

Abstract
Background
The most common inherited peripheral neuropathy is Charcot‐Marie‐Tooth disease (CMT), with a prevalence of 1/2500. Other symptoms can be associated to the condition, such as hearing loss. Currently, no global hearing impairment assessment has been determined, and the physiopathology is not well known.
Methods
The aim of the study was to analyze among a French series of 3,412 patients with inherited peripheral neuropathy (IPN), the ones who also suffer from hearing loss, to establish phenotype‐genotype correlations. An NGS strategy for IPN one side and nonsyndromic hearing loss (NSHL) on the other side, were performed.
Results
Hearing loss (HL) was present in only 44 patients (1.30%). The clinical data of 27 patients were usable. Demyelinating neuropathy was diagnosed in 15 cases and axonal neuropathy in 12 cases. HL varied from mild to profound. Five cases of auditory neuropathy were noticed. Diagnosis was made for 60% of these patients. Seven novel pathogenic variants were discovered in five different genes: PRPS1; MPZ; SH3TC2; NEFL; and ABHD12. Two patients with PMP22 variant, had also an additional variant in COCH and MYH14 respectively. No pathogenic variant was found at the DFNB1 locus. Genotype‐phenotype correlations do exist, especially with SH3TC2, PRPS1, ABHD12, NEFL, and TRPV4.
Conclusion
Involvement of PMP22 is not enough to explain hearing loss in patients suffering from IPN. HL can be due to cochlear impairment and/or auditory nerve dysfunction. HL is certainly underdiagnosed, and should be evaluated in every patient suffering from IPN.
Keywords: Charcot‐Marie‐Tooth, hearing loss, neuropathy, NGS
1. INTRODUCTION
The most common inherited peripheral neuropathy is Charcot‐Marie‐Tooth disease (CMT), with a prevalence of 1/2500. CMT has a wide range of phenotypes and is genetically heterogeneous. PMP22 duplication was the first identified pathogenic variant in 1992, and accounts for 15% of CMT patients (Timmerman et al., 1992). More than 90 genes are involved in the different types, which can be demyelinating, axonal, or intermediate with variable inheritance and expression. Other symptoms can be associated to the condition, such as scoliosis or hearing loss. Currently, no global hearing impairment assessment has been determined, and the physiopathology is not well‐known. The hypothesis of retrocochlear dysfunction has been suggested (Anzalone, Nuhanovic, Olund, & Carlson, 2018). It is therefore supposed that profound hearing impairment could be the result of cochlear nerve desynchronization, leading to auditory neuropathy.
Almost 10% of the French population suffers from hearing loss, which can be sensorineural, conductive, or mixed. Sensorineural hearing loss can be due to a virus (e.g. CytoMegaloVirus), environment (e.g. noise exposure), or genetic factors. Congenital hearing loss represents more than 50% of sensorineural hearing loss. More than 100 genes have been identified to be responsible for NSHL, and more still for syndromic hearing loss. Genetic hearing loss is most of the time a monogenic disease.
The aim of the study was to analyze a French series of patients suffering from inherited peripheral neuropathy associated with hearing loss, in order to establish phenotype‐genotype correlations.
2. MATERIALS AND METHODS
2.1. Patients
A French series of 3,412 patients suffering from IPN has been analysed thanks to medical records and a clinical questionnaire, so as to identify patients presenting IPN and hearing loss. The 3,412 patients had been selected on clinical and inheritance criterion, and all have been genetically screened.
Phenotypes were screened on the basis of clinical data and electroneuromyograms (ENMG) for IPN, and audiograms, OtoAcoustic Emissions (OAE), and Auditory Brainstem Responses (ABR) for hearing loss.
Peripheral blood samples of the patients were collected on EDTA tubes after giving their informed consent. The protocol was in accordance with French ethical legislation and Helsinki declaration.
2.2. Pathogenic variant detection
Genomic DNA was extracted by standard methods (Illustra DNA Extraction kit BACC3, GEHC). For neuropathy screening, a Next Generation Sequencing (NGS) strategy was implemented using a 92‐gene custom panel designed for CMT and associated neuropathies diagnosis (Table S1). It included the 44 known CMT genes, 27 genes involved in HSN (Hereditary Sensitive Neuropathy) and HMN (Hereditary Motor Neuropathy) and 21 other genes of interest involved in neuropathies of differential diagnosis. The amplified library was prepared with Ion P1 HiQ Template OT2 200 kit (Ampliseq Custom [Life technologies]), sequenced on Proton sequencer (Life technologies), and mapped to the human reference sequence GHCh37. Variants were assessed with Alamut Mutation Interpretation Software (Interactive Biosoftware, Rouen, France). Databases such as ExAC Genome browser (http://exac.broadinstitute.org), dbSPN135 (National Center for Biotechnology Information [NCBI], Bethesda, Maryland, USA, http://www.ncbi.nlm.nih.gov/projects/SPN/), ClinVar (www.ncbi.nlm.nih.gov/clinvar), and HGMD (www.hgmd.cf.ac.uk) were also screened. In silico studies were performed thanks to Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg), UMD‐Predictor (http://umd-predictor.eu/), and Mutation Taster (http://www.mutationtaster.org/). Pathogenic variants of interest were verified by Sanger sequencing using forward and reverse primer pairs. Data have been submitted into a freely accessible public database, namely LOVD at https://databases.lovd.nl/shared/genes/DMD.
For hearing loss screening, MLPA and Sanger sequencing for GJB2 and GJB6 were performed for all the deaf patients. A NGS strategy was performed on a 63‐gene custom panel designed for hearing loss in 8 selected patients (Baux et al., 2017) (Table S2). Variants of interest were verified by Sanger sequencing using forward and reverse primer pairs.
Literature analysis has identified 36 genes described to be involved in both IPN and hearing loss (Table 1). These 36 genes are all present in the 92‐gene custom panel designed for CMT and IPN.
Table 1.
The 36 genes of interest involved in both IPN and HL and their reference sequence
| Genes described to be involved in NP + HL | |||||||
|---|---|---|---|---|---|---|---|
| AARS | NM_001605.2 | DNAJB2 | NM_006736.5 | PEX12 | NM_000286.2 | SH3TC2 | NM_024577.3 |
| ABHD12 | NM_015600.4 | INF2 | NM_022489.3 | PEX7 | NM_000288.3 | SLC5A7 | NM_021815.2 |
| AIFM1 | NM_004208.3 | KIF5A | NM_004984.2 | PHYH | NM_001323082.1 | SLC25A46 | NM_138773.2 |
| DNMT1 | NM_001130823.1 | MFN2 | NM_014874.3 | PMP22 | NM_000304.2 | SOX10 | NM_006941.3 |
| FIG4 | NM_014845.5 | MPZ | NM_001315491.1 | POLG | NM_001126131.1 | SPTLC1 | NM_001281303.1 |
| GBE1 | NM_000158.3 | MYH14 | NM_001145809.1 | PRPS1 | NM_002764.3 | SURF1 | NM_003172.3 |
| GJB1 | NM_000166.5 | NDRG1 | NM_006096.3 | SBF2 | NM_030962.3 | TRPV4 | NM_021625.4 |
| GJB3 | NM_001005752.1 | NEFL | NM_006158.3 | SCN9A | NM_002977.3 | TTR | NM_000371.3 |
| GLA | NM_000169.2 | PDK3 | NM_001142386.2 | SETX | NM_015046.5 | TYMP | NM_001113755.1 |
3. RESULTS
3.1. Clinical description
Among the series of 3,412 patients, we had the information in medical records and clinical questionnaire of hearing loss associated with IPN in only 44 patients (1.30%). The clinical data of 27 patients, 15 women and 12 men, were usable for this study. The clinical description is presented in Table 2. The mean age was 60.77 years (from 10 to 90).
Table 2.
Phenotypes of our 27 patients
| Patient | Polyneuropathy | Hearing loss | Other symptoms | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
Reference Family |
Patient (gender/age in years) |
Form (Fam/spo)/ AD, AR or X‐linked |
Neuropathy | Pes cavus | VCM (m/s) | Age at onset (years) | Degree | Age at onset (years) | |
| I | F, 87 | Spo/ NA | Sensori‐motor Demyelinating | Y | 48 | 12 | NC | NC | Urinary incontinence, Small legs |
| II | F, 90 | Spo/ NA | Sensori‐motor Axonal | Y | NC | 61 | Severe AN | 68 | / |
| III | M, 80 | Fam/ AD | Sensori‐motor Axonal | Y | 56 | 59 | Moderate | NC | Cataracts, Retinal detachment |
| IV | M, 35 | Spo/ NA | Sensori‐motor Axonal | N | 20–30 | 8 | Profound | 1 | Optic Neuropathy, Balance disorder |
| V | F, 86 | Fam/AD | Sensori‐motor Demyelinating | Y | 18 | 68 | Progressive | NC | Balance disorder |
| VI | M, 88 | Fam/AD | Sensori‐motor Demyelinating | Y | 31 | 72 | NC | NC | Ataxia, Alzheimer disease |
| VII | F, 60 | Fam/AD | Demyelinating | NC | NC | NC | Moderate | NC | Balance disorder |
| VIII | F, 47 | Spo/NA | Sensori‐motor Demyelinating | Y | 27 | 4 | NC | NC | Optic Neuropathy |
| IX | F, 47 | Fam/AD | Sensori‐motor Axonal | Y | NC | 44 | Moderate | 35 | Pain, Chronic Respiratory Insufficiency |
| X | M, 69 | Fam/AD | Sensori‐motor Demyelinating | NC | NC | NC | NC | NC | Severe form |
| X1 | F, 68 | Fam/AD | Sensori‐motor Axonal | Y | 43 | 35 | Moderate and progressive | 35 | / |
| XII | M, 33 | Fam/AD | Sensori‐motor Demyelinating | Y | 36 | 20 | Severe AN | 5 | Tomacular neuropathy |
| XIII | F, 34 | Spo/ NA | Sensori‐motor Demyelinating | Y | NC | 12 | Profound | 1 | Pain, Primary amenorrhea, oesophagus atresia |
| XIV | F, 68 | Fam/AD | Sensori‐motor Demyelinating | NC | 30 | 50 | Severe to Profound AN | 50 | Balance disorders, Cochlear Implantation |
| XV | M, 29 | Spo/NA | Sensori‐motor Axonal | Y | 58 | < 5 | NC | < 5 | / |
| XVI | F, 75 | Spo/NA | Sensori‐motor Demyelinating | Y | 51 | < 5 | NC | NC | / |
| XVII | F, 68 | Spo/NA | Sensori‐motor Demyelinating | Y | 34–37 | 9 | Moderate | 60 | Scoliosis |
| XVIII | M, 68 | Fam/AD | Sensori‐motor Axonal | Y | NC | 65 | Moderate AN | 62 | Bilateral Vocal cord Paresis |
| XIX | F, 69 | Fam/AD | Sensori‐motor Axonal | Y | NC | 45 | Moderate | 45 | |
| XX | M, 83 | Fam/AR | Sensori‐motor Demyelinating | Y | 31 | 73 | Moderate | 60 | |
| XXI | F, 10 | Fam/AD | Sensori‐motor Demyelinating | Y | NC | 2 | Moderate | 1 | / |
| XXII | F, 80 | Spo/ NA | Sensori‐motor Axonal | Y | NC | 45 | NC | NC | Scoliosis, Cataracts |
| XXIII | M, 19 | Spo/ NA | Sensori‐motor Axonal | Y | 52 | 11 | Mild | 6 | Urinary incontinence, wheelchair |
| XXIV | F, 71 | Spo/ NA | Sensori‐motor Demyelinating | Y | 41 | 45 | NC | < 5 | Scoliosis |
| XXV | M, 78 | Fam/AD | Sensori‐motor Axonal | Y | 46 | NC | NC | NC | Ataxia, Gougerot‐Sjogren |
| XXVI | M, 38 | Spo/ NA | Sensori‐motor Demyelinating | NC | 25–30 | 15 | Moderate to profound AN | 5 | Cataracts, Ataxia |
| XXVII | M, 61 | Spo/ NA | Sensori‐motor Axonal | Y | 44 | NC | Fluctuating | 54 | / |
Abbreviations: AN, auditory neuropathy; F, female; Fam, familial; M, male; NA, not Available; NC, not communicated; Spo, sporadic.
Demyelinating neuropathy was diagnosed in 15 cases and axonal neuropathy in 12 cases. Age at onset varied from 2 to 73 years. In case of early onset, both demyelinating and axonal forms were observed (Patients I, IV, VIII, XIII, XV, XVI, XVII, XXI, XXIII and XXVI).
Hearing loss varied from mild to profound, could be progressive, and five cases of auditory neuropathy (AN) were related (Patients II, XII, XIV, XVIII and XXVI). Endocochlear involvement was also present with absence of Otoacoustic Emissions as in case of patient XIV. Age at onset varied from 1 to 68. One patient has successful cochlear implantation (Patient XIV) and one patient has recently been assessed for a cochlear implant (Patient XII). They both suffered from AN. In case of early onset (n = 8 cases), HL was severe to profound in four cases (Patients IV, XII, XIII and XXVI).
IPN and HL can occur nearly simultaneously in some patients, as in the case with patients XIV or patient XIX, or closely as in patients IV, XI, XVIII, XXI or XXVI. In contrast, the two features occured with a delay of up to 40 years, in others as in patients XXIV, and also patients XII, XVII and XX. Most of the time, hearing loss preceded IPN by several decades.
The other major symptoms observed were linked to other cranial nerve disorder such as optic neuropathy (n = 2), or bilateral laryngeal nerve paresis (n = 1). Neurological features such as cerebellar ataxia (n = 3), proprioceptive balance disorders (n = 4), urinary incontinence (n = 2) and pain (n = 2) were observed. Ophthalmological conditions like cataracts (n = 3) or retinal detachment (n = 1) were also present. Scoliosis was present in three cases. 2.
3.2. Genetic testing
Thirteen sporadic and 14 familial cases were noted. Inheritance mode was in favour of an autosomal dominant (AD) way in 13 cases, and an autosomal recessive (AR) one in 1 case.
By screening the 36 genes known to be involved in both IPN and HL, pathogenic variants were identified in 16 patients out of 27 (59.26%): PMP22 (n = 5), SH3TC2 (n = 4), MPZ (n = 2), NEFL (n = 2), PRPS1 (n = 1), TRPV4 (n = 1), ABHD12 (n = 1) (Figure 1).
Figure 1.
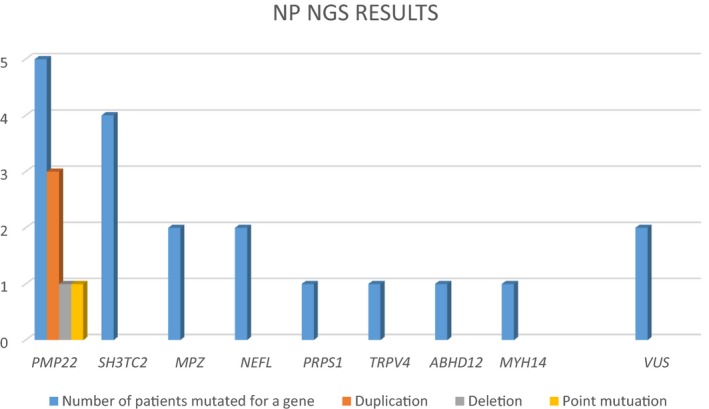
Representation of IPN NGS results of our series
The already known variants were: PMP22 duplication of 1.5Mb in three cases, PMP22 deletion of 1.4Mb in one case, PMP22 variant p.(Leu145Argfs*9), SH3TC2 variants c.2642A>G, p.(Asn881Ser); c.2860C>T, p.(Arg954*); c.3325C>T, p.(Arg1109Ter); c.3596G>A, p.(Trp1199*), NEFL variant c.293A>C, p.(Asn98Ser) and TRPV4 variant c.694C>T, p.(Arg232Cys).
As a consequence, seven novel variants, that could be classified as pathogenic or probably pathogenic, were discovered in five different genes PRPS1, MPZ, SH3TC2, NEFL and ABHD12. These variants were all absent from the different databases, and in silico studies were in favour of pathogenic variants (Tables 3 and 4).
-
‐
c.202A>T, p.(Met68Leu) in PRPS1: it was found in patient IV, who developed an X‐linked and axonal form of neuropathy. Family segregation was in accordance with a carrier mother (Figure 2).
-
‐
c.437T>C, p.(Val146Ala) and c.418T>C, p.(Ser140Pro) in MPZ: these two variants were present in patients XI and XVI respectively. Autosomal dominant transmission was suspected in one case. Family segregation was concordant as seen on pedigrees in Figure 2.
-
‐
c.3377T>C, p.(Leu1126Pro) and c.3617C>A, p.(Ala1206Asp) in SH3TC2: patient XVII developed an autosomal recessive form of demyelinating neuropathy and was associated with the known variant c.2860C>T, p.(Arg954*). Family segregation could not have been performed because parents’ DNA was not available.
-
‐
c. 3617C>A, p.(Ala1206Asp) in SH3TC2: Patient XX had an autosomal recessive form of demyelinating neuropathy associated with moderate hearing loss. His sister only developed peripheral neuropathy, but her DNA was not available.
-
‐
‐c.269A>G, p.(Glu90Gly) in NEFL: patient XIX had an autosomal dominant form of axonal neuropathy. Family segregation was concordant in the son presenting this pathogenic variant associated with IPN and hearing loss.
-
‐
c.379_385delAACTACTinsGATTCCTTATATACCATTGTAGTCTTACTGCTTTTGGTGAACACA, p.(Asn127Aspfs*23) in ABHD12: patient XXVI presented that homozygous variant. Family segregation was concordant, each asymptomatic parent presenting the heterozygous pathogenic variant.
Table 3.
Genotypes of our 27 patients
| Patient | First gene identified | Second gene identified | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference Family | Patient (gender/age in years) |
Form (Fam/spo)/AD, AR or X‐linked |
Gene | Mutation type | Zygosity | Nucleotide change | Amino acid change Localization | dbSNP | Gene | Mutation type | Zygosity | Nucleotide change | Amino acid change Localization | dbSNP |
| I | F, 87 | Spo/ NA | SPTLC1 | deletion | htz | c.‐35delCCGCTTCCTTCCGGAAGGCGGGTCACAAG | NF | / | / | / | / | / | ||
| II | F, 90 | Spo/ NA | / | / | / | / | / | / | / | / | / | / | ||
| III | M, 80 | Fam/ AD | / | / | / | / | / | / | / | / | / | / | ||
| IV | M, 35 | Spo/ NA | PRPS1 | missense | hemizygous | c.202A>T | p.(Met68Leu) | NF | / | / | / | / | / | |
| V | F, 86 | Fam/AD | PMP22 | duplication of 1.5 Mb | htz | / | / | N2G | N2G | N2G | N2G | N2G | ||
| VI | M, 88 | Fam/AD | DNMT1 | missense | htz | c.1250C>T | p.(Ala417Val) | NF | / | / | / | / | / | |
| VII | F, 60 | Fam/AD | PMP22 | duplication of 1.5 Mb | htz | / | / | N2G | N2G | N2G | N2G | N2G | ||
| VIII | F, 47 | Spo/NA | SH3TC2 | missense | HMZ | c.3325C>T | p.(Arg1109*) | rs80338934 | / | / | / | / | / | |
| IX | F, 47 | Fam/AD | / | / | / | / | / | / | / | / | / | / | ||
| X | M, 69 | Fam/AD | PMP22 | duplication of 1.5 Mb | htz | / | / | N2G | N2G | N2G | N2G | N2G | ||
| X1 | F, 68 | Fam/AD | MPZ | missense | htz | c.437T>C | p.(Val146Ala) | NF | N2G | N2G | N2G | N2G | N2G | |
| XII | M, 33 | Fam/AD | PMP22 | deletion of 1.4 Mb | htz | / | / | MYH14 | htz | missense | c.1067C>T | p.(Thr356Met) | rs151082668 | |
| XIII | F, 34 | Spo/ NA | / | / | / | / | / | / | / | / | / | / | ||
| XIV | F, 68 | Fam/AD | PMP22 | deletion | htz | c.434delT | p.(Leu145Argfs*9) | rs863225029 | COCH | htz | missense | c.326T>C | p.(Ile109Thr) | rs1219089 |
| XV | M, 29 | Spo/NA | / | / | / | / | / | / | / | / | / | / | ||
| XVI | F, 75 | Spo/NA | MPZ | missense | htz | c.418T>C | p.(Ser140Pro) | NF | N2G | N2G | N2G | N2G | N2G | |
| XVII | ♀, 68 | Spo/NA | SH3TC2 | missense x2 | htz x2 | c.2860C>T + c.3377T>C | p.(Arg954Ter) + p.(Leu1126Pro) | rs80338933 + NF | / | / | / | / | / | |
| XVIII | ♂, 68 | Fam/AD | TRPV4 | missense | htz | c.694C>T | p.(Arg232Cys) | rs387906904 | / | / | / | / | / | |
| XIX | ♀, 69 | Fam/AD | NEFL | missense | htz | c.269A>G | p.(Glu90Gly) | NF | N2G | N2G | N2G | N2G | N2G | |
| XX | ♂, 83 | Fam/AR | SH3TC2 | missense | HMZ | c.3617C>A | p.(Ala1206Asp) | NF | / | / | / | / | / | |
| XXI | ♀, 10 | Fam/AD | NEFL | missense | htz | c.293A>G | p.(Asn98Ser) | rs58982919 | / | / | / | / | / | |
| XXII | ♀, 80 | Spo/ NA | / | / | / | / | / | / | / | / | / | / | ||
| XXIII | ♂, 19 | Spo/ NA | / | / | / | / | / | / | / | / | / | / | ||
| XXIV | ♀, 71 | Spo/ NA | SH3TC2 | missense x2 | htz x2 | c.2642A>G + c.3596G>A | p.(Asn881Ser) + p.(Trp1199*) | rs80338930 + rs761972717 | / | / | / | / | / | |
| XXV | ♂, 78 | Fam/AD | / | / | / | / | / | / | / | / | / | / | ||
| XXVI | ♂, 38 | Spo/ NA | ABHD12 | deletion‐insertion | HMZ | c.379_385delAACTACTinsGATTCCTTATATACCATTGTAGTCTTACTGCTTTTGGTGAACACA | p.(Asn127Aspfs*23) | NF | / | / | / | / | / | |
| XXVII | ♂, 61 | Spo/ NA | / | / | / | / | / | / | / | / | / | / | ||
Abbreviations: hmz, homozygous; htz, heterozygous; N2G, no second Gene; NA, not available; NF, not found.
Table 4.
In silico studies of the seven novel pathogenic variants found with IPN‐NGS
| Genes | Mutations | Amino Acid change | Polyphen | SIFT | Mutation Taster | UMD‐Predictor | ExAC |
|---|---|---|---|---|---|---|---|
| ABHD12 | c.379_385delAACTACTinsGATTCCTTATATACCATTGTAGTCTTACTGCTTTTGGTGAACACA | p.(Asn127Aspfs*23) | / | / | / | / | Not found |
| MPZ | c.418T>C | p.(Ser140Pro) | 0.903 possibly damaging | 0.03 deleterious | deleterious | 87 pathogenic | Not found |
| c.437T>C | p.(Val146Ala) | 0.767 possibly damaging | 0.01 deleterious | deleterious | 75 pathogenic | Not found | |
| NEFL | c.269A>G | p.(Glu90Gly) | 0.999 probably damaging | 0 deleterious | / | / | Not found |
| PRPS1 | c.202A>T | p.(Met68Leu) | 0.208 benign | 0.06 tolerated | deleterious | 72 probably pathogenic | Not found |
| c.3377T>C | p.(Leu1126Pro) | 0.992 probably damaging | 0 deleterious | deleterious | 84 pathogenic | Not found | |
| SH3TC2 | c.3617C>A | p.(Ala1206Asp) | 0.759 possibly damaging | 0 deleterious | deleterious | 84 pathogenic | Not found |
Figure 2.
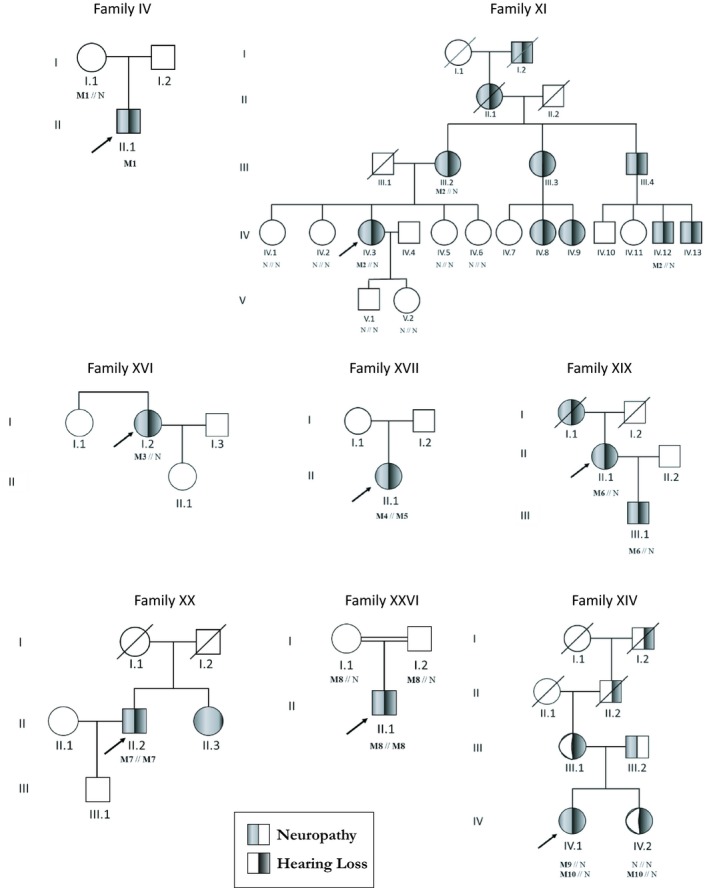
Pedigrees and associated variants ‐ N: Normal allele; M1: c.202A>T, p.(Met68Leu) in PRPS1; M2: c.437T>C, p.(Val146Ala) in MPZ; M3: c.418T>C, p.(Ser140Pro) in MPZ; M4: c.2860C>T, p.(Arg954Ter) in SH3TC2; M5: c.3377T>C, p.(Leu1126Pro) in SH3TC2; M6: c.269A>G, p.(Glu90Gly) in NEFL; M7: c.3617C>A, p.(Ala1206Asp) in SH3TC2; M8: c.379_385delAACTACTinsGATTCCTTATATACCATTGTAGTCT, p.(Asn127Aspfs*23) in ABHD12; M9: c.434delT, p.(Leu145Argfs*9) in PMP22; M10: c.336T>C, p.(Ile109Thr) in COCH
Another rare heterozygous variant in MYH14, c.1067C>T, p.(Thr356Met) was found in patient XII, already as being carrier of a 1.4Mb deletion of PMP22. MYH14 was found once in ExAc. The patient had presented a tomacular neuropathy since the age of 20. Severe auditory neuropathy started at the age of 5. Although family segregation was not possible, this variant seems to be potentially pathogenic.
Two additional variants were classified as variants of unknown significance (VUS): patient I presented a novel variant in SPTCL1, c. ‐35delCCGCTTCCTTCCGGAAGGCGGGTCACAAG, located in the promotor that could prevent SPTLC1 expression. However, segregation analysis was not possible, and we cannot conclude for this patient. For patient VI, we found a variant of uncertain significance: c.1250C>T, p.(Ala417Val) in DNMT1. It was not present in ExAc. However, this variant involves a residue which is not well conserved among species, and seems to be likely benign.
No other potential pathogenic variant was identified in any other screened genes.
Analysis at the DFNB1 locus did not reveal any pathogenic variant in the 27 patients.
In addition, NGS of the HL‐gene panel was performed in eight selected patients (Patients V, VII, X, XI, XII, XIV, XVI, XIX) carrying IPN and rearrangements in genes frequently involved (PMP22, MPZ and NEFL). This revealed a known pathogenic variant in COCH, c.326T>C, p.(Ile109Thr) (Bae et al., 2014; Pauw et al., 2007) for patient XIV who had a point deletion in PMP22 (patient XIV with c.434delT, p.[Leu145Argfs*9]). This female patient developed both demyelinating sensori‐motor neuropathy and progressive severe to profound auditory neuropathy at 50. Balance disorders were also reported. Family segregation was concordant, as her sister presented only hearing loss, associated with the COCH pathogenic variant.
3.3. Phenotype‐Genotype correlations
Hearing loss was mild or moderate in one case of PMP22 duplication, in cases of variants in SH3TC2 (n = 2), NEFL (n = 2), MPZ (n = 1), TRPV4 (n = 1). By contrast, hearing impairment was profound to severe in one case of 1.4Mb deletion PMP22, in cases of variants in PRPS1 and in ABHD12.
Hearing loss could develop simultaneously with neuropathy, in some patients with pathogenic variants in PRPS1 (n = 1), NEFL (n = 2), MPZ (n = 1), TRPV4 (n = 1); or at a distance in some cases of variants in SH3TC2 (n = 2) and ABHD12 (n = 1). Hearing loss occurrence varied widely with PMP22.
In our series, auditory neuropathy was found in five cases: three cases of PMP22 (1.4Mb deletion, point pathogenic variant), one case due to TRPV4 and one case due to ABHD12. For patient II, no pathogenic variant was identified. Endocochlear hearing loss was observed in patients with variants in PRPS1, MPZ, SH3TC2, NEFL and PMP22 (duplication).
In case of AR demyelinating IPN, SH3TC2 seems to be the most frequent cause (n = 4). This corresponds to CMT4C or AR‐CMTde‐SH3TC2 (Mathis et al., 2015 ). The frequent association with deafness and/or scoliosis in CMT4C may be a clue for the diagnosis.
Patients who develop polyneuropathy associated with sensorineural hearing loss and optic atrophy during childhood, with an X‐linked inheritance, such as Patient IV, should be assessed for PRPS1. PRPS1 pathogenic variants lead to CMTX5, a rare condition with only seven variants already reported. Our variant c.202A>T, p.(Met68Leu) is novel.
Pathogenic variants in NEFL responsible for CMT are rare and associated to various phenotypes. However, hearing loss is often linked to neuropathy, up to 64% of cases, especially with the pathogenic variants p.(Glu90Lys) and p.(Asn98Ser) (Likar et al., 2018), as it was the case in our p.(Glu90Gly) pathogenic variant. All these pathogenic variants are located in the head domain or in the two ends of the rod domain.
ABHD12 pathogenic variants lead to a rare phenotype named PHARR syndrome (MIM612674), which is a neurodegenerative disease including demyelinating Polyneuropathy, Hearing loss, cerebellar Ataxia, Retinis pigmentosa and early‐onset Cataract (PHARR). Patient XVI presented demyelinating Polyneuropathy, Hearing loss with auditory neuropathy, Ataxia and Cataracts.
TRPV4 is responsible for CMT2C or AD‐dHMN‐TRPV4. The phenotype is characterized by association with vocal cord and/or diaphragm paresis, and hearing loss (Dyck et al., 1994; Landoure et al., 2012). Patient XVIII’s phenotype corresponds to that clinical presentation.
MPZ variants causing axonal neuropathy are often associated with other features, such as hearing loss, or pupil abnormality. A characteristic audiogram of gentle slope curve towards the high frequencies is seen in patients suffering from CMT and sensorineural hearing loss. This was observed in patient XI.
Auditory neuropathy has been associated to PMP22 variants. As observed in our study, hearing loss associated to neuropathy due to PMP22 is very variable.
4. DISCUSSION
Through the analysis of the literature, we identified 36 genes that have been described to be involved in both IPN and hearing loss. They were all present in the 92 genes custom panel designed for CMT and associated neuropathies diagnosis. They consist in 16 CMT genes, 4 HMN genes, 2 HSN genes, and 14 other IPN genes, mostly syndromic forms. 1.
In our series of 27 patients suffering from both IPN and hearing loss, a molecular diagnosis was made in 16 patients, thus in approximately 60%. Hearing impairment is probably underdiagnosed among PN population. Those 27 patients have been found thanks to medical records and clinical questionnaires among a French series of 3,412 patients suffering from IPN.
In our study, SH3TC2 seems to be the most frequent gene involved in autosomal recessive demyelinating IPN, CMT4C or AR‐CMTde‐SH3TC2, as among 350 patients tested with IPN NGS, 13 had a pathogenic variants in this gene, and four patients were reported deaf. Hearing loss is the most frequent cranial nerve pathology (Azzedine, LeGuern, & Salih, 2008; Piscosquito et al., 2016; Yger et al., 2012). Scoliosis is present in more than one third of this population (Claramunt et al., 2007). Hearing loss frequency (in the patients from our series with variants in SH3TC2) is statistically different from that in the general population, showing that the pathogenic variant in SH3TC2 is directly responsible for hearing loss. We report two novel variants, c.3377T>C, p.(Leu1126Pro) associated with the already known variant, c.2860C>T, p.(Arg954*) and c.3617C>A, p.(Ala1206Asp).
For ABHD12, hearing loss is almost constant and is the first clinical sign, starting in the late teens. It is progressive and varies from moderate to profound. IPN is the most variable symptom.
PRPS1 is linked to three different phenotypes, always associated with hearing loss: CMTX5, DFNX1 and Arts syndrome. These three clinical presentations tend to overlap (Nishikura et al., 2018). In our series of 350 patients tested with NGS, only one patient was diagnosed with this gene, which is a novel hemizygous variant, c.202A>T, p.(Met68Leu). This variant is predicted as pathogenic.
For NEFL, hearing loss is associated with IPN in case of the following variants: (p.(Glu90Lys), p.(Asn98Ser), p.(Asn98Thr), p.(Leu268Pro), p.(Cys322_Asn326del), p.(Glu396Lys)) (Abe et al., 2009; Fabrizi et al., 2007; Horga et al., 2017; Silvera et al., 2013; Zuchner et al., 2004); and also with our new pathogenic variant: c.269A>G, p.(Glu90Gly). The seven heterozygous variants, including ours, are located on « hot spots» of the protein and seem directly linked to the hearing loss observed in the patients. A tonal audiogram with a moderate slope on the high frequencies is characteristic of variants p.(Asn98Ser) and p.(Glu90Gly) (Likar et al., 2018). The same audiogram was also observed in patient XIX.
TRPV4 is responsible for CMT2C or AD‐dHMN‐TRPV4. The phenotype is characterized by vocal cord paresis and/or diaphragm paresis, and hearing loss (Dyck et al., 1994; Landoure et al., 2012). Patient XVIII’s phenotype corresponds to this description. In our French series, five patients were detected with a variant in TRPV4, but only one of them was referred with diagnosed hearing loss.
Interestingly in our series of 3,412 IPN patients, 60 patients were mutated in MPZ. Nevertheless, only two patients were reported with hearing impairment (3.33%). Hearing loss frequency does not seem to be statistically different from that in the general population, suggesting that pathogenic variants in MPZ may not be the real cause of hearing loss in these patients who are susceptible to carry additional pathogenic variants in HL genes.
Hearing loss has also been described in association with duplication, deletion or point pathogenic variants of the PMP22 gene (Luigetti, Zollino, Conti, Romano, & Sabatelli, 2013). PMP22 is a major protein expressed in compact myelin of peripheral nerves as well as cranial nerves. Hearing loss in CMT patients is reported with point pathogenic variants or deletions in the transmembrane domain of PMP22, which is in close proximity to the extracellular component of this protein. It has been suggested that pathogenic variants at this site could cause defective interactions with other proteins in Schwann cells, which may result in hypo‐ or demyelination of the peripheral nerves, including the auditory nerve (Postelmans & Stokroos, 2006). Demyelination of the auditory nerve may also be a plausible mechanism to explain the retrocochlear involvement (Verhagen et al., 2005). In addition, endo and retrocochlear hearing loss has been observed in patient presenting the point variant c.193G>T, p.(Val65Phe) (Postelmans & Stokroos, 2006). However, while PMP22 duplication is responsible for 60% of CMT1, the AD demyelinating type, only few patients in fact suffer from hearing loss. In our series of 3,412 patients, 784 patients were mutated in PMP22 (23%) and we had information on associated hearing loss in only 5 of them (0.05%), presenting duplications (n = 3), a large deletion (n = 1) or 1 basepair deletion (n = 1). This 0.05% proportion is statistically different from the rate in the general population, with 10% of hearing loss. Nevertheless, the IPN and CMT populations are younger. It seems difficult to conclude that variations in PMP22 could protect from hearing loss. We therefore think it is probably underdiagnosed. The rarity of severe hearing loss in families with PMP22 pathogenic variants could rather suggest that most PMP22 pathogenic variants have minimal or no effects on hearing loss occurrence. As a consequence, hearing loss in that population could be due to other genes, as we started to point out for two of our patients, with a pathogenic variant in COCH and a suspected one in MYH14.
The COCH gene is responsible for DFNA9, which consists in post‐lingual progressive hearing loss with vestibular dysfunction, such as Meniere‐like diseases (Manolis et al., 1996). The cochline protein is detected in spindle‐shaped cells located along nerve fibers between the auditory ganglion and the sensory epithelium. Patient XIV presented with progressive severe to profound hearing loss, with desynchronised ABR and absent Acoustic Oto Emission. It is the first case to be reported with IPN so far. She also suffered from balance disorders, which could be due to vestibular dysfunction, as the penetrance is very variable.
Indeed, proprioceptive balance disorders or cerebellar ataxia could be misdiagnosed with vestibular dysfunction. Clinical examination is difficult in those patients suffering from IPN. Therefore, vestibular investigations should be performed in IPN patients suffering from balance disturbances.
MYH14 can lead to two different conditions: DFNA4 with progressive non syndromic hearing loss starting in the first or second decade of life and leading to severe to profound hearing loss in the fourth decade of life (Firstly described by Mirghomizadeh et al., 2002); or to IPN associated with myopathy, hoarseness, and hearing loss (Choi et al., 2011). This phenotype is only reported in one article. Patient XII had presented severe auditory neuropathy starting at the age of 5 and a tomacular neuropathy since the age of 20. No hoarseness or dysphony was reported. A rare heterozygous variant in MYH14, c.1067C>T, p.(Thr356Met), that could potentially explain hearing loss, was found in addition to the 1.4Mb deletion of PMP22 that explains the IPN. That is in accordance with our hypothesis that PMP22 is not responsible for hearing loss. MYH14 could nevertheless be also responsible for IPN. Actually, only one case has been reported with IPN and hearing loss (Choi et al., 2011), and two articles have been published about hearing loss with the same pathogenic variants (Chen et al., 1995; Mirghomizadeh et al., 2002).We can wonder whether a founder effect exists, or if a pathogenic variant in a HL gene close to MYH14 exists.
Hearing loss is reported regularly in patients suffering from IPN. The pathogenesis of hearing loss in those patients is uncertain, even though the cranial nerves are part of the peripheral nervous system and wrapped by Schwann cells. The hypothesis of retrocochlear dysfunction has been suggested and profound hearing loss is supposed to be due to desynchronization of the cochlear nerve (Anzalone et al., 2018). However, in our study we have shown that hearing impairment could be endocochlear, and not only due to AN, as it was the case for patients XIV. In our series, we noticed that both auditory nerve and cochlear dysfunctions were present, as auditory neuropathy was found in five cases: two cases of PMP22 (1.4Mb deletion, point pathogenic variant), one case due to TRPV4 and one case due to ABHD12 (molecular diagnosis was not made for the last one); and endocochlear hearing loss was observed in patients with variants in PRPS1, MPZ, SH3TC2, NEFL and PMP22 (duplication). That was also demonstrated by Kovach et al. (2002) in a patient presenting a variant in PMP22. However in most studies, there is a lack of information concerning testing to clearly distinguish cochlear and neuronal components.
To our knowledge, only three patients suffering from IPN and AN received a cochlear implant (absence of information about CMT type or hearing loss type by Anzalone et al., 2018; auditory neuropathy and absence of variant in PMP22 or GJB1 by Goswamy, Bruce, Green, & O’Driscoll, 2012; cochlear and auditory nerve dysfunction with a point pathogenic variant in PMP22, c.193G>T, p.(Val65Phe) by Postelmans et al., 2006). Our patient XIV also benefited from this surgery. Cochlear implant can recreate synchronous neuronal activity through the electrostimulation, and thus improves speech understanding. However, progress is slower than in other patients with cochlear implant. Patients describe a final significant benefit.
Moreover, hearing loss can precede, occur at the same time or follow IPN. It can be progressive, and the severity varies from mild to profound. We suggest that audiologic assessment should be made in all patients suffering from IPN, and vice versa, patients suffering from hearing loss should be tested for neuropathic involvement, associated with NGS screening of a large panel including genes involved in syndromic pathologies. Indeed, the delay between the different symptoms can be very long (up to 40 years) and a large NGS screening could help to find the gene involved and so to improve the care of the patient. This is for instance the case of Perrault syndrome type II (MIM# 233400), a rare autosomal recessive condition, characterized by sensorineural hearing loss, gonadic dysgenesis in males and females, and neurological features such as developmental delay or intellectual disability, cerebellar ataxia, motor and sensory peripheral neuropathy. As in patients suffering from IPN and hearing loss, the delay between the onset of the two or more symptoms can be up to 40 years, which leads to underdiagnose this phenotype if the involved genes are not tested in “hearing loss” NGS screening (Lerat et al., 2016).
To our knowledge, it is the first time that hearing loss screening has been carried out by MLPA and Sanger sequencing for GJB2 and GJB6. Analysis at the DFNB1 locus did not reveal any pathogenic variant for all diagnosed and known deaf patients. It is also the first time that HL NGS was tested on this population. However, only eight cases could be tested by HL‐NGS because of availability of the analysis. It would have been more significant to test all the 27 patients suffering from IPN and HL by HL‐NGS so as to give better genotype‐phenotype correlations.
Another possibility to explain both IPN and hearing loss is the presence of modifier genes that will induce that particular phenotype. That is why, for unsolved cases, WES could be very useful to identify new candidate genes for IPN and hearing loss, so as to improve diagnosis and patient care.
In addition, to better understand the physiopathology of neuropathies associated with hearing loss, animal models e.g. in rats and mice, should be developed. Indeed, it could be interesting to perform a biopsy of the auditory nerve and cochlea of wild‐type and affected animals in order to localize accurately, for example by immunochemistry the proteins involved in those two features. However, murine phenotype might be different as the organization differs.
5. CONCLUSION
Through an NGS strategy, we have been able to establish a molecular diagnosis in almost 60% of the cases presenting IPN associated with HL. As a consequence, a precise description of the phenotype can help molecular investigations. PMP22, and in a lesser proportion MPZ, involvement is not enough to explain hearing loss in patients suffering from hereditary peripheral neuropathy. Hearing loss can be due to cochlear impairment and/or auditory nerve dysfunction. As HL is certainly underdiagnosed in IPN patients, we suggest that audiologic tests should be systematically performed in these patients and their DNA should be screened with large NGS panels. This would enhance the diagnosis, help to better understand the physiopahology of IPN + HL and eventually improve patient's care.
CONFLICT OF INTEREST
None.
Supporting information
ACKNOWLEDGMENTS
We are thankful to the nucleic acid platform BISCEM, University of Limoges, France. We also thank Sylvie Gautier for English revision.
Lerat J, Magdelaine C, Roux A‐F, et al. Hearing loss in inherited peripheral neuropathies: Molecular diagnosis by NGS in a French series. Mol Genet Genomic Med. 2019;7:e839 10.1002/mgg3.839
REFERENCES
- Abe, A. , Numakura, C. , Saito, K. , Koide, H. , Oka, N. , Honma, A. , … Hayasaka, K. (2009). Neurofilament light chain polypeptide gene mutations in Charcot‐Marie‐Tooth disease: Nonsense mutation probably causes a recessive phenotype. Journal of Human Genetics, 54, 94–97. 10.1038/jhg.2008.13 [DOI] [PubMed] [Google Scholar]
- Anzalone, C. L. , Nuhanovic, S. , Olund, A. P. , & Carlson, M. L. (2018). Cochlear implantation in charcot‐marie‐tooth disease: Case report and review of the literature. Case Reports in Medicine, 2018, 1760978 10.1155/2018/1760978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzedine, H. , LeGuern, E. , & Salih, M. A. (2008). Charcot‐Marie‐Tooth Neuropathy Type 4C In: Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J., Stephens K., & Amemiya A., (eds). GeneReviews®. Seattle, WA: University of Washington. [Google Scholar]
- Bae, S.‐H. , Robertson, N. G. , Cho, H.‐J. , Morton, C. C. , Jung, D. J. , Baek, J. , … Kim, U. K. (2014). Identification of pathogenic mechanisms of COCH mutations, abolished cochlin secretion, and intracellular aggregate formation: Genotype‐phenotype correlations in DFNA9 deafness and vestibular disorder. Human Mutation, 35, 1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baux, D. , Vaché, C. , Blanchet, C. , Willems, M. , Baudoin, C. , Moclyn, M. , … Roux, A. F. (2017). Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing‐impaired patients. Scientific Reports, 7, 16783 10.1038/s41598-017-16846-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, A. H. , Ni, L. , Fukushima, K. , Marietta, J. , O’Neill, M. , Coucke, P. , … Smith, R. J. (1995). Linkage of a gene for dominant non‐syndromic deafness to chromosome 19. Human Molecular Genetics, 4, 1073–1076. 10.1093/hmg/4.6.1073 [DOI] [PubMed] [Google Scholar]
- Choi, B.‐O. , Kang, S. H. , Hyun, Y. S. , Kanwal, S. , Park, S. W. , Koo, H. , … Chung, K. W. (2011). A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14. Human Mutation, 32, 669–677. 10.1002/humu.21488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claramunt, R. , Sevilla, T. , Lupo, V. , Cuesta, A. , Millán, J. M. , Vílchez, J. J. , … Espinós, C. (2007). The p. R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot‐Marie‐Tooth disease type 4. Clinical Genetics, 71, 343–349. 10.1111/j.1399-0004.2007.00774.x [DOI] [PubMed] [Google Scholar]
- Dyck, P. J. , Litchy, W. J. , Minnerath, S. , Bird, T. D. , Chance, P. F. , Schaid, D. J. , & Aronson, A. E. (1994). Hereditary motor and sensory neuropathy with diaphragm and vocal cord paresis. Annals of Neurology, 35, 608–615. 10.1002/ana.410350515 [DOI] [PubMed] [Google Scholar]
- Fabrizi, G. M. , Cavallaro, T. , Angiari, C. , Cabrini, I. , Taioli, F. , Malerba, G. , … Rizzuto, N. (2007). Charcot‐Marie‐Tooth disease type 2E, a disorder of the cytoskeleton. Brain, 130, 394–403. 10.1093/brain/awl284 [DOI] [PubMed] [Google Scholar]
- Goswamy, J. , Bruce, I. A. , Green, K. M. J. , & O’Driscoll, M. P. (2012). Cochlear implantation in a patient with sensori‐neural deafness secondary to Charcot‐Marie‐Tooth disease. Cochlear Implants International, 13, 184–187. 10.1179/1754762811Y.0000000021 [DOI] [PubMed] [Google Scholar]
- Horga, A. , Laurà, M. , Jaunmuktane, Z. , Jerath, N. U. , Gonzalez, M. A. , Polke, J. M. , … Reilly, M. M. (2017). Genetic and clinical characteristics of NEFL-related Charcot-Marie-Tooth disease. Journal of Neurology, Neurosurgery, and Psychiatry, 88(7), 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach, M. J. , Campbell, K. C. M. , Herman, K. , Waggoner, B. , Gelber, D. , Hughes, L. F. , & Kimonis, V. E. (2002). Anticipation in a unique family with Charcot‐Marie‐Tooth syndrome and deafness: Delineation of the clinical features and review of the literature. American Journal of Medical Genetics, 108, 295–303. 10.1002/ajmg.10223 [DOI] [PubMed] [Google Scholar]
- Landouré, G. , Sullivan, J. M. , Johnson, J. O. , Munns, C. H. , Shi, Y. , Diallo, O. , … Sumner, C. J. (2012). Exome sequencing identifies a novel TRPV4 mutation in a CMT2C family. Neurology, 79, 192–194. 10.1212/WNL.0b013e31825f04b2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat, J. , Jonard, L. , Loundon, N. , Christin‐Maitre, S. , Lacombe, D. , Goizet, C. , … Marlin, S. (2016). An application of NGS for molecular investigations in perrault syndrome: Study of 14 families and review of the literature. Human Mutation, 37, 1354–1362. 10.1002/humu.23120 [DOI] [PubMed] [Google Scholar]
- Likar, T. , Hasanhodžić, M. , Teran, N. , Maver, A. , Peterlin, B. , & Writzl, K. (2018). Diagnostic outcomes of exome sequencing in patients with syndromic or non‐syndromic hearing loss. PLoS ONE, 13, e0188578 10.1371/journal.pone.0188578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luigetti, M. , Zollino, M. , Conti, G. , Romano, A. , & Sabatelli, M. (2013). Inherited neuropathies and deafness caused by a PMP22 point mutation: A case report and a review of the literature. Neurological Sciences, 34, 1705–1707. 10.1007/s10072-012-1277-5 [DOI] [PubMed] [Google Scholar]
- Manolis, E. N. , Yandavi, N. , Nadol, J. B. , Eavey, R. D. , McKenna, M. , Rosenbaum, S. , … Seidman, J. G. (1996). A gene for non‐syndromic autosomal dominant progressive postlingual sensorineural hearing loss maps to chromosome 14q12‐13. Human Molecular Genetics, 5, 1047–1050. 10.1093/hmg/5.7.1047 [DOI] [PubMed] [Google Scholar]
- Mathis, S. , Goizet, C. , Tazir, M. , Magdelaine, C. , Lia, A.‐S. , Magy, L. , & Vallat, J. M. (2015). Charcot‐Marie‐Tooth diseases: An update and some new proposals for the classification. Journal of Medical Genetics, 52, 681–690. 10.1136/jmedgenet-2015-103272 [DOI] [PubMed] [Google Scholar]
- Mirghomizadeh, F. , Bardtke, B. , Devoto, M. , Pfister, M. , Oeken, J. , König, E. , … Blin, N. (2002). Second family with hearing impairment linked to 19q13 and refined DFNA4 localisation. European Journal of Human Genetics, 10, 95–99. 10.1038/sj.ejhg.5200769 [DOI] [PubMed] [Google Scholar]
- Nishikura, N. , Yamagata, T. , Morimune, T. , Matsui, J. , Sokoda, T. , Sawai, C. , … Maruo, Y. (2018). X‐linked Charcot‐Marie‐Tooth disease type 5 with recurrent weakness after febrile illness. Brain and Development, 41, 201–204. 10.1016/j.braindev.2018.08.006 [DOI] [PubMed] [Google Scholar]
- Pauw, R. J. , Huygen, P. L. M. , Collin, R. W. J. , Cruysberg, J. R. M. , Hoefsloot, L. H. , Kremer, H. , & Cremers, C. W. (2007). Phenotype description of a novel DFNA9/COCH mutation, I109T. Annals of Otology, Rhinology, and Laryngology, 116, 349–357. 10.1177/000348940711600506 [DOI] [PubMed] [Google Scholar]
- Piscosquito, G. , Saveri, P. , Magri, S. , Ciano, C. , Gandioli, C. , Morbin, M. , … Pareyson, D. (2016). Screening for SH3TC2 gene mutations in a series of demyelinating recessive Charcot‐Marie‐Tooth disease (CMT4). Journal of the Peripheral Nervous System, 21, 142–149. 10.1111/jns.12175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postelmans, J. T. F. , & Stokroos, R. J. (2006). Cochlear implantation in a patient with deafness induced by Charcot‐Marie‐Tooth disease (hereditary motor and sensory neuropathies). Journal of Laryngology and Otology, 120, 508–510. 10.1017/s0022215106000727 [DOI] [PubMed] [Google Scholar]
- Sivera, R. , Sevilla, T. , Vílchez, J. J. , Martínez‐Rubio, D. , Chumillas, M. J. , Vázquez, J. F. , … Espinós, C. (2013). Charcot‐Marie‐Tooth disease: Genetic and clinical spectrum in a Spanish clinical series. Neurology, 81, 1617–1625. 10.1212/WNL.0b013e3182a9f56a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman, V. , Nelis, E. , Van Hul, W. , Nieuwenhuijsen, B. W. , Chen, K. L. , Wang, S. , … Van Broeckhoven, C. (1992). The peripheral myelin protein gene PMP‐22 is contained within the Charcot‐Marie‐Tooth disease type 1A duplication. Nature Genetics, 1, 171–175. 10.1038/ng0692-171 [DOI] [PubMed] [Google Scholar]
- Verhagen, W. I. M. , Huygen, P. L. M. , Gabre??ls‐Festen, A. A. W. M. , Engelhart, M. , van Mierlo, P. J. W. B. , & van Engelen, B. G. M. (2005). Sensorineural hearing impairment in patients with Pmp22 duplication, deletion, and frameshift mutations. Otology & Neurotology, 26, 405–414. 10.1097/01.mao.0000169769.93173.df [DOI] [PubMed] [Google Scholar]
- Yger, M. , Stojkovic, T. , Tardieu, S. , Maisonobe, T. , Brice, A. , Echaniz‐Laguna, A. , … Dubourg, O. (2012). Characteristics of clinical and electrophysiological pattern of Charcot‐Marie‐Tooth 4C. Journal of the Peripheral Nervous System, 17, 112–122. 10.1111/j.1529-8027.2012.00382.x [DOI] [PubMed] [Google Scholar]
- Züchner, S. , Mersiyanova, I. V. , Muglia, M. , Bissar‐Tadmouri, N. , Rochelle, J. , Dadali, E. L. , … Vance, J. M. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nature Genetics, 36, 449–451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials