

Abstract
Background
Preaxial polydactyly type IV, also referred as polysyndactyly, has been described in a few syndromes. We present three generations of a family with preaxial polydactyly type IV and other clinical features of Greig cephalopolysyndactyly syndrome (GCPS).
Methods and results
Sequencing analysis of the GLI3 coding region identified a novel donor splice site variant NC_000007.14(NM_000168.6):c.473+3A>T in the proband and the same pathogenic variant was subsequently identified in other affected family members. Functional analysis based on Sanger sequencing of the proband's complementary DNA (cDNA) sample revealed that the splice site variant c.473+3A>T disrupts the original donor splice site, thus leading to exon 4 skipping. Based on further in silico analysis, this pathogenic splice site variant consequently results in a truncated protein NP_000159.3:p.(His123Argfs*57), which lacks almost all functionally important domains. Therefore, functional cDNA analysis confirmed that the haploinsufficiency of the GLI3 is the cause of GCPS in the affected family members.
Conclusion
Despite the evidence provided, pathogenic variants in the GLI3 do not always definitely correlate with syndromic or nonsyndromic clinical phenotypes associated with this gene. For this reason, further transcriptomic and proteomic evaluation could be suggested.
Keywords: functional cDNA analysis, GLI3, Greig cephalopolysyndactyly syndrome, polysyndactyly, preaxial polydactyly type IV
1. INTRODUCTION
Polydactyly is one of the major types of hand and foot malformations. It can be associated with other clinical features as part of a syndrome or can occur as an isolated condition. Polydactylies manifest in a wide spectrum, from a normal number of digits with broad distal phalanges or extra bones to complete duplication of the digital ray.
According to the Temtamy and McKusick classification scheme, polydactyly can be divided into several types (Temtamy & McKusick, 1978). Postaxial polydactyly (MIM 174200; ORPHA 3047) presents with an extra digit on the ulnar or fibular side of the extremities and is subdivided into type A, which includes a well‐formed extra digit, and type B, which may present as small skin tag attached to the medial border of the fifth finger. Mesoaxial polydactyly includes additional fingers in the central part of the hand. Preaxial polydactyly is diagnosed when the extra digit is on the radial or tibial side of the extremities and consists of several types. In type I (MIM 174400; ORPHA 93339), only thumbs or halluxes are duplicated; type II (MIM 174500; ORPHA 93336) includes triphalangeal thumbs; and type III (MIM 174600; ORPHA 93337) is characterized by absent thumbs with one or two extra preaxial digits.
Preaxial polydactyly can also present with other types of digital malformations, most commonly syndactyly. Polysyndactyly is referred as preaxial polydactyly type IV (MIM 174700; ORPHA 93338). The feet are usually more severely affected, with complete duplication of the hallux and variable syndactyly of other digits, while in the hands, the thumbs may be broad or bifid and often accompanied by cutaneous syndactyly of other fingers and postaxial polydactyly type B. Type IV preaxial polydactyly is therefore sometimes referred as crossed polydactyly. These hand and foot malformations have been described in Greig cephalopolysyndactyly syndrome (GCPS; MIM 175700; ORPHA 380), Pallister‐Hall syndrome (PHS; MIM 146510; ORPHA 672), and Acrocallosal syndrome (MIM 200990; ORPHA 36).
Research has found that more than 100 genes are associated with human polydactyly, including the GLI3 (GLI‐Kruppel Family Member 3; MIM 165240) and SHH (Sonic hedgehog; MIM 600725) genes, which are the most reported (Xiang et al., 2017). The SHH‐GLI3 pathway is essentially important during early development of different tissues, including the neural tube, craniofacial structure, and limbs (Motoyama, 2006).
In this study, we present three generations of a family with type IV preaxial polydactyly and other clinical features of GCPS caused by novel splice site variant NC_000007.14(NM_000168.6):c.473+3A>T in the GLI3, the consequences and the possible pathomechanism of which were evaluated by functional gene analysis.
2. PATIENTS AND METHODS
2.1. Clinical evaluation of the patients
Patient 1 (IV‐1), a 6‐year‐old girl, was an only child of unrelated Lithuanian parents (Figure 1a). She was born from the first pregnancy, which was complicated by an imminent abortion during the seventh week. The proband was born at 39 weeks of gestation by normal delivery. At birth, her weight was 3,500 g (50th centile), her length was 52 cm (50–75th centile), and her Apgar score was 9 at 1 min and 10 at 5 min. The newborn was found to have an accessory finger attached with a skin bundle to the ulnar border of the fifth finger on both hands and asymmetrical complete cutaneous syndactyly. On the right hand, the proband had partial syndactyly of the second to fourth fingers (Figure 1b) and there was complete syndactyly of the second to fifth fingers of the left hand. Additionally, preaxial polydactyly of the feet and complete symmetrical syndactyly of toes 1–3 were observed (Figure 1c,d). Congenital anomalies of her hands were surgically corrected at the age of 1–4 years. Vision disorders were suspected when she reached the age of 4 years. She was diagnosed with congenital chorioretinitis, macular dystrophy, astygmatismus myopicus, and strabismus. During a physical examination at the age of 6 years, her head circumference was 55 cm (>97th centile), her height was 122 cm (50–75th centile), and her weight 21 kg (25–50th centile). The phenotype was remarkable for macrocephaly, deformities, and scars on hands after surgery, broad thumbs, and syndactyly of toes 1–3.
Figure 1.
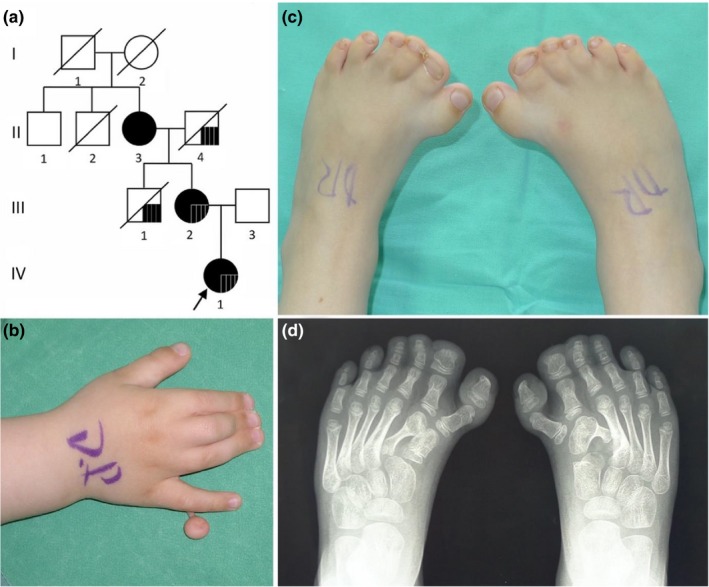
(a) Genealogy of the family. Black symbols denote patients with analyzed variants in GLI3. Black square in II‐4, III‐1, III‐2, IV‐1 family members denotes congenital chorioretinitis. (b, c) Photographs of hand and feet and (d) X‐ray of feet of the patient IV‐1 before the surgery at age of 1 year
Patient 2 (III‐2; mother of patient 1) was born from the first pregnancy by natural vaginal delivery (Figure 1a). Her birth weight was 4,600 g (>97th centile), and her length was 55 cm (97th centile). She had postaxial polydactyly type B on both hands, syndactyly of fingers 3–5 on the right hand and syndactyly of fingers 1–5 on the left hand. She also had syndactyly and preaxial polydactyly on her feet. The first surgical procedure was initiated at the age of 6 months and later surgical procedures were performed at 6‐month intervals. At age of 3 years, vision problems manifested and myopic astigmatism was diagnosed. At present, she is diagnosed with bilateral degeneration of the peripheral retina. Family history disclosed that this disorder was inherited from her father. Due to frequent headaches, a brain CT, which revealed mild hydrocephalus, was performed at age of 27 years. As measured during a physical examination that took place when she was 32 years of age, her head circumference was 61 cm (>97th centile), her height was 172 cm, and her weight was 75 kg. She has scars and deformations on her hands and feet after surgical treatment and syndactyly of the second and third digits of her feet. Furthermore, foot surgery caused iatrogenic pain.
Patient 3 (II‐3; grandmother of patient 1) was born from the first pregnancy, which had no complications (Figure 1a). Her birth weight was 4,000 g (90th centile) and she had polysyndactyly of her hands and feet. From the age of 6 months until 2 years of age, she underwent several surgical alterations of her hands. During her last physical examination at the age of 56 years, her head circumference was 57 cm (75th centile), her height was 164 cm, and her weight was 89 kg. She had scars on her hands from surgeries, camptodactyly of her fingers, broad thumbs, syndactyly of toes 1–4 and preaxial polydactyly of her feet.
2.2. Cell culture
A primary fibroblast cell line was obtained from patient II‐3's skin biopsy. The growing cell line was cultured in AmnioMAX C‐100 Basal Medium (Thermo Fisher Scientific), which was supplemented with AmnioMAX C‐100 Supplement (Thermo Fisher Scientific) and Amphotericin B (Gibco) according to the standard laboratory procedures for human cell cultures. The fibroblasts were cultured to the confluence of about 80%; adherent cells were then harvested. The final cell pellets were subsequently used for RNA isolation.
2.3. RNA extraction and reverse transcription reaction
Total RNA was isolated from the fibroblast cell line using RNeasy Mini Kit (Qiagen), while whole blood RNA was extracted using Tempus™ Blood RNA Tube and Tempus™ Spin RNA Isolation Kit (Thermo Fisher Scientific) according to the optimized manufacturers' protocols. Complementary DNA (cDNA) was synthesized from total RNA using a High‐Capacity RNA‐to‐cDNA Kit (Thermo Fisher Scientific) following manufacturer's protocol. Reverse transcription reactions were performed in a ProFlex polymerase chain reaction (PCR) system (Thermo Fisher Scientific) under the recommended conditions.
2.4. Polymerase chain reaction
Genomic DNA (gDNA) from the patient 1 (IV‐1), 2 (III‐2), 3 (II‐3), and healthy father (III‐3) of patient IV‐1 (Figure 1a) was isolated from peripheral blood leukocytes using standard phenol–chloroform extraction method.
Amplification using specific primers for the GLI3 was performed. Polymerase chain reaction primers were designed using the Primer3 ver0.4.0. (Whitehead Institute for Biomedical Research) and Primer‐BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) tools. Primers for the analysis of gDNA samples were designed to amplify a region spanning exons 4 and 14, while PCR amplifications of the cDNA sequence were performed using the same reverse primer designed on exon 5, but different forward primers, which were designed on the second to third exon junction and the third to fourth exon junction (Table S1). Polymerase chain reaction products were fractioned by agarose gel electrophoresis and visualized under UV light. The images were acquired using a gel documentation system with a transilluminator (E.A.S.Y. 442K, Herolab) and analyzed via E.A.S.Y. Win 32 image analysis software (Herolab).
2.5. Sanger sequencing
Sanger sequencing of the GLI3 gene was conducted for patient IV‐1. In addition, gDNA samples from three other available family members (III‐2, II‐3, III‐3) were analyzed by Sanger sequencing for family segregation analysis. In order to elucidate the pathogenicity of the detected donor splice site variant, functional analysis by cDNA Sanger sequencing was performed for patient II‐3, who has the same clinical features as patient IV‐1. Both strands of PCR products were sequenced with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). Capillary electrophoresis was carried out with an ABI3130xl Genetic Analyzer (Thermo Fisher Scientific). Fluorescent signals were analyzed with Sequence Analysis v5.1 software (Thermo Fisher Scientific). The sequences were aligned with the reference sequence of the GLI3 (NCBI: NM_000168.6) gene.
2.6. In silico analysis
In silico, Mutation Taster (http://www.mutationtaster.org/) and Human Splicing Finder (http://www.umd.be/HSF3/) databases were used for predicting splice site alterations. Possible variant's effect on GLI3 (UniProtKB: P10071) protein was predicted using different tools and databases storing information on the sequences and structures of proteins such as ExPASy Bioinformatics Resource Portal (https://www.expasy.org/), Pfam 32.0 database (https://pfam.xfam.org/), and UniProt (https://www.uniprot.org/).
3. RESULTS
Sanger sequencing analysis of the GLI3 coding region identified a novel heterozygous 5′ splice site NC_000007.14(NM_000168.6):c.473+3A>T variant and heterozygous missense NM_000168.6:c.2179G>A variant (NP_000159.3:p.(Gly727Arg); rs121917710) in patient IV‐1. Family segregation analysis subsequently revealed the splice site c.473+3A>T variant in the affected family members III‐2 and II‐3, while missense NM_000168.6:c.2179G>A variant was inherited from the healthy father of patient IV‐1 (Figure 1a).
The donor splice site variant c.473+3A>T was not described in the 1000 Genomes Project or ExAC database. Additionally, it was not found in our local whole exome database (98 exomes). In silico analysis with Human Splicing Finder predicted that this variant would most probably disrupt splicing of exon 4 to exon 5, and Mutation Taster suggested that the variant would lead to the loss of the splice‐donor site. In order to confirm the pathogenicity of the novel c.473+3A>T variant, functional analysis of patient II‐3's cDNA sample was performed. First, cDNA, synthesized from patient II‐3's RNA, which was extracted from a total blood sample, was analyzed. To avoid gDNA contamination, the primer pair was designed on the cDNA sequence to anneal on the junction of exons 3 and 4 (forward primer) and on exon 5 (reverse primer). The expected amplification fragment size of this primer pair was 166 nt in length (amplicon #3; Table S1). However, agarose gel electrophoresis did not fraction any PCR product in both patient II‐3's and control individual's sample, thus suggesting that this may be due to low GLI3 expression in the whole blood sample (Figure 2a). Further analysis of cDNA synthesized from patient II‐3's RNA sample extracted from a fibroblast cell line was performed. To get a longer amplicon, another forward primer annealing on the junction of exons 2 and 3 with the same reverse primer on exon 5 was used. The expected size of the amplification fragment of this primer pair was 419 nt (amplicon #4; Table S1). The results of gel electrophoresis revealed an amplification product of 166 nt (amplicon #3), thus confirming successful amplification of the patient's cDNA but not excluding the possibility of only wild type allele amplification. The forward primer of amplicon #3 was designed to be complementary to the junction of exons 3 and 4 in the cDNA. For this reason, if exon 4 was skipped, there would be no possibility for the mutated allele to be amplified. Further analysis of the longer amplicon #4, which is 419 nt in length, confirmed this assumption, because the mutated cDNA produced an additional amplicon of about 313 nt in length (Figure 2b). The additional band was about 106 nt shorter than the band of the wild type allele, thus corresponding to the length of exon 4.
Figure 2.
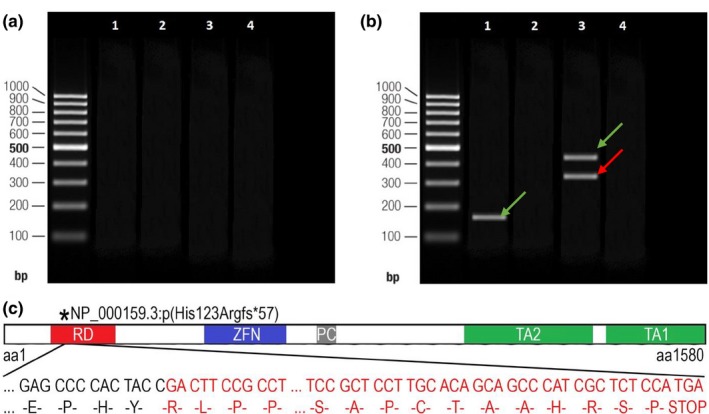
Polymerase chain reaction (PCR) amplifications of patient II‐3's cDNA to verify the splice site NC_000007.14(NM_000168.6):c.473+3A>T variant of the GLI3 visualized on agarose gel and the consequences of this variant. (a) Lanes 1–2 show the results of the amplification of amplicon #3 of cDNA synthesized from the RNA sample extracted from patient II‐3's total blood sample. Lanes 3–4 correspond to the amplification of amplicon #3 performed on the blood cDNA of the healthy control individual without c.473+3A>T variant (control sample). Lanes 2 and 4 correspond to the PCR reaction products of negative control. A 100 bp DNA ladder was used as the size marker. (b) Lanes 1–4 show the results of the amplification of cDNA synthesized from the RNA sample that was extracted from patient II‐3's fibroblast cell line. Lane 1 shows one band corresponding to a shorter amplicon (#3) of wild type allele of patient II‐3's cDNA sample (166 nt); lane 3 shows the two bands derived from the amplification of the cDNA of patient II‐3 (313 nt and 419 nt reflecting normal and mutated alleles [amplicon #4]); and lanes 2 and 4 correspond to the PCR reaction products of negative control. The green arrow indicates the amplicon of the wild type allele, while the red one indicates the amplicon of the mutated allele. (c) A schematic view of the truncated GLI3 protein and its functional domains (part of the frameshifted sequence of exon 5 has been omitted for clarity): the red square indicates the repressor domain (RD; aa106‐aa263), the blue square indicates the zinc finger domain (ZFN; aa462‐aa645), the gray square indicates the proteolytic cleavage site (PC; aa703‐aa740), and green squares indicate both transactivation domain 2 (TA2; aa1044‐aa1322) and transactivation domain 1 (TA1; 1376‐aa1580; according to Ito et al., 2018)
To verify these findings, PCR products were analyzed by standard Sanger sequencing. The analysis of the GLI3 coding sequence revealed that the donor splice site variant led to the skipping of exon 4. Bioinformatics analysis demonstrated that the alternative splicing is predicted to cause a translational frameshift of 57 amino acids and formation of premature termination codon, thus resulting in truncated protein NP_000159.3:p.(His123Argfs*57). According to the Pfam and UniProt database, truncated GLI3 (UniProtKB: P10071) protein lacks part of its repressor domain (RD; aa106‐aa263), zinc finger domain (ZFN; aa462‐aa645), proteolytic cleavage site (PC; aa703‐aa740), transactivation domain 2 (TA2; aa1044‐aa1322), and transactivation domain 1 (TA1; 1376‐aa1580; Figure 2c; Ito et al., 2018).
4. DISCUSSION
Pathogenic variants in the GLI3 manifest in several clinical phenotypes, including GCPS and PHS. The main clinical features of PHS are hypothalamic hamartoma, central and postaxial polydactyly, bifid epiglottis, imperforate anus, and renal abnormalities (Hall et al., 1980). Greig cephalopolysyndactyly syndrome is a distinct clinical entity, mainly characterized by preaxial polydactyly or mixed pre‐ and postaxial polydactyly, true widely spaced eyes, and macrocephaly (Biesecker, 2008).
The clinical features of affected individuals varied within our family. Two of three patients with GCPS had both polysyndactyly of their hands and feet and craniofacial abnormalities and fulfilled criteria suggested by Biesecker (2008). Craniofacial anomalies included macrocephaly in both patients and internal hydrocephalus in patient III‐2. Only isolated polysyndactyly (preaxial polydactyly type IV), but not macrocephaly, was observed in patient II‐3. Additionally, patient III‐2 displayed macrosomia at birth. Interestingly, Demurger et al. observed macrosomia in at least 13% of GCPS cases in their series (Demurger et al., 2015). Chorioretinitis in patients IV‐1 and III‐2 was inherited from patient III‐2's father, who is unaffected with GCPS and therefore not associated with the pathogenic variant of GLI3.
The GLI3 (UniProtKB: P10071) protein, a zinc finger transcription factor, is a downstream mediator of the SHH pathway, either activating or repressing the transcription of target genes of the SHH pathway during early development (Cohen, 2010). The balance between the full‐length GLI3 activator (GLI3A) and truncated repressor (GLI3R) is important for the regulation of digit number and identity (Litingtung, Dahn, Li, Fallon, & Chiang, 2002). The GLI3 protein contains several functional domains, including RD, ZFN, PC, TA2, and TA1 (Dai et al., 1999; Ito et al., 2018; Wang, Fallon, & Beachy, 2000). Multiple biological functions of GLI3 are related to the different phenotypes caused by variants in GLI3 (Biesecker & Johnston, 2005). To date, more than 200 different GLI3 variants have been reported on the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk). Most of these variants—including deletions, translocations, frameshifts and nonsense, missense, and splice site variants—cause GCPS, while the PHS mutational spectrum is smaller (Johnston et al., 2005). Manifestation of one or another syndrome depends on the mutation site in the gene. Pallister‐Hall syndrome is generally caused by truncating variants that are located in the middle third of the GLI3 and generate a constitutive repressor allele (Al‐Qattan, Shamseldin, Salih, & Alkuraya, 2017; Crapster, Hudgins, Chen, & Gomez‐Ospina, 2017; Johnston et al., 2010). At the same time, a greater spectrum of pathogenic variants located before 1998 nt and after 3481 nt of GLI3 cDNA cause loss of function and manifest in GCPS (Furniss, Critchley, Giele, & Wilkie, 2007; Johnston et al., 2010; Kalff‐Suske et al., 1999). The underlying mechanism of the influence of the location of the variants on different syndromes is still unknown (Ni et al., 2019). The haploinsufficiency of GLI3 is however proposed to be the main mechanism of the pathogenesis of GCPS. This suggestion has been confirmed by studies of an animal model, in which Gli3 loss of function in mice exhibits GCPS‐specific features: polydactyly and several craniofacial abnormalities (Quinn, Haaning, & Ware, 2012; Veistinen et al., 2012).
A considerable amount of pathogenic splice site variants affects mRNA translation, mainly by skipping the adjacent exon or misreading the affected intron (Acedo et al., 2012; Furuya & Nakatani, 2013). A pathogenic 5′ donor splice site variant usually retains an intron, while a variant in the 3′ acceptor splice site causes exon skipping. For example, FLCN studies confirmed that all acceptor splice site variants lead to exon skipping (Furuya et al., 2018). There are, however, some studies contradicting the statement about the consequences of 5′ splice site variants (Diederichs et al., 2016). Two independent studies demonstrated that the donor splice site variants lead to the skipping of exon 10 of the CETP (Sakai, Santamarina‐Fojo, Yamashita, Matsuzawa, & Brewer, 1996) and the skipping of exon 3 of the FGA (Attanasio, David, & Neerman‐Arbez, 2003). Similarly, the functional analysis of patient II‐3's cDNA sample demonstrated that the novel splice site NC_000007.14(NM_000168.6):c.473+3A>T variant, which cosegregates with disease in the family, led to the skipping of exon 4 of the GLI3. In silico, the alternative splicing is predicted to result in the truncated protein NP_000159.3:p.(His123Argfs*57), which lacks part of the RD domain, as well as the ZFN, PC, and two TA domains (Figure 2c). The loss of the ZFN is suggested to be critical for the GCPS phenotypic manifestation, because this domain is fundamentally important for gene transcription, translation, cytoskeleton organization, epithelial development, cell adhesion, protein folding, and chromatin remodeling during early development (Laity, Lee, & Wright, 2001). The ZFN, which is highly conserved between evolutionary distinct species, is especially important for limb development (Al‐Qattan et al., 2017; Crapster et al., 2017).
The second variant, c.2179G>A (Gly727Arg), detected in patient IV‐1 has been inherited from her healthy father. This missense variant was reported by Radhakrishna et al. (1999). In four generations of the family, eight individuals were affected with dominant postaxial polydactyly type A and type B, but none of them had syndactyly or any recognizable cranial anomaly (Radhakrishna et al., 1999). More recent data demonstrate that variant c.2179G>A has been observed in healthy individuals (the allele frequency in the 1000 Genomes Project—0.003, ExAC—0.006, our local whole exome database—2/75), and it is therefore likely that this variant has no deleterious effect on the function of the GLI3 protein.
Based on these findings and the clinical features of the affected individuals of our family, a novel heterozygous 5′ splice site NC_000007.14(NM_000168.6):c.473+3A>T variant resulting in haploinsufficiency of the GLI3 causes GCPS syndrome. Despite the evidence that is provided, pathogenic variants in the GLI3 do not always definitely correlate with syndromic (GCPS, PHH) or nonsyndromic clinical phenotypes associated with this gene. For this reason, further transcriptomic and proteomic evaluation could be suggested.
CONFLICTS OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
We are very thankful to the family for their contribution. This research was funded by a grant (No. S‐MIP‐17‐19/LSS‐150000‐1179) from the Research Council of Lithuania.
Siavrienė E, Mikštienė V, Radzevičius D, et al. Novel GLI3 variant causes Greig cephalopolysyndactyly syndrome in three generations of a Lithuanian family. Mol Genet Genomic Med. 2019;7:e878 10.1002/mgg3.878
REFERENCES
- Acedo, A. , Sanz, D. J. , Duran, M. , Infante, M. , Perez‐Cabornero, L. , Miner, C. , & Velasco, E. A. (2012). Comprehensive splicing functional analysis of DNA variants of the BRCA2 gene by hybrid minigenes. Breast Cancer Research, 14, R87 10.1186/bcr3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Qattan, M. M. , Shamseldin, H. E. , Salih, M. A. , & Alkuraya, F. S. (2017). GLI3‐related polydactyly: A review. Clinical Genetics, 92(5), 457–466. 10.1111/cge.12952 [DOI] [PubMed] [Google Scholar]
- Attanasio, C. , David, A. , & Neerman‐Arbez, M. (2003). Outcome of donor splice site mutations accounting for congenital afibrinogenemia reflects order of intron removal in the fibrinogen alpha gene (FGA). Blood, 101, 1851–1856. 10.1182/blood-2002-03-0853 [DOI] [PubMed] [Google Scholar]
- Biesecker, L. G. (2008). The Greig cephalopolysyndactyly syndrome. Orphanet Journal of Rare Diseases, 3, 10 10.1186/1750-1172-3-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker, L. G. , & Johnston, J. (2005). Syndromic and non‐syndromic GLI3 phenotypes. Clinical Genetics, 68, 284 10.1111/j.1399-0004.2005.0485a.x [DOI] [PubMed] [Google Scholar]
- Cohen, M. M. Jr (2010). Hedgehog signaling update. American Journal of Medical Genetics Part A, 152A, 1875–1914. 10.1002/ajmg.a.32909 [DOI] [PubMed] [Google Scholar]
- Crapster, J. A. , Hudgins, L. , Chen, J. K. , & Gomez‐Ospina, N. (2017). A novel missense variant in the GLI3 zinc finger domain in a family with digital anomalies. American Journal of Medical Genetics Part A, 173(12), 3221–3225. 10.1002/ajmg.a.38415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, P. , Akimaru, H. , Tanaka, Y. , Maekawa, T. , Nakafuku, M. , & Ishii, S. (1999). Sonic Hedgehog‐induced activation of the Gli1 promoter is mediated by GLI3. The Journal of Biological Chemistry, 274, 8143–8152. 10.1074/jbc.274.12.8143 [DOI] [PubMed] [Google Scholar]
- Demurger, F. , Ichkou, A. , Mougou‐Zerelli, S. , Le Merrer, M. , Goudefroye, G. , Delezoide, A. L. , … Attie‐Bitach, T. (2015). New insights into genotype‐phenotype correlation for GLI3 mutations. European Journal of Human Genetics, 23(1), 92–102. 10.1038/ejhg.2014.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs, S. , Bartsch, L. , Berkmann, J. C. , Frose, K. , Heitmann, J. , Hoppe, C. , … Wullenkord, R. (2016). The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non‐coding RNA and synonymous mutations. EMBO Molecular Medicine, 8(5), 442–457. 10.15252/emmm.201506055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furniss, D. , Critchley, P. , Giele, H. , & Wilkie, A. O. (2007). Nonsense‐mediated decay and the molecular pathogenesis of mutations in SALL1 and GLI3. American Journal of Medical Genetics Part A, 143A, 3150–3160. 10.1002/ajmg.a.32097 [DOI] [PubMed] [Google Scholar]
- Furuya, M. , Kobayashi, H. , Baba, M. , Ito, T. , Tanaka, R. , & Nakatani, Y. (2018). Splice‐site mutation causing partial retention of intron in the FLCN gene in Birt‐Hogg‐Dube syndrome: A case report. BMC Medical Genomics, 11(1), 42 10.1186/s12920-018-0359-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya, M. , & Nakatani, Y. (2013). Birt‐Hogg‐Dube syndrome: Clinicopathological features of the lung. Journal of Clinical Pathology, 66(3), 178–186. 10.1136/jclinpath-2012-201200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J. G. , Pallister, P. D. , Clarren, S. K. , Beckwith, J. B. , Wiglesworth, F. W. , Fraser, F. C. , … Reed, S. D. (1980). Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly—A new syndrome? Part I: Clinical, causal, and pathogenetic considerations. American Journal of Medical Genetics, 7(1), 47–74. 10.1002/ajmg.1320070110 [DOI] [PubMed] [Google Scholar]
- Ito, S. , Kitazawa, R. , Haraguchi, R. , Kondo, T. , Ouchi, A. , Ueda, Y. , & Kitazawa, S. (2018). Novel GLI3 variant causing overlapped Greig cephalopolysyndactyly syndrome (GCPS) and Pallister‐Hall syndrome (PHS) phenotype with agenesis of gallbladder and pancreas. Diagnostic Pathology, 13(1), 1 10.1186/s13000-017-0682-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , Olivos‐Glander, I. , Killoran, C. , Elson, E. , Turner, J. T. , Peters, K. F. , … Biesecker, L. G. (2005). Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister‐Hall syndromes: Robust phenotype prediction from the type and position of GLI3 mutations. American Journal of Medical Genetics, 76(4), 609–622. 10.1086/429346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , Sapp, J. C. , Turner, J. T. , Amor, D. , Aftimos, S. , Aleck, K. A. , … Biesecker, L. G. (2010). Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Human Mutation, 31(10), 1142–1154. 10.1002/humu.21328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalff‐Suske, M. , Wild, A. , Topp, J. , Wessling, M. , Jacobsen, E. M. , Bornholdt, D. , … Grzeschik, K. H. (1999). Point mutations throughout the GLI3 gene cause Greig cephalopolysyndactyly syndrome. Human Molecular Genetics, 8(9), 1769–1777. 10.1093/hmg/8.9.1769 [DOI] [PubMed] [Google Scholar]
- Laity, J. H. , Lee, B. M. , & Wright, P. E. (2001). Zinc finger proteins: New insights into structural and functional diversity. Current Opinion in Structural Biology, 11, 39–46. 10.1016/s0959-440x(00)00167-6 [DOI] [PubMed] [Google Scholar]
- Litingtung, Y. , Dahn, R. D. , Li, Y. , Fallon, J. F. , & Chiang, C. (2002). Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature, 418, 979–983. 10.1038/nature01033 [DOI] [PubMed] [Google Scholar]
- Motoyama, J. (2006). Essential roles of Gli3 and sonic hedgehog in pattern formation and developmental anomalies caused by their dysfunction. Congenital Anomalies, 46(3), 123–128. 10.1111/j.1741-4520.2006.00114.x [DOI] [PubMed] [Google Scholar]
- Ni, F. , Han, G. , Guo, R. , Cui, H. , Wang, B. , & Li, Q. (2019). A Novel frameshift mutation of GLI3 causes isolated postaxial polydactyly. Annals of Plastic Surgery, 82, 570–573. 10.1097/SAP.0000000000001685 [DOI] [PubMed] [Google Scholar]
- Quinn, M. E. , Haaning, A. , & Ware, S. M. (2012). Preaxial polydactyly caused by Gli3 haploinsufficiency is rescued by Zic3 loss of function in mice. Human Molecular Genetics, 21(8), 1888–1896. 10.1093/hmg/dds002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna, U. , Bornholdt, D. , Scott, H. S. , Patel, U. C. , Rossier, C. , Engel, H. , … Antonarakis, S. E. (1999). The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type‐IV and postaxial polydactyly type‐A/B; no phenotype prediction from the position of GLI3 mutations. The American Journal of Human Genetics, 65(3), 645–655. 10.1086/302557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, N. , Santamarina‐Fojo, S. , Yamashita, S. , Matsuzawa, Y. , & Brewer, H. B. Jr (1996). Exon 10 skipping caused by intron 10 splice donor site mutation in cholesteryl ester transfer protein gene results in abnormal downstream splice site selection. Journal of Lipid Research, 37, 2065–2073. [PubMed] [Google Scholar]
- Temtamy, S. A. , & McKusick, V. A. (1978). The genetics of hand malformations. Birth Defects Original Article Series, 4(3), i–xviii,1–619. [PubMed] [Google Scholar]
- Veistinen, L. , Takatalo, M. , Tanimoto, Y. , Kesper, D. A. , Vortkamp, A. , & Rice, D. P. (2012). Loss‐of‐function of Gli3 in mice causes abnormal frontal bone morphology and premature synostosis of the interfrontal suture. Frontiers in Physiology, 3, 121 10.3389/fphys.2012.00121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Fallon, J. F. , & Beachy, P. A. (2000). Hedgehog‐regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell, 100, 423–434. 10.1016/s0092-8674(00)80678-9 [DOI] [PubMed] [Google Scholar]
- Xiang, Y. , Jiang, L. , Wang, B. , Xu, Y. , Cai, H. , & Fu, Q. (2017). Mutational screening of GLI3, SHH, preZRS, and ZRS in 102 Chinese children with nonsyndromic polydactyly. Developmental Dynamics, 246(5), 392–402. 10.1002/dvdy.24488 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials