

Abstract
Background
Angelman Syndrome (AS) is a neurodevelopmental disorder with core features of intellectual disability, speech impairment, movement disorders, and a unique behavioral profile. Typically, AS results from absent maternal expression of UBE3A, but some individuals have imprinting defects in a portion of their cells. These individuals are mosaic for normal and defective UBE3A expression, resulting in mosaic AS (mAS) with a partial loss of gene expression.
Methods
This study aims to contrast the mAS phenotype to that of AS. Clinical characteristics of mAS were obtained from a parental survey of 22 mAS patients and from the Angelman Natural History study. These were contrasted with those of AS using historical data.
Results
Developmental delay was present in nearly all mAS patients, whereas the core features of AS were reported in less than 40%. While language and ability to manage activities of daily living were markedly improved over that expected in AS, mAS patients demonstrated a high incidence of behavioral challenges.
Conclusion
Clinical work‐up of an individual with developmental delay, hyperactivity, anxiety, and an uncharacteristically happy demeanor should prompt methylation studies to rule out mAS. We expand the phenotypic spectrum of AS to include features that overlap with Prader‐Willi such as hyperphagia.
Keywords: Angelman syndrome, epilepsy, imprinting, mosaic Angelman syndrome, mosaicism
What this paper adds.
We expand the clinical phenotype of individuals with AS.
Specific SNRPN methylation testing should be considered in individuals with developmental delay, happy demeanor, and behavioral concerns even in the absence of seizures and presence of speech.
Anxiety and ADHD remain significant challenges for mAS patients.
1. INTRODUCTION
Angelman Syndrome (AS) (OMIM #105830) is a neurodevelopmental disorder with clinical features characterized by severe developmental delay/intellectual disability, speech impairment, movement disorders or ataxia, and a unique behavioral profile with an excitable, smiling, and happy demeanor. In addition, the majority of AS patients are noted to be microcephalic by 2 years of age and exhibit epilepsy and characteristically abnormal electroencephalogram (EEG) patterns (Fiumara, Pittala, Cocuzza, & Sorge, 2010; Saitoh, 2015). Epilepsy in AS is common and may be particularly medication resistant (Thibert et al., 2009).
AS was first linked to chromosome abnormalities at 15q11.2‐q13 (Magenis, Brown, Lacy, Budden, & LaFranchi, 1987) with further refinement leading to association with the UBE3A gene (OMIM #601623) (Kishino, Lalande, & Wagstaff, 1997). UBE3A is one of a handful of genes which are imprinted in the central nervous system. Imprinting occurs via regulation at the imprinting control center located within the SNRPN (OMIM #182279) transcript that prevents the maternal allele from re‐imprinting to indicate maternal inheritance (Buiting et al., 1995; Glenn, Porter, Jong, Nicholls, & Driscoll, 1993; Özçelik et al., 1992; Reed & Leff, 1994). Thus, deficient expression of the maternal copy of UBE3A results in loss of the UBE3A protein and AS. Mutations in chromosome 15q11‐q13 are mediated by nonhomologous recombination, de novo in the majority of cases, and are classified based upon the size of the deletion. Type I deletions are the larger of the two common deletions (approximately 6 megabases [Mb] in size) and are clinically associated with a more severe AS phenotype. Type II deletions are approximately 5 Mb in size. Uniparental disomy (UPD), a condition where two paternal copies of UBE3A are inherited, both of which are epigenetically silenced, is a less common cause of AS (Williams, Driscoll, & Dagli, 2010). UPD is associated with a less severe phenotype (Gentile et al., 2010; Saitoh, 2015). Additional mechanisms by which one can have AS include an imprinting defect, most commonly a primary epimutation or in about 10% of cases mutations (primarily deletions) in SNRPN, and lastly, mutations in the maternally inherited copy of UBE3A (Aypar, Hoppman, Thorland, & Dawson, 2016).
Mosaicism refers to the presence of two different populations of cells. It occurs when there is a postzygotic change in a single cell that then divides to form a unique cell population. Cases of mosaic AS (mAS) have been described previously in the literature (Aypar et al., 2016; Camprubí et al., 2007; Fairbrother et al., 2015; Gillessen‐Kaesbach et al., 1999; Lawson‐Yuen, Wu, Lip, Sahoo, & Kimonis, 2006; Le Fevre et al., 2017; Nazlican et al., 2004). mAS is caused by cell populations with lack of expression of maternally inherited UBE3A and is detected by methylation studies that reveal partial methylation of the maternal allele (Aypar et al., 2016; Camprubí et al., 2007; Fairbrother et al., 2015; Le Fevre et al., 2017; Nazlican et al., 2004). In this case, somatic mosaicism with loss of maternal 15q11.2q13 (SNRPN) methylation pattern is caused by a post‐zygotic imprint maintenance error after primary cell division (Aypar et al., 2016; Camprubí et al., 2007; Fairbrother et al., 2015). Another possible mechanism of mosaicism would include failed trisomy rescue in the case of uniparental disomy. Mosaic Angelman appears to be characterized by a milder phenotype; for example, the characteristic expressive language delay is milder with greater attainment of meaningful words (Le Fevre et al., 2017). We aim to profile the mAS phenotype in a larger collection of individuals to encourage clinicians to consider a lower threshold for adding this diagnosis to the differential and to discuss mAS in the context of piloting and developing meaningful outcome measures for upcoming clinical trials to impact the lives of individuals with Angelman syndrome.
2. MATERIALS AND METHODS
2.1. Ethical policies and ethical compliance
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Review Board at Vanderbilt University Medical Center (IRB #170990) and consistent with the ethical standards outlined by the 1964 Helsinki declaration and its later amendments.
2.2. Data collection methods
Anonymous surveys were administered to caretakers of individuals with mAS through social media or in follow‐up to confirmed diagnoses in the AS clinic at Vanderbilt University Medical Center. Data were collated and included in the full data set. Four additional patients were identified from a cohort of 302 patients enrolled in the Angelman Natural Study (NHS), an observational study over 8 years of the developmental progress, behavior, and medical morbidity of individuals with AS. Raw scores for neuropsychological testing were compared between mAS and AS patients at ages 4–6 years of age, a time when data were available for three mAS patients. Comparison of specific behaviors was obtained from data recorded at the patients’ first or second follow‐up visit to the NHS. Results of genetic testing, including chromosomal microarray (CMA), methylation‐specific polymerase chain reaction (MS‐PCR), and methylation‐specific multiplex ligation‐dependent probe amplification (MS‐MLPA) were viewed by a geneticist to confirm the molecular diagnosis.
2.3. Statistical analysis
Age to walk data were compared using t‐test to historical data from the literature for typically developing children (Poranen‐Clark et al., 2015) and by one‐sample t‐test for boys and girls with AS (Leitner & Smith, 1996). Signs and symptoms of mAS were contrasted with the incidence of the core symptoms of AS (Williams et al., 2006) by chi‐square. Given the limitations of parental survey data, we additionally compared our findings to historical data in mAS patients described by Le Fevre et al. (2017). Comparison between our cohort and the 28 patients described by Le Fevre et al. (2017) was with chi‐square. Comparison of specific behaviors from mAS patients and AS patients from within the NHS was with the student's t‐test or with Fisher's exact test. As multiple data types from a variety of sources were used and data were complementary in nature (Figure 4), raw p‐values are provided without adjustment for the false discovery rate. GraphPad Prism and GraphPad QuickCalcs (La Jolla, CA) were utilized for statistical analysis and generation of figures.
Figure 4.
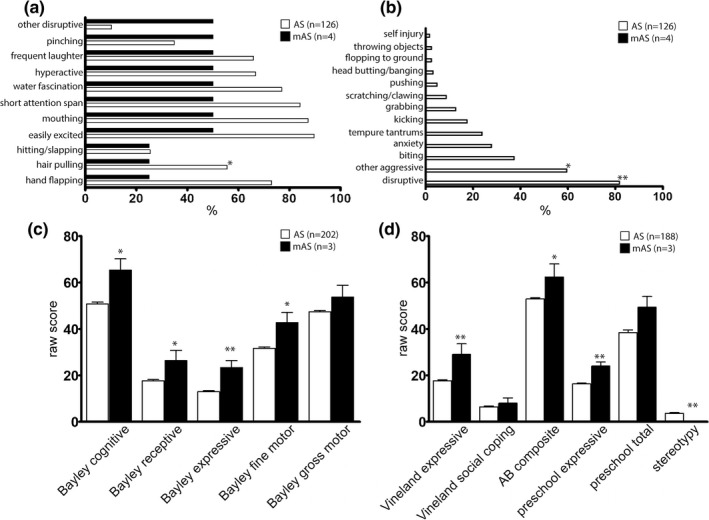
Behavioral characteristics of mAS patients contrasted to typical AS patients identified as part of the Angelman Syndrome Natural History trial. Behavioral characteristics occurring in greater than 25% of mAS patients (a). Behaviors present in AS patients but not reported in mAS patients (b). *p < 0.05, **p < 0.01 by Fisher's exact test. Neuropsychological and language raw scores from mAS and AS patients from between 4–6 years of age (c and d). n = 3–4 for mAS, n = 126–202 for AS. *p < 0.05, **p < 0.01 by Student's t test or one‐sample t test
3. RESULTS
To characterize the clinical manifestations of mAS, anonymous surveys were administered to caregivers of mAS patients. There were a total of 22 respondents to the survey. The mechanism of mosaicism in all cases was an imprinting defect. The patients described in the responding group were 59% male with a mean current age of 12 years (Table 1). Average age of diagnosis was 4 years and 4 months. The majority of patients were diagnosed via methylation‐specific polymerase chain reaction (MS‐PCR), confirming only the diagnosis of mAS. Of the 22 respondents, 28% were diagnosed using methylation‐specific multiplex ligation‐dependent probe amplification (MS‐MLPA) allowing for a more quantitative analysis of percent mosaicism. The average percent mosaicism within these individuals showed 42% methylation. Due to the limited number of individuals with quantitative analysis of methylation, a correlation between percent mosaicism and phenotypic severity could not be performed.
Table 1.
Demographics and behavioral characteristics of mAS patients
| AS consensus criteria (%) |
mAS n (%) mean ± SD/range |
mAS with known %mosaicism n (%) mean ± SD/range |
p‐value | |
|---|---|---|---|---|
| Gender | ||||
| Female | 9 (41) | 1 (20) | ||
| Male | 13 (59) | 4 (80) | ||
| Total | 22 | 5 | ||
| Current age (years) | 12 (3–30) | 12 (6–20) | ||
| Age diagnosis (years) | 4y 4m. ± 3y 2m | 5y ± 2y 6m | ||
| % Mosaicism | ||||
| Known | 5 (24) | |||
| % Mosaicism if known | 42 ± 22 | |||
| MRI/CNS findings | ||||
| Mild cortical atrophy/dysmyelination | 2 (29)a | 1 (50)a | 0.51 | |
| No atrophy/dysmyelination | 5 (71)a | 1 (50)a | 0.51 | |
| MRI not done | 4 (18) | 3 (60) | ||
| Not known/reported | 11 (50) | 0 (0) | ||
| Flat occiput | 20–80 | 7 (32) | 1 (20) | |
| Hyperactivity | 13 (59) | 2 (40) | 0.39 | |
| Treated with | ||||
| Stimulant | 3 | |||
| Guanfacine | 3 | 1 | ||
| Behavioral approaches | 2 | |||
| Outside play | 1 | |||
| Proprioceptive sensory diet | 1 | |||
| Erythromycin | 1 | |||
| Other behaviors | ||||
| Food seeking | 20–80 | 18 (82) | 3 (60) | 0.2 |
| Attraction/fascination with water | 20–80 | 11 (50) | 3 (60) | 0.65 |
| Attraction/fascination with crinkly items | 20–80 | 10 (45) | 1 (20) | 0.26 |
| Excessive chewing/mouthing | 20–80 | 9 (41) | 2 (40) | 0.99 |
| ‐outgrew | 1 (5) | 1 (20) | ||
| ADLs | ||||
| Feed themselves | 22 (100) | 5 (100) | b | |
| Brush teeth | 18 (82) | 4 (80) | 0.91 | |
| Shower independently | 14 (64) | 4 (80) | 0.46 | |
| Answer questions | 11 (50) | 4 (80) | 0.18 | |
| Ride a 2‐wheeled bike unassisted | 10 (45) | 3 (60) | 0.5 | |
| Do house cleaning chores | 8 (36) | 2 (40) | 0.85 | |
| Do laundry | 5 (23) | 2 (40) | 0.37 | |
| Toilet trained | ||||
| Daytime | 19 (86) | 5 (100) | 0.37 | |
| Nighttime | 11 (50) | 3 (60) | 0.65 |
Denotes % of those for which testing was done with results known by respondent.
Denotes inability to do chi‐square due to expected value of 0.
Developmental delay without regression, absence of language, ataxia of gait, and tremulousness of limbs are key features of AS, present in virtually all patients (Williams et al., 2006). Global developmental delay was the most common finding in the mAS cohort, reported in 90% of patients, which is a slight, but significant reduction versus what is reported in AS (p < 0.0001) (Figure 1a). In contrast with AS, less than 15% of respondents endorsed severe developmental delay by 6–12 months of age in the mAS cohort (p < 0.0001). In contrast with the marked language impairment in AS, the majority of mAS patients endorsed speaking >20 words (p < 0.001). Ataxia of gait and tremulousness of limbs, key features of AS, were endorsed in fewer than 33% of (p < 0.0001).
Figure 1.
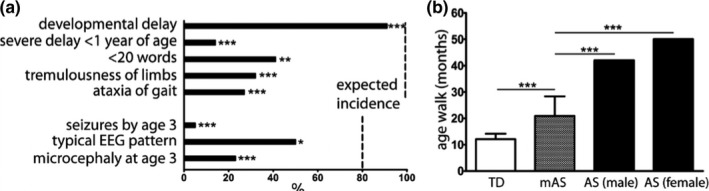
Reduced incidence of core features of AS in mAS patients. Incidence of core features present in mAS contrasted with expected incidence in AS based on consensus criteria (a). Data represents % patients with each feature. Data compared to expected incidence by chi‐square. Dashed line denotes expected incidence. Average age of mAS patients (n = 22) to walk in contrast with historic data in TD children (Poranen‐Clark et al., 2015) (n = 398) and AS patients (Leitner & Smith, 1996) (n = 10 males, 5 females) (b). TD and mAS data represent mean ± SD with AS data representing historic mean. Data compared with Student's t‐test (TD vs. mAS) and one‐sample t‐test (mAS vs. AS). *p < 0.05, ***p < 0.0001
Epilepsy is highly prevalent in AS patients, seen in over 80% of patients by 3 years of age. An equal proportion of AS patients demonstrate an abnormal EEG pattern with high amplitude slow spike‐and‐wave discharges. In the mAS cohort, seizures were rarely reported, with seizures prior to the age of 3 years endorsed in 5% of patients (p < 0.0001) (Figure 1a). The typical background EEG pattern often seen in AS patients, consisting of high amplitude slow spike‐and‐wave discharges was present in 54% of mAS patients who had EEG data reported, a significant reduction compared with that reported in AS (p < 0.0184).
All mAS patients could walk, though the average time to walking of 22 months was significantly delayed in contrast with a published cohort of typically developing children (Poranen‐Clark et al., 2015) (Figure 1b). In contrast, the time to walk of 22 months was reduced from published data in AS, which reported walking by 42 months in boys and by 50 months in girls (Leitner & Smith, 1996).
Given the limitations of parental survey based data, we compared our findings to the historical mAS data reported by Le Fevre et al. (2017). When compared to the historic AS data we used above, similar reductions in incidence of core features of ataxia, seizures, microcephaly, and word number were seen in the Le Fevre et al. (2017) cohort as were seen in ours (Table 2) with the exception of incidence of abnormal EEG findings. This may reflect an increased incidence of all EEG abnormalities versus the specific presence of the notched‐delta pattern. When core clinical feature data from our cohort of patients was compared directly to Le Fevre et al.’s cohort (Le Fevre et al., 2017), only the reported incidence of seizures differed between groups, with only one patient in our cohort reporting seizures (Table 3).
Table 2.
Historical mAS cohort reported by Le Fevre et al., (2017) contrasted to expected incidence in AS
| Core clinical features of AS |
Historical incidence in AS % |
Le Fevre et al., 2017 mAS cohort Present/total (%) |
p‐value |
|---|---|---|---|
| Word number < 20 | 100 | 14/26 (46)a | <0.0001 |
| ataxia | 100 | 11/25 (44) | <0.0001 |
| Seizures by age 3 | >80 | 8/28 (29) | <0.0001 |
| Microcephaly | >80 | 3/26 (12) | <0.0001 |
| Typical EEG pattern | >80 | 7/11 (64)b | 0.1748 |
Denotes that Le Fevre data (Le Fevre et al., 2017) represents word number <10.
Denotes numbers for an “abnormal EEG” and not “typical” AS EEG pattern.
Table 3.
Clinical characteristics of historic and current mAS clinical features
| Clinical Feature |
Le Fevre et al., 2017 Present/total (%) |
Carson and Duis, 2019 Present/total (%) |
p‐value |
|---|---|---|---|
| Word number | >10, 12/26 (46) | >20, 13/22 (59) | 0.18 |
| Age of onset walking (years) | 1.5 (range 1–3) | 1.7 (range 1–3) | |
| Walked by age 5 | 24/24 (100) | 22/22 (100) | a |
| Ataxia | 11/25 (44) | 6/21 (29) | 0.10 |
| Seizures | 8/28 (29) | 1/22 (5) | <0.0001 |
| Microcephaly | 3/26 (12) | 5/22 (23) | 0.16 |
| Abnormal EEG | 7/11 (64) | 7/16 (44) | 0.19 |
| Hypopigmentation | 2/11 (18) | 5/22 (23) | 0.70 |
| Obesity | 8/25 (32) | 14/22 (64) | 0.0009 |
| Hyperphagia | 11/20 (55) | 18/22 (82) (food seeking) | 0.0017 |
| Feeding problem | 7/24 (29) (Neonatal) | 2/22 (9) | 0.0006 |
| Behavioral problems | 4/5 (80) |
21/22 (95) anxiety 13/22 (59) hyperactivity |
0.12 |
| Sleeping Difficulties | 5/6 (83) | 16/22 (73) | 0.56 |
| Facial dysmorphic features | 3/9 (33) | 7/22 (32) | 0.93 |
Denotes inability to do chi‐square do to expected value of 0.
Mild cortical atrophy or dysmyelination are the most common structural brain abnormalities reported in AS. Mild cortical atrophy or dysmyelination was reported in 29% of mAS patients who had MRI imaging (Table 1). In 68% of patients, either MRIs had never been done or the findings were not known to the respondents. While microcephaly occurs in more than 80% of patients with AS, the incidence of microcephaly in the mAS cohort was reduced to 19% (Figure 1a).
Additional clinical features common in AS were also endorsed in patients with mAS. The three most prominent findings included abnormal sleep/wake cycles and decreased sleep in 73% of patients, followed by obesity in 64% of patients and heat sensitivity in 45% of patients (Figure 2a). Constipation, a common problem in AS patients was also noted in the mAS cohort, with 72% of patients endorsing constipation (Figure 2b).
Figure 2.
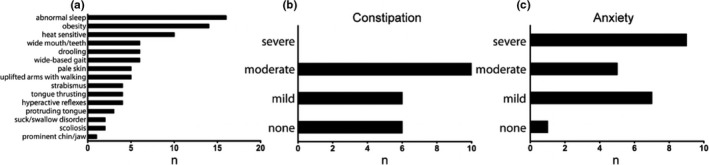
Frequency of AS associated features in mAS patients. Incidence of associated features in mAS patients (a). Incidence and severity of constipation (b) and anxiety (c) in mAS patients. Data represents % of patients (n = 22). (#) is raw number
Along with epilepsy and sleep disturbances, behavioral challenges are frequently reported in patients with AS. Anxiety was the most frequently endorsed behavior reported in our cohort, present to some degree in 95% of mAS patients and rated as severe in 43% of patients. Hyperactivity was commonly endorsed, reported in 59% of patients. Of the patients with hyperactivity, 92% received some form of therapy, with the most common treatments being stimulants and guanfacine, followed by behavioral approaches. Additional behaviors commonly reported in our mAS cohort included food seeking behaviors (82%) fascination with water (50%) or fascination with items such as plastic making “crinkly” noises (45%).
Functionally, as noted above, the majority of mAS patients could speak greater than 20 words, with nearly 20% of patients using over 1,000 words (Figure 3a). The ability to write was endorsed in the majority of mAS patients though only ~5% of patients attained the ability to write a sentence (Figure 3b). Educationally, 5% of patients were in regular classes with 36% of patients participating in regular classes, but with additional assistance (mainstreamed) (Figure 3c). 36% of patients were primarily in special needs programs. The remainder of patients were either homeschooled, in day programs, or data were not reported. In addition, the majority of patients were able to feed themselves, brush their teeth, shower independently, and answer questions (Table 1). 86% of patients were toilet trained during the daytime, though 50% continued to have difficulty at night. The ability to participate in house cleaning chores was seen in 36% of patients with 23% of patients able to do laundry.
Figure 3.
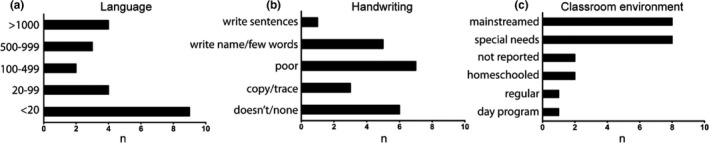
Characteristics of communication and education in mAS patients. Words known by mAS patients (a). Ability of mAS patients to communicate through writing (b). Type of classroom environment which mAS patients attend (c). Data represents raw # of patients, n = 22
When our findings were contrasted with the published findings by Le Fevre et al. (2017), obesity and hyperphagia were present in both cohorts of patients but were significantly increased in our cohort relative to Le Fevre et al. (2017) (Table 3). A significant difference in feeding problems were present in Le Fevre et al. (2017) cohort which was not reported herein, though this most likely reflects the nature of the question, as Le Fevre et al. (2017) group asked specifically about neonatal feeding problems. No significant differences in incidence of hypopigmentation, behavioral problems, sleeping difficulties, or facial dysmorphic features were seen in our mAS cohort versus Le Fevre's (Le Fevre et al., 2017).
Data from the Angelman Natural History study were used to compare with the findings from survey data. As expected, many of the common behaviors seen in AS were present in mAS patients, with pinching, laughing, hyperactivity, fascination with water, short attention span, mouthing of objects, and easy excitability reported in 50% of mAS patients (Figure 4). Many behaviors reported in AS were not reported in the small mAS cohort in the NHS, though statistically significant differences were only seen with respect to hair pulling, disruptive behaviors, and general aggressive behaviors. Neurocognitive raw scores were significantly better for mAS patients compared to AS patients with the most notable differences in expressive language. We also saw a decreased incidence of stereotypies. Gross motor skills, social coping skills, and preschool total language were not significantly different between the mAS and typical AS patients.
4. DISCUSSION
The data reported herein represent the largest single cohort of mAS patients and expands our knowledge of the clinical characteristics of mAS (Aypar et al., 2016; Fairbrother et al., 2015; Le Fevre et al., 2017). Our findings demonstrate that while the overall phenotype of mAS is less severe than that of AS, significant challenges remain.
The consistent features of AS defined by the consensus criteria include severe developmental delay, movement or a balance disorder, behavioral uniqueness, and marked speech impairment (Saitoh, 2015), features seen in nearly 100% of patients. These consistent features were far less common in mAS. While developmental delay was present in nearly all patients, it was rated as severe in less than 20% of patients. Additionally, the core feature of gait ataxia was far less common that expected in AS, a finding which likely contributes to the earlier age of ambulation.
Expressive language is very limited in patients with AS, as exemplified recently in a study where 5/6 AS patients were in the 1st percentile of expressive language when rated by the Bayley scale (Grieco et al., 2018). In contrast, the majority of mAS patients demonstrated expressive language, with nearly 1 in 5 patients speaking more than 1,000 words. Expressive language though remains a challenge in mAS patient as exemplified by the finding that 38% of patients speak less than 20 words. The overall ability to communicate language through writing was severely restricted in our mAS cohort, with only 1 of the 22 patients being able to write sentences.
Seizures are common in AS, with multiple studies supporting that over 80% of AS patients have epilepsy, with nearly one in four having seizures in the first year of life (Sueri et al., 2017; Williams et al., 2010). Consistent with this, over 80% of patients have epileptiform discharges on EEG or a stereotypical delta frequency activity. Though seizure frequency decreases over time, the typical EEG pattern persists (Sueri et al., 2017; Vendrame et al., 2012). In contrast with these findings, seizures were rare in our mAS cohort, though over 50% of mAS patients demonstrated EEG findings consistent with AS.
Our data demonstrate that many clinical features of mAS are mild in contrast with typical AS, including overall function and ability of patients to participate in self‐care. Given that three patients in our cohort were 6 years of age or younger, we may have underestimated the overall functional abilities of our mAS cohort. Despite the better overall functioning, behavioral challenges remain significant, occurring at least as frequently as in typical AS, and sometimes (e.g. anxiety) more frequently.
Our cohort of mAS patients presented an overall milder phenotype than those reported by Aypar et al., who reported microcephaly in 66% of patients, gait ataxia in 75% of patients, and seizures in 40% of patients (Aypar et al., 2016). Obesity and hyperphagia were less commonly reported in the Aypar cohort. While genetic data were reported for all patients in the Aypar study, the study was limited with clinical data in only 50% of patients.
Given that we are reporting survey data in a rare subset of a rare disease, we do acknowledge the inherent biases that may occur in the study of rare disease. Self‐selection bias may occur due to parents’ interest in research studies or effort to seek out specialty clinics (Hammer, Prel, & Blettner, 2009). Reporting and ascertainment biases suffer the same concerns as self‐selection bias but with additional challenges inherent to computer‐based surveys, including prevalence of social media or limitations imposed by baseline computer literacy or access (Slater & Kiran, 2016). In the study of rare diseases with observational designs, such concerns are not just limited to smaller studies but also may apply to inclusion in disease registries (Cole et al., 2011). There is the potential for overlap in cases reported in our cohort and cases reported in other studies as well. Through parental report, we know of one confirmed patient that was previously described by Fairbrother et al (Lawson‐Yuen et al., 2006). To address these concerns, we compared our results to previously published data on mAS and examined patient characteristics from mAS patients identified as part of the Angelman NHS. Our results were similar to those reported by Le Fevre et al. (2017) with a few noted differences. The frequency of seizures in our cohort is notably lower than that described by Le Fevre et al. (2017), who reported seizures in 29% of mAS patients. The frequency of abnormal EEGs though was similar between studies. Obesity and hyperphagia/food seeking were present in both groups of patients, but were both significantly increased in our mAS cohort when compared to Le Fevre et al. (2017). Given the similarities between our study, the NHS data and from Le Fevre et al. (2017), we do not feel that either self‐selection bias or reporting bias has significantly skewed our findings. Our study is limited by a lack of qualitative data to estimate level of mosaicism, which could correlate with skill level and possibly awareness and ability to act on hyperphagia. However, these data are most helpful from the CNS and it is unlikely we will be able to quantify percentage of mosaicism in this most relevant tissue.
Our experience in AS suggests that hyperphagia is more prevalent in the AS population than previously reported, and that it is particular to carbohydrates. Data show increased incidence of obesity in patients with the milder AS phenotype associated with uniparental disomy or imprinting defects (Lossie et al., 2001), but obesity appears less severe than seen in Prader‐Willi syndrome (PWS). The underlying genetic mechanisms leading to hyperphagia in mAS or in subsets of AS are not clear. One proposed hypothesis is that hyperphagia due to chromosome 15 anomalies is at least in part an obsessive‐compulsive (OC) type behavior and not solely a genetically driven lack of satiety. The other genes in the distal chromosome 15q11.2‐q13 region include gamma aminobutyric acid (GABA) receptor subunits GABRB3, GABRA5, and GABRG3, which are not imprinted. Alterations in the GABAergic systems are associated with hunger (Turenius et al., 2009), anxiety (Hodges et al., 2014), OC symptoms, and addiction (Stephens, King, Lambert, Belelli, & Duka, 2017). While hypoactivity may contribute to obesity in PWS (Dhar et al., 2004), nearly 60% of our patients were reported to be hyperactive and perhaps this sheds some light on differences in metabolic phenotype in AS compared to PWS. Metabolically defining AS by molecular subtype is an important future direction given this expanded phenotypic feature noted in mAS.
Our data support the idea that mAS presents with a variable phenotype, with key features being mild‐moderate global developmental delay, anxiety, hyperactivity, constipation and retained, but limited speech. While the presence of language and absence of seizures may drive clinicians away from AS as a plausible diagnosis, the abnormal EEG findings present in over 50% of patients may aid in suggesting mAS as a possible diagnosis.
Overall these data encourage specialists to broaden the clinical features of AS. While the American College of Medical Genetics (ACMG) now recognizes a chromosome microarray (CMA) as the first‐tier genetic test for individuals with developmental disabilities, AS‐specific SNRPN methylation testing typically is not ordered unless a diagnosis of AS is suspected based on the core features previously described (Manning, & Hudgins, 2010). A CMA could potentially detect a mosaic 15q11.2–13 deletion and if probes containing single nucleotide polymorphisms (SNPs) are included, cases of mosaic UPD due to failed trisomy rescue during meiosis II (isodisomy), depending on the level of mosaicism within an individual. However, cases of mAS due to a meiosis I error (heterodisomy) without recombination and visible regions of isodisomy, or due to an imprinting defect would not be detected using a CMA. Given that all reported cases in this study were a result of an imprinting defect, there is a large potential for missed diagnoses of mAS using a CMA as a first line test in the absence of SNRPN methylation testing. Based on this data as well as previous studies, specialists should consider ordering SNRPN‐specific methylation testing for patients with uncharacteristically happy demeanors, hyperactivity, anxiety, and developmental delay despite a lack of seizures and the presence of speech, especially in the context of a normal CMA.
Furthermore, these data suggest that there may be clinical benefit from partial re‐activation of UBE3A in the central nervous system, with the caveat that clinically significant behavioral symptoms may persist. With ongoing research into various therapies and methodologies to restore partial UBE3A activity, studies such as this provide insight into various biomarkers and phenotypic traits that could be analyzed once this is achieved.
CONFLICT OF INTEREST
Authors have no conflicts of interest to declare.
ACKNOWLEDGMENTS
The authors thank the patients and their families for participating in research and making this work possible. We also thank the Angelman Syndrome Foundation for their generous support of the Angelman Syndrome Clinic at The Monroe Carell Jr. Children's Hospital at Vanderbilt. We thank the investigators in the NIH RDCRN Angelman Syndrome sub‐consortium who obtained the normative data used in this study: Rene Barbieri‐Welge (Rady Children's Hospital San Diego), Sarika Peters and Rachel Hundley (Vanderbilt University Medical Center), Carlos Bacino and Lisa Noll (Texas Children's Hospital), Steven Skinner and Lucia Horowitz (Greenwood Genetic Center), Logan Wink (Cincinnati Children's Hospital Medical Center), Wen‐Hann Tan, Jennifer Gentile, and Anjali Sadhwani (Boston Children's Hospital).
Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of Mosaic Angelman Syndrome. Mol Genet Genomic Med. 2019;7:e837 10.1002/mgg3.837
REFERENCES
- Aypar, U. , Hoppman, N. L. , Thorland, E. C. , & Dawson, D. B. (2016). Patients with mosaic methylation patterns of the Prader‐Willi/Angelman Syndrome critical region exhibit AS‐like phenotypes with some PWS features. Molecular Cytogenetics, 9(26), 1–6. 10.1186/s13039-016-0233-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buiting, K. , Saitoh, S. , Gross, S. , Dittrich, B. , Schwartz, S. , Nicholls, R. D. , & Horsthemke, B. (1995). Inherited microdeletions in the Angelman and Prader‐Willi syndromes define an imprinting centre on human chromosome 15. Nature Genetics, 9(4), 395–400. 10.1038/ng0495-395 [DOI] [PubMed] [Google Scholar]
- Camprubí, C. , Coll, M. D. , Villatoro, S. , Gabau, E. , Kamli, A. , Martínez, M. J. , … Guitart, M. (2007). Imprinting center analysis in Prader‐Willi and Angelman syndrome patients with typical and atypical phenotypes. European Journal of Medical Genetics, 50(1), 11–20. 10.1016/j.ejmg.2006.10.001 [DOI] [PubMed] [Google Scholar]
- Cole, J. A. , Taylor, J. S. , Hangartner, T. N. , Weinreb, N. J. , Mistry, P. K. , & Khan, A. (2011). Reducing selection bias in case‐control studies from rare disease registries. Orphanet Journal of Rare Diseases, 6(61), 1–7. 10.1186/1750-1172-6-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar, M. S. , Sommardahl, C. S. , Kirkland, T. , Nelson, S. , Donnell, R. , Johnson, D. K. , & Castellani, L. W. (2004). Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. The Journal of Nutrition, 134(4), 799–805. 10.1093/jn/134.4.799 [DOI] [PubMed] [Google Scholar]
- Fairbrother, L. C. , Cytrynbaum, C. , Boutis, P. , Buiting, K. , Weksberg, R. , & Williams, C. (2015). Mild Angelman syndrome phenotype due to a mosaic methylation imprinting defect. American Journal of Medical Genetics Part A, 167(7), 1565–1569. 10.1002/ajmg.a.37058 [DOI] [PubMed] [Google Scholar]
- Fiumara, A. , Pittala, A. , Cocuzza, M. , & Sorge, G. (2010). Epilepsy in patients with Angelman syndrome. Italian Journal of Pediatrics, 36(31), 1–6. 10.1186/1824-7288-36-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile, J. K. , Tan, W.‐H. , Horowitz, L. T. , Bacino, C. A. , Skinner, S. A. , Barbieri‐Welge, R. , … Peters, S. U. (2010). A neurodevelopmental survey of Angelman syndrome with genotype‐phenotype correlations. Journal of Developmental and Behavioral Pediatrics, 31(7), 592–601. 10.1097/DBP.0b013e3181ee408e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillessen‐Kaesbach, G. , Demuth, S. , Thiele, H. , Theile, U. , Lich, C. , & Horsthemke, B. (1999). A previously unrecognised phenotype characterised by obesity, muscular hypotonia, and ability to speak in patients with Angelman syndrome caused by an imprinting defect. European Journal of Human Genetics, 7(6), 638–644. 10.1038/sj.ejhg.5200362 [DOI] [PubMed] [Google Scholar]
- Glenn, C. C. , Porter, K. A. , Jong, M. T. , Nicholls, R. D. , & Driscoll, D. J. (1993). Functional imprinting and epigenetic modification of the human SNRPN gene. Human Molecular Genetics, 2(12), 2001–2005. 10.1093/hmg/2.12.2001 [DOI] [PubMed] [Google Scholar]
- Grieco, J. C. , Bahr, R. H. , Schoenberg, M. R. , Conover, L. , Mackie, L. N. , & Weeber, E. J. (2018). quantitative measurement of communication ability in children with Angelman syndrome. Journal of Applied Research in Intellectual Disabilities, 31(1), e49–e58. 10.1111/jar.12305 [DOI] [PubMed] [Google Scholar]
- Hammer, G. P. , du Prel, J.‐B. , & Blettner, M. (2009). Avoiding bias in observational studies: Part 8 in a series of articles on evaluation of scientific publications. Deutsches Arzteblatt International, 106(41), 664–668. 10.3238/arztebl.2009.0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges, L. M. , Fyer, A. J. , Weissman, M. M. , Logue, M. W. , Haghighi, F. , Evgrafov, O. , … Hamilton, S. P. (2014). Evidence for linkage and association of GABRB3 and GABRA5 to panic disorder. Neuropsychopharmacology, 39(10), 2423–2431. 10.1038/npp.2014.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino, T. , Lalande, M. , & Wagstaff, J. (1997). UBE3A/E6‐AP mutations cause Angelman syndrome. Nature Genetics, 15(1), 70–73. 10.1038/ng0197-70 [DOI] [PubMed] [Google Scholar]
- Lawson‐Yuen, A. , Wu, B. L. , Lip, V. , Sahoo, T. , & Kimonis, V. (2006). Atypical cases of Angelman syndrome. American Journal of Medical Genetics Part A, 140(21), 2361–2364. 10.1002/ajmg.a.31481 [DOI] [PubMed] [Google Scholar]
- Le Fevre, A. , Beygo, J. , Silveira, C. , Kamien, B. , Clayton‐Smith, J. , Colley, A. , … Dudding‐Byth, T. (2017). Atypical Angelman syndrome due to a mosaic imprinting defect: Case reports and review of the literature. American Journal of Medical Genetics Part A, 173(3), 753–757. 10.1002/ajmg.a.38072 [DOI] [PubMed] [Google Scholar]
- Leitner, R. P. , & Smith, A. (1996). An Angelman syndrome clinic: Report on 24 patients. Journal of Paediatrics and Child Health, 32(2), 94–98. 10.1111/j.1440-1754.1996.tb00902.x [DOI] [PubMed] [Google Scholar]
- Lossie, A. C. , Whitney, M. M. , Amidon, D. , Dong, H. J. , Chen, P. , Theriaque, D. , … Driscoll, D. J. (2001). Distinct phenotypes distinguish the molecular classes of Angelman syndrome. Journal of Medical Genetics, 38(12), 834–845. 10.1136/jmg.38.12.834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magenis, R. E. , Brown, M. G. , Lacy, D. A. , Budden, S. , & LaFranchi, S. (1987). Is Angelman syndrome an alternate result of del(15)(q11q13)? American Journal of Medical Genetics, 28(4), 829–838. 10.1002/ajmg.1320280407 [DOI] [PubMed] [Google Scholar]
- Manning, M. , & Hudgins, L. (2010). Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genetics in Medicine, 12, 742–745. 10.1097/GIM.0b013e3181f8baad [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazlican, H. , Zeschnigk, M. , Claussen, U. , Michel, S. , Boehringer, S. , Gillessen‐Kaesbach, G. , … Horsthemke, B. (2004). Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Human Molecular Genetics, 13(21), 2547–2555. 10.1093/hmg/ddh296 [DOI] [PubMed] [Google Scholar]
- Özçelik, T. , Leff, S. , Robinson, W. , Donlon, T. , Lalande, M. , Sanjines, E. , … Francke, U. (1992). Small nuclear ribonucleoprotein polypeptide N (SNRPN), an expressed gene in the Prader‐Willi syndrome critical region. Nature Genetics, 2(4), 265–269. 10.1038/ng1292-265 [DOI] [PubMed] [Google Scholar]
- Poranen‐Clark, T. , von Bonsdorff, M. B. , Lahti, J. , Räikkönen, K. , Osmond, C. , Rantanen, T. , … Eriksson, J. G. (2015). Infant motor development and cognitive performance in early old age: The Helsinki Birth Cohort Study. Age (Dordrecht, Netherlands), 37(3), 44 10.1007/s11357-015-9785-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, M. L. , & Leff, S. E. (1994). Maternal imprinting of human SNRPN, a gene deleted in Prader‐Willi syndrome. Nature Genetics, 6(2), 163–167. 10.1038/ng0294-163 [DOI] [PubMed] [Google Scholar]
- Saitoh, S. (2015). Clinical, molecular, and neurophysiological features in Angelman syndrome. Journal of Pediatric Epilepsy, 4(1), 17–22. 10.1055/s-2003-42213 [DOI] [Google Scholar]
- Slater, M. , & Kiran, T. (2016). Measuring the patient experience in primary care: Comparing e‐mail and waiting room survey delivery in a family health team. Canadian Family Physician, 62(12), e740–e748. Retrieved from https://www.cfp.ca/content/62/12/e740.long. [PMC free article] [PubMed] [Google Scholar]
- Stephens, D. N. , King, S. L. , Lambert, J. J. , Belelli, D. , & Duka, T. (2017). GABAA receptor subtype involvement in addictive behaviour. Genes, Brain, and Behavior, 16(1), 149–184. 10.1111/gbb.12321 [DOI] [PubMed] [Google Scholar]
- Sueri, C. , Ferlazzo, E. , Elia, M. , Bonanni, P. , Randazzo, G. , Gasparini, S. , … Aguglia, U. (2017). Epilepsy and sleep disorders improve in adolescents and adults with Angelman syndrome: A multicenter study on 46 patients. Epilepsy & Behavior, 75, 225–229. 10.1016/j.yebeh.2017.07.041 [DOI] [PubMed] [Google Scholar]
- Thibert, R. L. , Conant, K. D. , Braun, E. K. , Bruno, P. , Said, R. R. , Nespeca, M. P. , & Thiele, E. A. (2009). Epilepsy in Angelman syndrome: A questionnaire‐based assessment of the natural history and current treatment options. Epilepsia, 50(11), 2369–2376. 10.1111/j.1528-1167.2009.02108.x [DOI] [PubMed] [Google Scholar]
- Turenius, C. I. , Htut, M. M. , Prodon, D. A. , Ebersole, P. L. , Ngo, P. T. , Lara, R. N. , … Stanley, B. G. (2009). GABA(A) receptors in the lateral hypothalamus as mediators of satiety and body weight regulation. Brain Research, 1262(16‐24), 10.1016/j.brainres.2009.01.016 [DOI] [PubMed] [Google Scholar]
- Vendrame, M. , Loddenkemper, T. , Zarowski, M. , Gregas, M. , Shuhaiber, H. , Sarco, D. P. , … Kothare, S. V. (2012). Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy & Behavior, 23(3), 261–265. 10.1016/j.yebeh.2011.11.027 [DOI] [PubMed] [Google Scholar]
- Williams, C. A. , Beaudet, A. L. , Clayton‐Smith, J. , Knoll, J. H. , Kyllerman, M. , Laan, L. A. , … Wagstaff, J. (2006). Angelman syndrome 2005: Updated consensus for diagnostic criteria. American Journal of Medical Genetics Part A, 140(5), 413–418. 10.1002/ajmg.a.31074 [DOI] [PubMed] [Google Scholar]
- Williams, C. A. , Driscoll, D. J. , & Dagli, A. I. (2010). Clinical and genetic aspects of Angelman syndrome. Genetics in Medicine, 12(7), 385–395. 10.1097/GIM.0b013e3181def138 [DOI] [PubMed] [Google Scholar]