLEDGEMENTS
The authors would like to thank all the individuals who participated in this study.
Zhao Y, Zhang Y, Wang X, Zhang L. Variant analysis in Chinese families with hereditary hemorrhagic telangiectasia. Mol Genet Genomic Med. 2019;7:e893 10.1002/mgg3.893
Yuan Zhang and Luo Zhang contributed equally to the study.
Funding information
This work was supported by the National Key R&D Program of China (2016YFC0905200), the program for Changjiang Scholars and Innovative Research Team (IRT13082), the National Natural Science Foundation of China (81420108009, 81630023, 81400470 and 81570895), Capital's Funds for Health Improvement and Research (2018‐2‐1092, 2016‐4‐1092), Beijing Health Bureau Program for High Level Talents (2014‐3‐015), Beijing Municipal Administration of Hospitals’ Mission Plan (SML20150203), Beijing Advanced Innovation Center For Food Nutrition And Human Health (Beijing Technology and Business University, 20181045) and Beijing Natural Science Foundation (7164247).
Contributor Information
Yuan Zhang, Email: summer_zhang1211@126.com.
Luo Zhang, Email: dr.luozhang@139.com.
REFERENCES
- Abdalla, S. A. , Cymerman, U. , Johnson, R. M. , Deber, C. M. , & Letarte, M. (2003). Disease‐associated mutations in conserved residues of ALK‐1 kinase domain. European Journal of Human Genetics, 11(4), 279–287. 10.1038/sj.ejhg.5200919 [DOI] [PubMed] [Google Scholar]
- Abdalla, S. A. , Cymerman, U. , Rushlow, D. , Chen, N. , Stoeber, G. P. , Lemire, E. G. , & Letarte, M. (2005). Novel mutations and polymorphisms in genes causing hereditary hemorrhagic telangiectasia. Human Mutation, 25(3), 320–321. 10.1002/humu.9312 [DOI] [PubMed] [Google Scholar]
- Abdalla, S. A. , Geisthoff, U. W. , Bonneau, D. , Plauchu, H. , McDonald, J. , Kennedy, S. , … Letarte, M. (2003). Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. Journal of Medical Genetics, 40(7), 494–502. 10.1136/jmg.40.7.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayrak‐Toydemir, P. , Mao, R. , Lewin, S. , & McDonald, J. (2004). Hereditary hemorrhagic telangiectasia: An overview of diagnosis and management in the molecular era for clinicians. Genetics in Medicine, 6(4), 175–191. https://doi.org/10.109701.GIM.0000132689.25644.7C [DOI] [PubMed] [Google Scholar]
- Bayrak‐Toydemir, P. , McDonald, J. , Markewitz, B. , Lewin, S. , Miller, F. , Chou, L.‐S. , … Mao, R. (2006). Genotype‐phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. American Journal of Medical Genetics. Part A, 140(5), 463–470. 10.1002/ajmg.a.31101 [DOI] [PubMed] [Google Scholar]
- Berg, J. N. , Gallione, C. J. , Stenzel, T. T. , Johnson, D. W. , Allen, W. P. , Schwartz, C. E. , … Marchuk, D. A. (1997). The activin receptor‐like kinase 1 gene: Genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. American Journal of Human Genetics, 61(1), 60–67. 10.1086/513903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossler, A. D. , Richards, J. , George, C. , Godmilow, L. , & Ganguly, A. (2006). Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): Correlation of genotype with phenotype. Human Mutation, 27(7), 667–675. 10.1002/humu.20342 [DOI] [PubMed] [Google Scholar]
- Brusgaard, K. , Kjeldsen, A. D. , Poulsen, L. , Moss, H. , Vase, P. , Rasmussen, K. , … Hørder, M. (2004). Mutations in endoglin and in activin receptor‐like kinase 1 among Danish patients with hereditary haemorrhagic telangiectasia. Clinical Genetics, 66(6), 556–561. 10.1111/j.1399-0004.2004.00341.x [DOI] [PubMed] [Google Scholar]
- Chen, Y.‐J. , Yang, Q.‐H. , Liu, D. , Liu, Q.‐Q. , Eyries, M. , Wen, L. , … Jing, Z.‐C. (2013). Clinical and genetic characteristics of Chinese patients with hereditary haemorrhagic telangiectasia‐associated pulmonary hypertension. European Journal of Clinical Investigation, 43(10), 1016–1024. 10.1111/eci.12138 [DOI] [PubMed] [Google Scholar]
- Cymerman, U. , Vera, S. , Karabegovic, A. , Abdalla, S. , & Letarte, M. (2003). Characterization of 17 novel endoglin mutations associated with hereditary hemorrhagic telangiectasia. Human Mutation, 21(5), 482–492. 10.1002/humu.10203 [DOI] [PubMed] [Google Scholar]
- Cymerman, U. , Vera, S. , Pece‐Barbara, N. , Bourdeau, A. , White, R. I. Jr , Dunn, J. , & Letarte, M. (2000). Identification of hereditary hemorrhagic telangiectasia type 1 in newborns by protein expression and mutation analysis of endoglin. Pediatric Research, 47(1), 24–35. 10.1203/00006450-200001000-00008 [DOI] [PubMed] [Google Scholar]
- HHT mutation database. (Available at: http://www.arup.utah.edu/database/HHT/. Accessed October 30, 2017).
- Faughnan, M. E. , Palda, V. A. , Garcia‐Tsao, G. , Geisthoff, U. W. , McDonald, J. , & Proctor, D. D. , …, Zarrabeitia, R. ; HHT Foundation International ‐ Guidelines Working Group (2011). International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. Journal of Medical Genetics, 48(2), 73–87. 10.1136/jmg.2009.069013 [DOI] [PubMed] [Google Scholar]
- Fernandez‐L, A. , Sanz‐Rodriguez, F. , Zarrabeitia, R. , Perez‐Molino, A. , Morales, C. , Restrepo, C. M. , … Botella, L. M. (2006). Mutation study of Spanish patients with hereditary hemorrhagic telangiectasia and expression analysis of Endoglin and ALK1. Human Mutation, 27(3), 295 10.1002/humu.9413 [DOI] [PubMed] [Google Scholar]
- Gallione, C. J. , Richards, J. A. , Letteboer, T. G. W. , Rushlow, D. , Prigoda, N. L. , Leedom, T. P. , … Marchuk, D. A. (2006). SMAD4 mutations found in unselected HHT patients. Journal of Medical Genetics, 43(10), 793–797. 10.1136/jmg.2006.041517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govani, F. S. , & Shovlin, C. L. (2009). Hereditary haemorrhagic telangiectasia: A clinical and scientific review. European Journal of Human Genetics, 17(7), 860–871. 10.1038/ejhg.2009.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttmacher, A. E. , Marchuk, D. A. , & White, R. I. Jr (1995). Hereditary hemorrhagic telangiectasia. New England Journal of Medicine, 333(14), 918–924. 10.1056/NEJM199510053331407 [DOI] [PubMed] [Google Scholar]
- Harrison, R. E. , Flanagan, J. A. , Sankelo, M. , Abdalla, S. A. , Rowell, J. , Machado, R. D. , … Trembath, R. C. (2003). Molecular and functional analysis identifies ALK‐1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. Journal of Medical Genetics, 40(12), 865–871. 10.1136/jmg.40.12.865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimdal, K. , Dalhus, B. , Rodningen, O. K. , Kroken, M. , Eiklid, K. , Dheyauldeen, S. , … Kulseth, M. A. (2016). Mutation analysis in Norwegian families with hereditary hemorrhagic telangiectasia: Founder mutations in ACVRL1. Clinical Genetics, 89(2), 182–186. 10.1111/cge.12612 [DOI] [PubMed] [Google Scholar]
- Jarvik, G. P. , & Browning, B. L. (2016). Consideration of cosegregation in the pathogenicity classification of genomic variants. American Journal of Human Genetics, 98(6), 1077–1081. 10.1016/j.ajhg.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D. W. , Berg, J. N. , Baldwin, M. A. , Gallione, C. J. , Marondel, I. , Yoon, S.‐J. , … Marchuk, D. A. (1996). Mutations in the activin receptor‐like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nature Genetics, 13(2), 189–195. 10.1038/ng0696-189 [DOI] [PubMed] [Google Scholar]
- Johnson, D. W. , Berg, J. N. , Gallione, C. J. , McAllister, K. A. , Warner, J. P. , Helmbold, E. A. , … Marchuk, D. A. (1995). A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Research, 5(1), 21–28. 10.1101/gr.5.1.21 [DOI] [PubMed] [Google Scholar]
- Kjeldsen, A. D. , Brusgaard, K. , Poulsen, L. , Kruse, T. , Rasmussen, K. , Green, A. , & Vase, P. (2001). Mutations in the ALK‐1 gene and the phenotype of hereditary hemorrhagic telangiectasia in two large Danish families. American Journal of Medical Genetics, 98(4), 298–302. [DOI] [PubMed] [Google Scholar]
- Komiyama, M. , Ishiguro, T. , Yamada, O. , Morisaki, H. , & Morisaki, T. (2014). Hereditary hemorrhagic telangiectasia in Japanese patients. Journal of Human Genetics, 59(1), 37–41. 10.1038/jhg.2013.113 [DOI] [PubMed] [Google Scholar]
- Koressaar, T. , & Remm, M. (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics, 23(10), 1289–1291. 10.1093/bioinformatics/btm091 [DOI] [PubMed] [Google Scholar]
- Lesca, G. , Burnichon, N. , Raux, G. , Tosi, M. , Pinson, S. , Marion, M.‐J. , … Giraud, S. (2006). Distribution of ENG and ACVRL1 (ALK1) mutations in French HHT patients. Human Mutation, 27(6), 598 10.1002/humu.9421 [DOI] [PubMed] [Google Scholar]
- Lesca, G. , Olivieri, C. , Burnichon, N. , Pagella, F. , Carette, M.‐F. , Gilbert‐Dussardier, B. , … Plauchu, H. (2007). Genotype‐phenotype correlations in hereditary hemorrhagic telangiectasia: Data from the French‐Italian HHT network. Genetics in Medicine, 9(1), 14–22. https://doi.org/10.1097GIM.0b013e31802d8373 [DOI] [PubMed] [Google Scholar]
- Lesca, G. , Plauchu, H. , Coulet, F. , Lefebvre, S. , Plessis, G. , Odent, S. , … French Rendu‐Osler, N. (2004). Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. Human Mutation, 23(4), 289–299. 10.1002/humu.20017 [DOI] [PubMed] [Google Scholar]
- McAllister, K. A. , Grogg, K. M. , Johnson, D. W. , Gallione, C. J. , Baldwin, M. A. , Jackson, C. E. , …1994). Endoglin, a TGF‐beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature Genetics, 8(4), 345–351. 10.1038/ng1294-345 [DOI] [PubMed] [Google Scholar]
- McDonald, J. , Damjanovich, K. , Millson, A. , Wooderchak, W. , Chibuk, J. M. , Stevenson, D. A. , … Bayrak‐Toydemir, P. (2011). Molecular diagnosis in hereditary hemorrhagic telangiectasia: Findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clinical Genetics, 79(4), 335–344. 10.1111/j.1399-0004.2010.01596.x [DOI] [PubMed] [Google Scholar]
- McDonald, M. T. , Papenberg, K. A. , Ghosh, S. , Glatfelter, A. A. , Biesecker, B. B. , Helmbold, E. A. , Markel, D. S. , Zolotor, A. , McKinnon, W. C. , Vanderstoep, J. L. , Jackson, C. E. , Iannuzzi, M. , Collins, F. S. , Boehnke, M. , Porteous, M. E. , Guttmacher, A. E. , & Marchuk, D. A. , …1994). A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33‐34. Nature Genetics, 6(2), 197–204. 10.1038/ng0294-197 [DOI] [PubMed] [Google Scholar]
- Olivieri, C. , Pagella, F. , Semino, L. , Lanzarini, L. , Valacca, C. , Pilotto, A. , … Danesino, C. (2007). Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. Journal of Human Genetics, 52(10), 820–829. 10.1007/s10038-007-0187-5 [DOI] [PubMed] [Google Scholar]
- Pece, N. , Vera, S. , Cymerman, U. , White, R. I. Jr , Wrana, J. L. , & Letarte, M. (1997). Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest, 100(10), 2568–2579. 10.1172/jci119800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece‐Barbara, N. , Cymerman, U. , Vera, S. , Marchuk, D. A. , & Letarte, M. (1999). Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Human Molecular Genetics, 8(12), 2171–2181. 10.1093/hmg/8.12.2171 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte, C. , Geisthoff, U. , Lux, A. , Kupka, S. , Zenner, H. P. , Blin, N. , & Pfister, M. (2005). High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Human Mutation, 25(6), 595 10.1002/humu.9345 [DOI] [PubMed] [Google Scholar]
- Shovlin, C. L. (2010). Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Reviews, 24(6), 203–219. 10.1016/j.blre.2010.07.001 [DOI] [PubMed] [Google Scholar]
- Shovlin, C. L. , Guttmacher, A. E. , Buscarini, E. , Faughnan, M. E. , Hyland, R. H. , Westermann, C. J. J. , … Plauchu, H. (2000). Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu‐Osler‐Weber syndrome). American Journal of Medical Genetics, 91(1), 66–67. [DOI] [PubMed] [Google Scholar]
- Shovlin, C. L. , Hughes, J. , Tuddenham, E. , Temperley, I. , Perembelon, Y. , Scott, J. , … Seidman, J. G. (1994). A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nature Genetics, 6(2), 205–209. 10.1038/ng0294-205 [DOI] [PubMed] [Google Scholar]
- Torring, P. M. , Brusgaard, K. , Ousager, L. B. , Andersen, P. E. , & Kjeldsen, A. D. (2014). National mutation study among Danish patients with hereditary haemorrhagic telangiectasia. Clinical Genetics, 86(2), 123–133. 10.1111/cge.12269 [DOI] [PubMed] [Google Scholar]
- Trembath, R. C. , Thomson, J. R. , Machado, R. D. , Morgan, N. V. , Atkinson, C. , Winship, I. , … Morrell, N. W. (2001). Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. New England Journal of Medicine, 345(5), 325–334. 10.1056/NEJM200108023450503 [DOI] [PubMed] [Google Scholar]
- Vincent, P. , Plauchu, H. , Hazan, J. , Faure, S. , Weissenbach, J. , & Godet, J. (1995). A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Human Molecular Genetics, 4(5), 945–949. 10.1093/hmg/4.5.945 [DOI] [PubMed] [Google Scholar]
- Wooderchak‐Donahue, W. L. , McDonald, J. , Farrell, A. , Akay, G. , Velinder, M. , Johnson, P. , … Bayrak‐Toydemir, P. (2018). Genome sequencing reveals a deep intronic splicing ACVRL1 mutation hotspot in Hereditary Haemorrhagic Telangiectasia. Journal of Medical Genetics, 55(12), 824–830. 10.1136/jmedgenet-2018-105561 [DOI] [PubMed] [Google Scholar]
- Wooderchak‐Donahue, W. L. , McDonald, J. , O’Fallon, B. , Upton, P. D. , Li, W. , Roman, B. L. , … Bayrak‐Toydemir, P. (2013). BMP9 mutations cause a vascular‐anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. American Journal of Human Genetics, 93(3), 530–537. 10.1016/j.ajhg.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. S. , Yi, Y. , Peng, H. L. , Shen, J. K. , Xie, D. H. , & He, X. B. (2004). Clinical phenotypes, ALK1 gene mutation and level of related plasma proteins in Chinese hereditary hemorrhagic telangiectasia. Chin Med J (Engl), 117(6), 808–812. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials