

Abstract
Background
Intellectual disability (ID) is a feature of many rare diseases caused by thousands of genes. This genetic heterogeneity implies that pathogenic variants in a specific gene are found only in a small number of patients, and difficulties arise in the definition of prevailing genotype and characteristic phenotype associated with that gene. One of such very rare disorders is autosomal recessive ID type 66 (OMIM #618221) caused by defects in C12orf4. Up to now, six families have been reported with mostly truncating variants. The spectrum of the clinical phenotype was not emphasized in previous reports, and detailed phenotype was not always available from previous patients, especially from large cohort studies.
Methods
Exome sequencing was performed in a consanguineous Armenian family with two affected adult brothers.
Results
The patients carry a novel homozygous nonsense C12orf4 variant. The integration of previous data and phenotyping of the brothers indicate that the clinical picture of C12orf4 defects involves hypotonia in infancy, rather severe ID, speech impairment, and behavioral problems such as aggressiveness, unstable mood, and autistic features. Several other symptoms are more variable and less consistent.
Conclusion
This rather nonsyndromic and nonspecific clinical picture implies that additional patients with C12orf4 defects will likely continue to be identified using the “genotype‐first” approach, rather than based on clinical assessment. The phenotype needs further delineation in future reports.
Keywords: C12orf4, consanguinity, exome sequencing, intellectual disability
1. INTRODUCTION
Intellectual disability (ID) has a general prevalence exceeding 1%, and it represents a significant health and social burden (Maulik, Mascarenhas, Mathers, Dua, & Saxena, 2011). Due to the power of whole‐genome analysis methods, the number of genes associated with ID has been rapidly increasing in the recent past. The number of genes involved in autosomal recessive forms of ID (ARID) alone is estimated to be more than 2,000 (Jamra, 2018). The large genetic heterogeneity of monogenic forms of ID implies that only a very small number of patients carry causal variants in a specific gene. Each new case is thus valuable for better definition of the prevailing genotype (type of causal variants) and characteristic phenotype associated with that gene.
The C12orf4 gene was first suggested as a possible candidate gene for ARID in one Arab family in 2015 (Alazami et al., 2015). Clinical pictures of additional patients from a large Finnish pedigree and a Dutch family (Philips et al., 2017), and from a Turkish family (Reuter et al., 2017) were published later. Based on these reports the condition was listed in OMIM as autosomal recessive ID type 66 (#618221). Since then two additional families with affected individuals have been reported (Harripaul et al., 2018; Maddirevula et al., 2018), increasing the number of ARID families explained by variants in C12orf4 to six. All families were consanguineous, and most of the homozygous C12orf4 variants in affected individuals were truncating.
Here, we report an Armenian family where two affected sons of second‐cousin parents are homozygous for a novel nonsense variant in exon 4 of the C12orf4 gene. The brothers suffer from profound ID and show behavioral disturbances, in the absence of other significant symptoms. The phenotype information on patients identified in large cohort studies is often very limited (Harripaul et al., 2018; Maddirevula et al., 2018; Reuter et al., 2017). The present report contributes to the delineation of the genotype–phenotype correlation in this rare entity.
2. CASE PRESENTATION
The patients were two older sons of a healthy consanguineous Armenian couple (second cousins) who had three children. The youngest son in the family was unaffected. The mother and the farther were also unaffected (they had undergraduate and graduate education, respectively), and they had four and three siblings, respectively, each of whom had two to three unaffected children. Also the rest of the family had no history of cognitive or behavioral impairment. Karyotyping and SNP array analysis of the patients showed normal male karyotypes without any significant copy number variants.
The eldest brother (Patient Arm‐F5‐1, Figure 1a–c) is currently 28 years old. He was born from a normal pregnancy in the 38th gestational week with a weight of 3,900 g (75th centile) and length of 51 cm (62nd centile); head circumference (HC) at birth is unknown. He had no neonatal complications except unilateral cryptorchidism (which was surgically recovered at the age of 5 years). The boy showed hypotonia but no delay of motor development. He was able to sit without support at 6 months and walk at 1 year and 4 months of age, and he was able to talk since 1 year and 4 months of age. Prior to school age (7 years) psychomotor delay was not marked. However, the boy could not attend regular school due to learning difficulties and inadequate behavior, and he enrolled a special school at the age of 8 years. Medical examination at the age of 19 years revealed a weight of 65 kg (32nd centile), height of 186 cm (91st centile), and HC of 60.2 cm (>99th centile, +3.6 SD). Facial features of the patient included elongated triangular face with prominent frontal bossing, widow's peak, deep‐set small eyes, short eyebrows, narrow nasal base, short philtrum, narrow jaw, open mouth appearance, micrognathia, and protruding ears. His sexual development was normal. Ophthalmological and audiological examinations yielded no abnormal findings. Neurological examination revealed a delay of fine motor development and behavioral disturbances (unmotivated laugh and grimace). Psychiatric examination showed profound ID (IQ score of 16 assessed using SON‐R nonverbal test (Tellegen & Laros, 1993)). EEG analysis detected intracranial hypertension and a disorganized EEG pattern specific for delay with respect to the age: nonregular alpha rhythm, diffuse prevalence of beta rhythm, and synchronous theta‐beta bursts. When the boy was 21 the family moved abroad. This revealed his limited adaptive abilities and induced aggressiveness which disappeared when the family returned home. At the last examination at the age of 28 years the weight of the patient was 70 kg (52nd centile), height was 197 cm (>99th centile), and HC was 60.5 cm (>99th centile, +3.8 SD). His constitution was asthenic and his gait was normal. He had no eye contact and was shy, but had good memory, liked listening to music, and showed no aggressiveness. He did not attend any specialized center and showed very limited activity, mostly sitting in forward position with hands folded in his lap.
Figure 1.
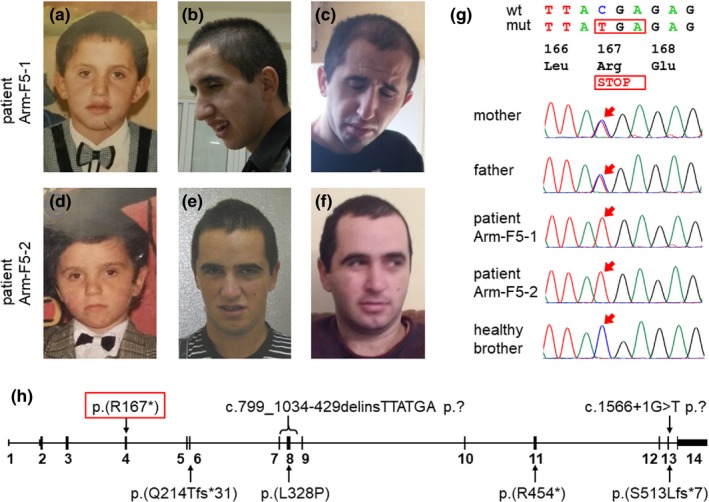
(a)–(f) Development of the facial phenotype of the patients with age: (a)–(c) Patient Arm‐F5‐1 at 7, 19, and 28 years of age; (d)–(f) Patient Arm‐F5‐2 at 7, 17.5, and 27 years of age. While the phenotype of both patients at a young age and of Patient Arm‐F5‐2 till young adulthood was not remarkable, Patient Arm‐F5‐1 developed an appearance with elongated triangular face, frontal bossing, deep‐set small eyes, narrow nasal base, short philtrum, narrow jaw, and micrognathia. (g) Sanger sequencing of the C12orf4 variant in the family. Wild type (wt) and mutated (mut) sequences and their protein translation are shown; arrows point to the mutated position. (h) Schematics of the C12orf4 gene with homozygous variants found in ARID families. The gene has 14 exons (13 coding) shown as black rectangles. The variant from the current family is framed in red. The exact effect of two variants at the protein level is unknown
His affected brother (Patient Arm‐F5‐2, Figure 1d‐f) is 27 years old. He was born from a normal pregnancy in the 39th gestational week with a weight of 3,800 g (69th centile), length of 50 cm (48th centile), and unknown HC. With the exception of hypotonia no neonatal complications were observed. He was able to sit without support at 6 months of age, started walking before 2 years of age, and could speak (although not clearly) at 2 years of age. Similarly to his older brother, he enrolled a special school at the age of 8 years. At examination at the age of 17.5 years his weight was 61 kg (35th centile), height was 171 cm (34th centile), and HC was 62 cm (>99th centile, +4.8 SD). He had no significant facial dysmorphic features and no ophthalmological or audiological abnormalities. His sexual development was normal. He suffered from profound ID (IQ of 19, SON‐R nonverbal test) and showed behavioral disturbances (arms swinging). EEG analysis revealed delay of cerebral rates, reduction of corticospinal activity, and intracranial hypertension. Similarly to his older brother, the stay abroad induced behavioral changes (namely aggressiveness; also, when he was 23 he did not get up from bed and did not eat for one month) which normalized after return. He started to attend a special center for ID patients and was reported to have a communicative and friendly personality. At the last examination at the age of 27 years the weight was 86 kg (86th centile), his height was 173 cm (31st centile), and HC was 62 cm (>99th centile, +4.8 SD). He had not been talking with others for 2 years and had periods of anorexia lasting up to 1 month. He expressed aggressive behavior which quickly changed to happiness and friendliness with no shyness, talked to himself and showed stereotypic movements. He was active, had normal gait, and could eat independently. He liked watching movies and fell asleep very late.
3. GENETIC ANALYSIS
The study was approved by the local ethics committees of the participating institutions, and informed consent was obtained from the family. Exome sequencing was performed on blood DNA samples of the parents and their three sons. SeqCap EZ MedExome Probes (Roche NimbleGen, Madison, WI, USA) were used for exome capture. The library construction, paired‐end sequencing, data processing, data analysis, and variant prioritization were performed as described previously (Prchalova et al., 2017). A homozygous truncating variant in C12orf4 exon 4 (chr12:4639042G>A (hg19) NM_020374.4:c.499C>T p.(Arg167*)) was identified in both patients. The parents were heterozygous and the unaffected youngest brother was homozygous for the normal allele. The results were confirmed using Sanger sequencing (Figure 1g) with the BigDye Terminator v3.1 Cycle Sequencing Kit and an ABI 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). No additional significant variants could be detected in the family.
4. DISCUSSION
Up to now, homozygous C12orf4 variants have been found in patients from six families including one large Finnish pedigree. Most of the variants were truncating: one nonsense variant (Reuter et al., 2017), two frameshift variants (Alazami et al., 2015; Harripaul et al., 2018), one splice donor variant (Maddirevula et al., 2018), and one genomic rearrangement affecting two coding exons (Philips et al., 2017); and only one variant was a missense change (Philips et al., 2017) (Table S1). The nonsense C12orf4 variant identified in the current family fits the prevailing variant spectrum; in fact, the variant is the most 5´ located truncating change among all variants (Figure 1h), and its effect may be among the most deleterious. All variants reported in patients are absent or are very rare in the gnomAD database, and they are never reported there in homozygous state. The C12orf4 gene seems to have lower tolerance to loss‐of‐function variation (31.4 variants expected, 14 observed), and among all loss‐of‐function and missense variants, just two, p.(Ile361Thr) and p.(Asp160Gly), are present in one homozygote each; otherwise no homozygotes are listed in gnomAD for any such variant (Lek et al., 2016). The low population frequency of deleterious C12orf4 variants is in accord with the fact that all seven families with pathogenic C12orf4 variants are consanguineous.
The phenotypes of individuals with biallelic C12orf4 defects include predominantly ID, usually at the more severe end of the spectrum, speech impairment, behavioral problems (such as aggressiveness, mood swings, and autistic features), and hypotonia in infancy. Other features such as variable facial dysmorphism, rigid gait, sleep problems, and hyperlaxity of joints are present much less consistently (Table S1). Both our patients showed macrocephaly but this feature may be familial (HC of their unaffected father and mother was 58.5 cm (>99th centile, +2.4 SD) and 56 cm (94th centile), respectively, while the unaffected youngest brother had HC of 54 cm (90th centile)). Also the previous patients with C12orf4 defects from whom sufficient clinical information was available did not show macrocephaly (Alazami et al., 2015; Maddirevula et al., 2018; Philips et al., 2017). Taken together, this rather nonsyndromic nature of the condition and its nonspecific clinical picture imply that additional patients will likely continue to be identified using the “genotype‐first” approach rather than based on clinical assessment.
The function of the C12orf4 gene and the mechanism how its defects cause ARID remain unknown. The product of the rat C12orf4 homologue has been found to be involved in mast cell degranulation (Mazuc et al., 2014), and an intronic SNP in C12orf4 has been associated with serum phosphorus levels in humans (Kestenbaum et al., 2010). Two rearrangements producing fusion genes of C12orf4 with another gene have been reported in cancer (Rudin et al., 2012; Yoshihara et al., 2015), a condition known to overlap in genetic etiology with neurodevelopmental disorders (Crawley, Heyer, & LaSalle, 2016). In the absence of functional data on C12orf4 itself, clues could be gained from its interaction partners. Interestingly, one of them is TBCK (Hein et al., 2015), the defects of which cause another rare type of ARID (Bhoj et al., 2016; Chong et al., 2016; Guerreiro et al., 2016), and the product of which is a TBC1 domain‐containing kinase involved in the regulation of mTOR signaling and control of cell proliferation and cytoskeleton. Similarly another C12orf4 interaction partner, PTPN23 (Hein et al., 2015), is an ARID candidate gene (Alazami et al., 2015; Hu et al., 2019), the product of which, a nonreceptor protein tyrosine phosphatase, may be involved in mRNA splicing, vesicular transport and regulation of cell proliferation by interacting with the Ras pathway. These interactions may indicate participation of C12orf4 in cellular pathways contributing to intellectual functioning. This together with the growing number of patients with biallelic defects in C12orf4 support the role of this gene in ARID.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank the family of the patients for cooperation. The work was supported by grant 17‐29423A from the Ministry of Health of the Czech Republic.
Hancarova M, Babikyan D, Bendova S, et al. A novel variant of C12orf4 in a consanguineous Armenian family confirms the etiology of autosomal recessive intellectual disability type 66 with delineation of the phenotype. Mol Genet Genomic Med. 2019;7:e865 10.1002/mgg3.865
Miroslava Hancarova and Davit Babikyan should be considered joint first author.
Funding information
Ministry of Health of the Czech Republic, Grant/Award Number: 17‐29423A.
REFERENCES
- Alazami, A. M. , Patel, N. , Shamseldin, H. E. , Anazi, S. , Al‐Dosari, M. S. , Alzahrani, F. , … Alkuraya, F. S. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole‐exome sequencing of prescreened multiplex consanguineous families. Cell Reports, 10(2), 148–161. 10.1016/j.celrep.2014.12.015 [DOI] [PubMed] [Google Scholar]
- Bhoj, E. J. , Li, D. , Harr, M. , Edvardson, S. , Elpeleg, O. , Chisholm, E. , … Hakonarson, H. (2016). Mutations in TBCK, encoding TBC1‐domain‐containing kinase, lead to a recognizable syndrome of intellectual disability and hypotonia. American Journal of Human Genetics, 98(4), 782–788. 10.1016/j.ajhg.2016.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. X. , Caputo, V. , Phelps, I. G. , Stella, L. , Worgan, L. , Dempsey, J. C. , … Doherty, D. (2016). Recessive inactivating mutations in TBCK, encoding a Rab GTPase‐activating protein, cause Severe infantile syndromic encephalopathy. American Journal of Human Genetics, 98(4), 772–781. 10.1016/j.ajhg.2016.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley, J. N. , Heyer, W. D. , & LaSalle, J. M. (2016). Autism and cancer share risk genes, pathways, and drug targets. Trends in Genetics, 32(3), 139–146. 10.1016/j.tig.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro, R. J. , Brown, R. , Dian, D. , de Goede, C. , Bras, J. , & Mole, S. E. (2016). Mutation of TBCK causes a rare recessive developmental disorder. Neurology Genetics, 2(3), e76 10.1212/NXG.0000000000000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harripaul, R. , Vasli, N. , Mikhailov, A. , Rafiq, M. A. , Mittal, K. , Windpassinger, C. , … Vincent, J. B. (2018). Mapping autosomal recessive intellectual disability: Combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Molecular Psychiatry, 23(4), 973–984. 10.1038/mp.2017.60 [DOI] [PubMed] [Google Scholar]
- Hein, M. Y. , Hubner, N. C. , Poser, I. , Cox, J. , Nagaraj, N. , Toyoda, Y. , … Mann, M. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell, 163(3), 712–723. 10.1016/j.cell.2015.09.053 [DOI] [PubMed] [Google Scholar]
- Hu, H. , Kahrizi, K. , Musante, L. , Fattahi, Z. , Herwig, R. , Hosseini, M. , … Najmabadi, H. (2019). Genetics of intellectual disability in consanguineous families. Molecular Psychiatry, 24(7), 1027–1039. 10.1038/s41380-017-0012-2 [DOI] [PubMed] [Google Scholar]
- Jamra, R. (2018). Genetics of autosomal recessive intellectual disability. Medizinische Genetik, 30(3), 323–327. 10.1007/s11825-018-0209-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestenbaum, B. , Glazer, N. L. , Köttgen, A. , Felix, J. F. , Hwang, S.‐J. , Liu, Y. , … Fox, C. S. (2010). Common genetic variants associate with serum phosphorus concentration. Journal of the American Society of Nephrology, 21(7), 1223–1232. 10.1681/ASN.2009111104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddirevula, S. , Alzahrani, F. , Al‐Owain, M. , Al Muhaizea, M. A. , Kayyali, H. R. , AlHashem, A. , … Alkuraya, F. S. (2018). Autozygome and high throughput confirmation of disease genes candidacy. Genetics in Medicine, 21(3), 736–742. 10.1038/s41436-018-0138-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik, P. K. , Mascarenhas, M. N. , Mathers, C. D. , Dua, T. , & Saxena, S. (2011). Prevalence of intellectual disability: A meta‐analysis of population‐based studies. Research in Developmental Disabilities, 32(2), 419–436. 10.1016/j.ridd.2010.12.018 [DOI] [PubMed] [Google Scholar]
- Mazuc, E. , Guglielmi, L. , Bec, N. , Parez, V. , Hahn, C. S. , Mollevi, C. , … Martineau, P. (2014). In‐cell intrabody selection from a diverse human library identifies C12orf4 protein as a new player in rodent mast cell degranulation. PLoS ONE, 9(8), e104998 10.1371/journal.pone.0104998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips, A. K. , Pinelli, M. , de Bie, C. I. , Mustonen, A. , Maatta, T. , Arts, H. H. , … Jarvela, I. (2017). Identification of C12orf4 as a gene for autosomal recessive intellectual disability. Clinical Genetics, 91(1), 100–105. 10.1111/cge.12821 [DOI] [PubMed] [Google Scholar]
- Prchalova, D. , Havlovicova, M. , Sterbova, K. , Stranecky, V. , Hancarova, M. , & Sedlacek, Z. (2017). Analysis of 31‐year‐old patient with SYNGAP1 gene defect points to importance of variants in broader splice regions and reveals developmental trajectory of SYNGAP1‐associated phenotype: Case report. BMC Medical Genetics, 18(1), 62 10.1186/s12881-017-0425-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter, M. S. , Tawamie, H. , Buchert, R. , Hosny Gebril, O. , Froukh, T. , Thiel, C. , … Abou Jamra, R. (2017). Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry, 74(3), 293–299. 10.1001/jamapsychiatry.2016.3798 [DOI] [PubMed] [Google Scholar]
- Rudin, C. M. , Durinck, S. , Stawiski, E. W. , Poirier, J. T. , Modrusan, Z. , Shames, D. S. , … Seshagiri, S. (2012). Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small‐cell lung cancer. Nature Genetics, 44(10), 1111–1116. 10.1038/ng.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellegen, P. , & Laros, J. (1993). The construction and validation of a nonverbal test of intelligence: The revision of the Snijders‐Oomen tests. European Journal of Psychological Assessment, 9(2), 147–157. [Google Scholar]
- Yoshihara, K. , Wang, Q. , Torres‐Garcia, W. , Zheng, S. , Vegesna, R. , Kim, H. , & Verhaak, R. G. (2015). The landscape and therapeutic relevance of cancer‐associated transcript fusions. Oncogene, 34(37), 4845–4854. 10.1038/onc.2014.406 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials