

Abstract
Background
OFD1 has long been recognized as the gene implicated in the classic dysmorphology syndrome, oral‐facial‐digital syndrome type I (OFDSI). Over time, pathogenic variants in OFD1 were found to be associated with X‐linked intellectual disability, Joubert syndrome type 10 (JBTS10), Simpson‐Golabi‐Behmel syndrome type 2 (SGBS2), and retinitis pigmentosa. Recently, OFD1 pathogenic variants have been implicated in primary ciliary dyskinesia (PCD), a disorder of the motile cilia with a phenotype that includes recurrent oto‐sino‐pulmonary infections, situs abnormalities, and decreased fertility.
Methods
We describe three male patients with PCD who were found to have hemizygous pathogenic variants in OFD1, further supporting that PCD is part of a clinical spectrum of OFD1‐related disorders. In addition, we provide a review of the available clinical literature describing patients with OFD1 variants and highlight the phenotypic variability of OFD1‐related disease.
Results
Some individuals with hemizygous OFD1 variants have PCD, either apparently isolated or in combination with other features of OFD1‐related disorders.
Conclusion
As clinicians consider the presence or absence of conditions allelic at OFD1, PCD should be considered part of the spectrum of OFD1‐related disorders. Understanding the OFD1‐related disease spectrum may allow for more focused genetic testing and more timely management of treatable sequelae.
Keywords: OFD1, Primary ciliary dyskinesia
1. INTRODUCTION
OFD1, residing on chromosome Xp22.2, has long been known to be the gene implicated in the classic dysmorphology syndrome, oral‐facial‐digital syndrome type I (OFDSI, MIM 311200) (Ferrante et al., 2001). Over recent years, multiple phenotypes have been identified as allelic to OFDSI including primary ciliary dyskinesia (PCD, MIM 244400). PCD is a disorder of the motile cilia with a phenotype that includes recurrent oto‐sino‐pulmonary infections, neonatal respiratory distress, bronchiectasis, situs abnormalities, and decreased fertility (Knowles, Daniels, Davis, Zariwala, & Leigh, 2013). Herein, we describe three individuals with PCD due to hemizygous loss‐of‐function in OFD1, supporting that PCD is part of a clinical spectrum of OFD1‐related disorders. To this end, we open with a brief description of clinical features known to be caused by OFD1 variants.
In 1954, OFDSI was described in girls who had dysmorphic features involving the mouth, face, and digits (Papillon‐Léage & Psaume, 1954). Classically, it was thought that OFDSI was inherited in an X‐linked dominant pattern and that it could be lethal in males (Gorlin, Anderson, & Scott, 1961); however, males with OFDSI have been described (Goodship, Platt, Smith, & Burn, 1991).
In 2006, a family was reported to have a distinct phenotype that segregated with an OFD1 frameshift variant (Budny et al., 2006). The male index patient had macrocephaly, intellectual disability, obesity, recurrent upper and lower airway infections, high‐arched palate, low‐set ears, and short fingers. Other affected males were noted to have chronic respiratory tract infections, and some had macrocephaly; heterozygous females were unaffected. The authors acknowledged that this was a different phenotype than OFDSI and had X‐linked recessive inheritance. Given the phenotypic overlap with Simpson‐Golabi‐Behmel syndrome (MIM 312870), this condition was recognized as Simpson‐Golabi‐Behmel Syndrome type 2 (SGBS2, MIM 300209). Due to chronic respiratory tract infections in affected individuals and abnormal high‐speed video microscopy (HSVM) of the nasal epithelium in the index case, PCD was recognized as part of this spectrum of disease. These data suggested that SGBS2 and PCD could exist concurrently in individuals with hemizygous loss‐of‐function in OFD1. This X‐linked inheritance of PCD differs from the vast majority of PCD, which is usually autosomal recessive.
Other conditions associated with OFD1 variants include Joubert syndrome type 10 (JBTS10, MIM 300804) (Coene et al., 2009) and non‐syndromic retinitis pigmentosa (RP23, MIM 300424) (Webb et al., 2012). Some individuals with OFD1 variants have overlapping features of OFDSI, JBTS10, and/or other associated symptoms (Field et al., 2012; Tsurusaki et al., 2013; Wentzensen et al., 2016).
Further characterizing a spectrum of disease associated with OFD1 variants, an affected patient and unrelated fetus with hemizygous loss‐of‐function were described as having features of SGBS2 and JBTS10 (Thauvin‐Robinet et al., 2013). A 13‐year‐old boy had intellectual disability, polydactyly, oculomotor apraxia, molar tooth sign (MTS), obesity, and chronic sinusitis and bronchitis. His nasal nitric oxide (nNO), a diagnostic tool for PCD, was 78 nl/min (a value <77 nl/min has a very good detection rate for PCD given a compatible clinical phenotype (Shapiro et al., 2016)). The fetus described in this case (Thauvin‐Robinet et al., 2013) was reported to be the first subject identified with an OFD1 variant who had situs inversus.
Clearly, there is variable expressivity observed in individuals with hemizygous loss of OFD1 function. In fact, some individuals appear to have features of multiple conditions that are associated with OFD1 variants. PCD is an important part of OFD1‐related disease, and the three patients described in this report contribute to our understanding of syndromic PCD and the OFD1 phenotype.
2. CASE REPORTS
2.1. Case 1
A 33‐year‐old man has a history of recurrent sinus infections and recurrent pneumonia. Sputum cultures have grown Nocardia farcinica, methicillin‐sensitive Staphylococcus aureus, and Mycobacterium chelonae. Computerized tomography (CT) imaging of the chest demonstrated bronchiectasis of the left lower lobe, inferior lingula, and medial segment of the right middle lobe. Spirometry results included forced vital capacity (FVC) 3.99 liters (L) (82% predicted), forced expiratory volume in the first second (FEV1) 2.62 L (65% predicted), and forced expiratory flow between 25 to 75% of FVC (FEF25‐75) of 1.59 L/s (38% predicted). There are no known situs abnormalities, and fertility status is unknown. There was no neonatal respiratory distress, and there were no significant episodes of early otitis media. There is no history of congenital heart disease or polycystic kidney disease. Electroretinograms did not demonstrate retinitis pigmentosa but were suspicious for retinal dystrophy. A nNO had been measured at 43 and 52 nl/min. Electron microscopy demonstrated normal ciliary ultrastructure (Figure 1).
Figure 1.
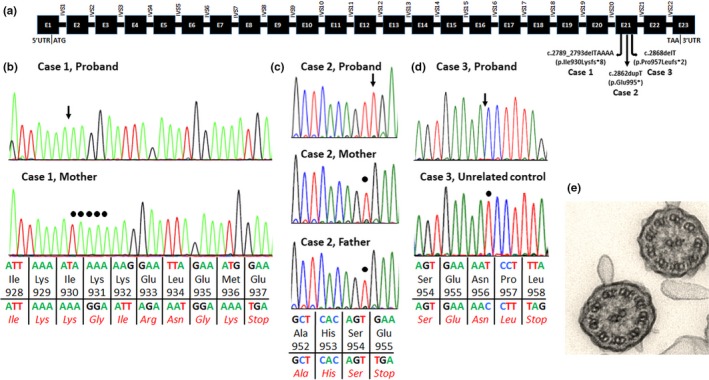
(a) The OFD1 transcript (NM_003611.2) is 3,651 base pairs and consists of 23 exons (represented by black rectangles), encoding a 1,012 amino acid protein. Exon 1 contains the entire 5′ untranslated region (5′UTR) and the start codon (ATG). Exon 23 contains the stop codon (TAA) and the entire 3′ untranslated region (3′UTR). The pathogenic hemizygous variants in the three cases described are all located in exon 21 and shown above. Introns (IVS) are represented by horizontal lines. (b‐d) Electropherograms for hemizygous variant in Case 1 affected (top) and wild‐type mother (bottom) (b), Case 2 affected (top) and wild‐type mother and father (bottom) (c), and Case 3 affected (top) and wild‐type unrelated control (bottom) (d). An arrow points to the pathogenic variant in the affected individual’s electropherogram, and a circle marks the wild‐type sequence at that nucleotide in the controls (parents in b and c; unrelated control in d). Wild‐type sequence, amino acid residue, and codon number are shown above the horizontal line. The sequence with the hemizygous variant and amino acid residues (in red) are shown below the horizontal line. (e) Transmission electron microscopy of the Case 1 proband demonstrating normal ciliary ultrastructure with the characteristic 9 + 2 microtubule pair organization and normal dynein arms
Dysmorphic features include macrocephaly, nasal shortening, mid‐face hypoplasia, alopecia, high‐arched palate, and postaxial polydactyly. BMI is 35 kg/m2. He has developmental delay and mild intellectual disability. He has a history of neonatal hypotonia, poor feeding as an infant with gastrostomy tube placement at age 3 months, gastroesophageal reflux, and episodes of apnea that began at age 3 weeks. CT imaging of the head as an infant demonstrated enlarged ventricles and an abnormal appearance of the white matter of the brain. Although he had nuclear magnetic resonance imaging (MRI) performed as an infant as part of a research study, neither the original images nor a radiologist report is available. He had transient hypoglycemia and transient hyperammonemia as an infant. As an adult, he had a relative decrease in non‐switched memory and switched memory B cells with normal immunoglobulin concentrations except for a slight increase in IgG subclass 4.
Multiple molecular tests were nondiagnostic. Ultimately, clinical exome sequencing identified a de novo hemizygous pathogenic OFD1 (NM_003611.2) variant: c.2789_2793delTAAAA (p.Ile930Lysfs*8). No other reported variants appeared to be disease‐causing in this individual. Specifically, a single variant was identified in DYNC1H1 (a gene associated with neurologic features and excluded as disease‐causing given the phenotype and inheritance from an asymptomatic father), only a single variant was identified in BBS7 (a gene associated with autosomal recessive disease), and two variants were identified in cis in CEP290 (a gene that is associated with disease only in the presence of biallelic variants). There were no other variants reported in genes known to be implicated in PCD, cystic fibrosis, or surfactant disorders.
2.2. Case 2
A 16‐year‐old male patient has a history of the following constellation of symptoms: neonatal respiratory distress following a full‐term gestation, recurrent ear infections (with tympanostomy tube placements, mastoidectomies, and mastoid debridement), recurrent sinusitis (with bilateral endoscopic maxillary antrostomies and bilateral endoscopic total and anterior–posterior ethmoidectomies), and recurrent pneumonia with a daily wet cough. At 9 months of age, he had acute respiratory failure, was intubated, and had tracheostomy placement with decannulation at 5 years of age. At age 15 years, chest CT showed no evidence of bronchiectasis. He is unable to cooperate with spirometry or measurement of nNO. Previous cultures demonstrated Pseudomonas aeruginosa, methicillin‐sensitive Staphylococcus aureus, and Candida. There are no situs abnormalities, and fertility status is unknown. He has not had electron microscopy assessment of ciliary ultrastructure or HSVM.
Dysmorphic features include macrocephaly, large forehead, deep‐set eyes, low nasal bridge, short and upturned nose, thick lips with a wide mouth, and postaxial polydactyly of the hands. BMI is 31 kg/m2. Neurologic features include intellectual disability. He is minimally verbal and minimally able to ambulate. He has a history of chronic cerebral atrophy, seizures, hypotonia, and apnea. He has gastroesophageal reflux and had a Nissen fundoplication and gastrostomy tube placement, and had dysphagia requiring a diet of pureed and soft foods. MRI of the brain demonstrated a large arachnoid cyst and mild to moderately enlarged ventricles, but no MTS. Other past medical history includes nephrocalcinosis requiring diuretics, atrial septal defect status post Amplatzer placement, iatrogenic adrenal insufficiency, and immunodeficiency with hypogammaglobulinemia and a deficiency of class switching memory B cells that has at times required intravenous immunoglobulins.
Genetic testing of 20 genes implicated in PCD and CFTR (Table 1) identified only a de novo pathogenic variant, c.2862dupT (p.Glu995*), in OFD1.
Table 1.
Genes analyzed for the presence of pathogenic/likely pathogenic variant
| Genes (alternative name) | Transcript Identifier | Disease association | Case 2: Ambry Panel | Case 3: Whole‐Exome Sequencing | |
|---|---|---|---|---|---|
| 1 | ARMC4 | NM_018076.2 | PCD | Yes | Yes |
| 2 | CCDC103 | NM_213607.1 | PCD | Yes | Yes |
| 3 | CCDC114 | NM_144577.3 | PCD | Yes | Yes |
| 4 | CCDC151 | NM_145045.4 | PCD | No | Yes |
| 5 | CCDC39 | NM_181426.1 | PCD | Yes | Yes |
| 6 | CCDC40 | NM_017950.3 | PCD | Yes | Yes |
| 7 | CCDC65 (DRC2) | NM_033124.4 | PCD | No | Yes |
| 8 | CCNO | NM_021147.4 | PCD | No | Yes |
| 9 | CFAP298 (C21orf59) | NM_021254.2 | PCD | No | Yes |
| 10 | CFAP300 (C11orf70) | NM_032930.2 | PCD | No | Yes |
| 11 | DNAAF1 (LRRC50) | NM_178452.4 | PCD | Yes | Yes |
| 12 | DNAAF2 (KTU) | NM_018139.2 | PCD | Yes | Yes |
| 13 | DNAAF3 | NM_001256714.1 | PCD | Yes | Yes |
| 14 | DNAAF4 (DYX1C1) | NM_130810.3 | PCD | No | Yes |
| 15 | DNAAF5 (HEATR2) | NM_017802.3 | PCD | Yes | Yes |
| 16 | DNAH1 | NM_015512.4 | PCD | No | Yes |
| 17 | DNAH11 | NM_001277115.1 | PCD | Yes | Yes |
| 18 | DNAH5 | NM_001369.2 | PCD | Yes | Yes |
| 19 | DNAH8 | NM_001206927.1 | PCD | No | Yes |
| 20 | DNAH9 | NM_001372.3 | PCD | No | Yes |
| 21 | DNAI1 | NM_012144.3 | PCD | Yes | Yes |
| 22 | DNAI2 | NM_023036.4 | PCD | Yes | Yes |
| 23 | DNAJB13 | NM_153614.2 | PCD | No | Yes |
| 24 | DNAL1 | NM_031427.3 | PCD | No | Yes |
| 25 | DRC1 (CCDC164) | NM_145038.3 | PCD | No | Yes |
| 26 | GAS8 | NM_001481.2 | PCD | No | Yes |
| 27 | GAS2L2 | NM_139285.3 | PCD | No | Yes |
| 28 | HYDIN | NM_032821.2 | PCD | No | Yes |
| 29 | LRRC56 | NM_198075 | PCD | No | Yes |
| 30 | LRRC6 | NM_012472.4 | PCD | Yes | Yes |
| 31 | MCIDAS | NM_001190787.1 | PCD | No | Yes |
| 32 | NME8 (TXNDC3) | NM_016616.4 | PCD | Yes | Yes |
| 33 | OFD1 | NM_003611.2 | PCD; OFD1 | Yes | Yes |
| 34 | PIH1D3 | NM_001169154.1 | PCD | No | Yes |
| 35 | RPGR | NM_000328.2 | PCD | Yes | Yes |
| 36 | RSPH1 | NM_080860.3 | PCD | No | Yes |
| 37 | RSPH3 | NM_031924.4 | PCD | No | Yes |
| 38 | RSPH4A | NM_001010892.2 | PCD | Yes | Yes |
| 39 | RSPH9 | NM_152732.4 | PCD | Yes | Yes |
| 40 | SPAG1 | NM_172218.2 | PCD | Yes | Yes |
| 41 | STK36 | NM_001243313 | PCD | No | Yes |
| 42 | TTC25 | NM_031421.2 | PCD | No | Yes |
| 43 | ZMYND10 | NM_015896.2 | PCD | No | Yes |
| 44 | CFTR | NM_000492.3 | Cystic Fibrosis | Yes | Yes |
| 45 | SFTPB | NM_000542.3 | SMDP1 | No | Yes |
| 46 | SFTPC | NM_001172410.1 | SMDP2 | No | Yes |
| 47 | ABCA3 | NM_001089.2 | SMDP3 | No | Yes |
| 48 | CSF2RA | NM_001161529.1 | SMDP4 | No | Yes |
| 49 | CSF2RB | NM_000395.2 | SMDP5 | No | Yes |
| 50 | SERPINA1 | NM_000295.4 | A1AT deficiency | No | Yes |
PCD, Primary ciliary dyskinesia (MIM# 244400); OFD1, Oral‐facial‐digital syndrome I (MIM# 311200); Cystic fibrosis (MIM# 219700); A1AT, Alpha‐1‐Antitrypsin deficiency (MIM# 613490); SMDP, Surfactant metabolism dysfunction protein [MIM#s for SFTPB (265120); SFTPC (610913); ABCA3 (610921; CSF2RA (300770); and CSF2RB (614370)].
2.3. Case 3
A 32‐year‐old man of Hispanic ethnicity was referred to pulmonology with the following history: recurrent sinus infections with three surgical interventions, recurrent pneumonia, and situs inversus totalis. CT imaging of the chest demonstrated bronchiectasis. Spirometry results were FVC 2.31 L (47.7% predicted), FEV1 0.98 L (25.0% predicted), and FEF25‐75 0.31 L/s (6.3% predicted). Sputum cultures grew H. influenza and oral flora. This patient is reported to have had two daughters, but it is not known if assisted reproductive technologies were used. DNA was not available for paternity testing, thus if donor sperm were not used, his daughters would be obligate carriers, information that would be useful to the family for reproductive counseling. However, we are unable to contact his family. There is no known history of congenital heart disease, polycystic kidney disease, or retinitis pigmentosa. The nNO level was 54.5 nl/min. Electron microcopy demonstrated normal ciliary ultrastructure.
This individual has no history of intellectual disability. Otherwise, details regarding his neurologic and ophthalmologic review of systems are not known.
As part of his PCD evaluation, exome sequencing was performed through the Yale Center for Mendelian Genomics (NIH/NHLBI Mendelian Exome Project) followed by analysis of genes associated with PCD and other respiratory conditions (Table 1). A hemizygous pathogenic OFD1 variant, c.2868delT (p.Pro957Leufs*2), was identified and parental specimens are not available for variant segregation analysis.
3. DISCUSSION
Few publications have suggested an OFD1 variant as a cause of PCD. As above, a family was characterized in which affected males have features of SGBS2 and PCD segregating with a hemizygous OFD1 variant (Budny et al., 2006). Previously, a family in which affected males had features of Simpson‐Golabi‐Behmel syndrome and macrocephaly and died of pneumonia was described with the phenotype segregating with a Xp22 locus (Brzustowicz, Farrell, Khan, & Weksberg, 1999), but no OFD1 genetic testing was reported in this family. As mentioned, a 13‐year‐old boy with features of intellectual disability, SGBS2, JBTS10, and PCD was described with the same hemizygous OFD1 variant as the individual described in Case 1 (c.2789_2793delTAAAA) (Thauvin‐Robinet et al., 2013). Notably, the individual described by Thauvin‐Robinet et al. had inherited maternally the c.2789_2793delTAAAA variant, whereas this was a de novo variant in Case 1. Clearly, these reports suggest an association of PCD with OFD1 variants, but very few individuals with PCD due to an OFD1 variant have been described.
As others have acknowledged, the PCD phenotype is clearly present in some individuals with OFD1 variants. As we characterize the spectrum of OFD1‐related disease, there are diagnostic implications. If syndromic features of OFD1‐related disease are found, then focused genetic testing can be considered rather than large panel testing for PCD or JS. Conversely, when a pathogenic OFD1 variant is identified, the patient should be evaluated for PCD. Indeed, the individual in Case 1 was diagnosed with PCD after his genetic testing returned and a referral for nNO testing was made.
Furthermore, the individuals described in this manuscript have some notable features that highlight the variable expressivity of OFD1‐related disorders (Table 2). All three individuals have symptoms that support a PCD phenotype, and the individuals in Cases 1 and 3 had nNO levels in the diagnostic range. As detailed, the dysmorphic features, cognitive function, neurologic symptoms, and other organ dysfunction observed in these three patients are varied. The individuals in Cases 1 and 2 have overlapping features of both SGB2 and JBTS10 although there is no MRI of brain available for Case 1, and imaging was negative for MTS in Case 2. They both have a history of polydactyly, obesity, macrocephaly, intellectual disability, hydrocephalus, hypotonia, and breathing abnormalities. Both Cases 1 and 2 demonstrate a deficiency of memory B cells, and Case 2 had hypogammaglobulinemia. It is not clear if this contributed to recurrent infections. To our knowledge, deficiency of memory B cells is not a characteristic finding in individuals with OFD1 variants, but this observation is noteworthy as we learn more about OFD1‐related disorders.
Table 2.
Clinical features of current cases and of OFD1‐associated syndromes
| Features | Case 1 | Case 2 | Case 3 | PCD | OFDSI | JBTS | SGBS |
|---|---|---|---|---|---|---|---|
| MIM # when condition is associated with OFD1 | N/A | N/A | N/A | Not assigned | 311200 | 300804 | 300209 |
| Gender | Male | Male | Male | Both | Classically female, but males have been described | Both (but JBTS10 is XLR) | Classically male |
| Ethnicity | English, Czechoslovakian | White | Hispanic | Multiple ethnicities | Multiple ethnicities | Multiple ethnicities | Multiple ethnicities |
| Inheritance pattern | XLR, de novo variant | XLR, de novo variant | XLR | AR (occasionally XLR) | Classically XLD | AR (occasionally XLR) | XLR |
| Variant | c.2789_2793delTAAAA (p.Ile930Lysfs*8) | c.2862dupT (p.Glu995*) | c.2868delT (p.Pro957Leufs*2) | N/A | N/A | N/A | N/A |
| Laterality defects | No | No | Situs inversus totalis | Yes | No | No | No |
| Oto‐sino‐pulmonary disease | Yes | Yes | Yes | Yes | No | No | No |
| Nasal nitric oxide (nL/min) | 43 and 52 | Not performed | 54.5 | Low | Presumed Normal | Presumed Normal | Presumed Normal |
| Neurologic symptoms | Hypotonia, poor feeding, apneic episodes | Hypotonia, apnea, dysphagia/feeding difficulties, seizures | None known | No | MRI findings as below | Hypotonia, poor feeding, irregular breathing. JSRD can include occipital encephalocele | Hypotonia, occasionally seizures |
| MRI of brain | Unknown | No MTS. Large arachnoid cyst, enlarged ventricles. | Unknown | Normal | Agenesis corpus callosum, porencephaly, hydrocephalus, polymicrogyria, Dandy‐Walker malformation | MTS, agenesis of corpus callosum | Occasionally CNS abnormalities (agenesis of corpus callosum, Chiari malformations, etc. |
| Macrocephaly | Yes | Yes | Not known | No | No | No | Yes |
| Dysmorphic features | Nasal shortening, mid‐face hypoplasia, high‐arched palate, postaxial polydactyly | Large forehead, deep‐set eyes, low nasal bridge, short and upturned nose, thick lips with wide mouth, postaxial polydactyly | None known | No | Oral frenuli, lobulated or bifid tongue, cleft lip/palate, dental anomalies, alar hypoplasia, lateral placement of inner canthi, digit anomalies | Broad forehead, arched eyebrows, ptosis, wide‐spaced eyes, open mouth, polydactyly | Coarse facies, hypertelorism, downslanting palpebral fissures, macroglossia, cleft lip/palate, digit anomalies, short webbed neck. |
| Intellectual disability | Yes | Yes | No | No | Yes | Yes | Yes |
| Renal anomalies | No | No | No | No | Polycystic kidney disease | JSRD can include cystic dysplasia or nephronophthisis | Occasionally such as cystic or large kidneys or duplication of renal pelvis |
| Congenital heart disease | No | ASD | No | Yes | No | No | Occasionally |
| Retinitis pigmentosa | No but abnormal electroretinogram | No | Not known | Not classically (occasionally with RPGR variants) | No | JSRD can include retinal dystrophy | No |
| Other features | Immunodeficiency as described in case | Immunodeficiency as described in case | Reported to have fathered children as described in case | Neonatal respiratory distress, infertility | Fibrocystic liver and pancreas | JRSD can include ocular coloboma among other ophthalmologic anomalies and hepatic fibrosis. | SGBS is associated with increased risk of specific tumors (but SGBS2 is not known to be) |
| References | Current study | Current study | Current study | Shapiro et al., 2016 | Jones, Jones, & Del Campo, 2013; Papillon‐Léage Mme & Psaume J. 1954 | Parisi, 2009 | Jones et al., 2013 |
Comparison of the phenotypes of Case 1, Case 2, Case 3, PCD, JBTS, and SGBS.
This table highlights some of the important characteristics of these conditions but is not meant to be a thorough summary of all phenotypic features. Individuals with features of these syndromes due to OFD1 variants may not fit well into the classic descriptions provided in the table (i.e., individuals with JBTS10 may have laterality defects, individuals with SGBS2 are not known to have the same cancer predisposition as individuals with SGBS, etc.).
Abbreviations not used previously: AR, autosomal recessive; JSRD, Joubert syndrome and related disorders; XLD, X‐linked dominant; XLR, X‐linked recessive.
Additionally, the patient described in Case 3 is reported to have had children. Because most men with PCD are infertile, this is an important finding. The range of male fertility in PCD is variable including immotile spermatozoa, oligozoospermia, azoospermia, and normal number of spermatozoa with normal or partially impaired motility (Munro et al., 1994). We are limited in interpreting the implications of his reported fertility because we do not know if assisted reproductive technologies were used; there is no molecular confirmation of paternity, and the fertility of men with OFD1 loss‐of‐function is not well understood. Seminal analysis of individuals with hemizygous loss‐of‐function variants in OFD1 may be useful in further describing OFD1‐related disorders.
We are still learning about the function of OFD1 and its role in ciliopathies. Tissue culture and animal studies provide insight into the function of OFD1. It is clear that OFD1 is located at the nucleus and the centrosome/basal body (Thauvin‐Robinet et al., 2013), that it regulates centriole length (Thauvin‐Robinet et al., 2014), and that OFD1 autophagy at centriolar satellites affects ciliogenesis (Tang et al., 2013). Antisense morpholinos in zebrafish lead to laterality defects and other anomalies (Ferrante et al., 2009). OFD1 knockout animals have laterality defects and paucity of embryonic node cilia as well as abnormal HOX gene expression in limb buds (Ferrante et al., 2006). Tissue culture and animal studies support the importance of OFD1 in ciliary function and development among other functions.
Although genotype–phenotype correlation of OFD1 variants is not completely understood, there have been studies that have characterized symptoms implicated by various pathogenic variants (Prattichizzo et al., 2008; Thauvin‐Robinet et al., 2006). Little is known regarding OFD1 variants that cause PCD. Note that the individual and the fetus with PCD‐like features described by Thauvin‐Robinet et al. both had a variant leading to a stop codon in exon 21 (Thauvin‐Robinet et al., 2013). A family and an unrelated individual have been described with JBTS10 due to exon 21 OFD1 variants leading to a stop codon; although it is unclear if affected people had PCD, individuals in the family had fatal recurrent infections, and the unrelated individual had recurrent middle‐ear infections (Coene et al., 2009). In light of these observations, it is pertinent that all three individuals described herein had pathogenic variants in exon 21 of OFD1 that led to a premature stop codon (Figure 1). We do recognize that other individuals with PCD‐like features did have pathogenic variants upstream of exon 21 (Budny et al., 2006; Tsurusaki et al., 2013; Wentzensen et al., 2016). Future characterization of PCD‐causing OFD1 variants will be useful.
Recognizing the clinical features of OFD1‐related disorders and making a genetic diagnosis could have significant therapeutic implications. Diagnosing PCD is of critical importance to allow for appropriate management. Expert consensus guidelines outline clinical recommendations for both otolaryngology care as well as routine and individualized care for pulmonary manifestations (Shapiro et al., 2016). Furthermore, evidence‐based therapeutic options for individuals with PCD are an active area of investigation. This research includes a phase 2, double‐blinded, placebo‐controlled randomized controlled trial evaluating multiple interventions (CLEAN‐PCD, ClinicalTrials.gov identifier NCT02871778). Clearly, recognizing the phenotype of individuals with OFD1‐related disorders can facilitate a diagnosis, and an accurate diagnosis of PCD has therapeutic implications.
Our summary points follow:
PCD is an important part of the phenotype in some patients with OFD1‐related disorders.
As has been suggested by other clinicians, we agree that individuals with OFD1 variants can have a spectrum of disease. The phenotype may include features of PCD, JBTS10, SGBS2, OFDSI, and RP23. It may be useful to consider the OFD1 phenotype as a spectrum of disease that can include some features of multiple classic conditions.
Understanding the phenotype of OFD1‐related disorders may allow for more focused genetic testing.
Any patient with a hemizygous, pathogenic OFD1 variant should be evaluated for possible PCD.
The phenotype of OFD1‐related disorders is expanding, and there are important implications for diagnosis and treatment.
INFORMED CONSENT
All three individuals provided informed consent. Cases 1 and 2 were enrolled through the UNC IRB. Case 3 was enrolled through the University of Washington IRB.
ACKNOWLEDGMENTS
We are grateful to the patients and family members, Michele Manion (founder of US PCD foundation), and the US PCD foundation. We thank Dr. Joao Pedro Lopes for interpreting a French article cited within this publication (Papillon‐Léage & Psaume J, 1954) and Dr. Katherine Dempsey for her thoughtful feedback of the manuscript. We thank Kimberly Burns from UNC for technical help. We thank Drs. Shrikant Mane and Francesc Lopez‐Giraldez, and Ms. Weilai Dong from Yale Center for Mendelian Genomics (UM1 HG006504) for providing whole‐exome sequencing and support. Funding support for research was provided to M.R.K, M.A.Z, and MR by the US NIH/ORDR/NHLBI grant 5U54HL096458‐11 and to M.R.K and M.A.Z by NIH‐NHLBI grant 5R01HL071798‐10. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Health. No authors had a conflict of interest to report.
Hannah WB, DeBrosse S, Kinghorn B, et al. The expanding phenotype of OFD1‐related disorders: Hemizygous loss‐of‐function variants in three patients with primary ciliary dyskinesia. Mol Genet Genomic Med. 2019;7:e911 10.1002/mgg3.911
Contributor Information
William B. Hannah, Email: william.hannah@duke.edu.
Maimoona A. Zariwala, Email: maimoona_zariwala@med.unc.edu.
REFERENCES
- Brzustowicz, L. M. , Farrell, S. , Khan, M. B. , & Weksberg, R. (1999). Mapping of a new SGBS locus to chromosome Xp22 in a family with a severe form of Simpson‐Golabi‐Behmel Syndrome. American Journal of Human Genetics, 65(3), 779–783. 10.1086/302527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budny, B. , Chen, W. , Omran, H. , Fliegauf, M. , Tzschach, A. , Wisniewska, M. , … Ropers, H.‐H. (2006). A novel X‐linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral‐facial‐digital type I syndrome. Human Genetics, 120(2), 171–178. 10.1007/s00439-006-0210-5 [DOI] [PubMed] [Google Scholar]
- Coene, K. L. M. , Roepman, R. , Doherty, D. , Afroze, B. , Kroes, H. Y. , Letteboer, S. J. F. , … de Brouwer, A. P. M. (2009). OFD1 is mutated in X‐linked Joubert syndrome and interacts with LCA5‐encoded lebercilin. American Journal of Human Genetics, 85(4), 465–481. 10.1016/j.ajhg.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante, M. I. , Feather, S. A. , Bulfone, A. , Wright, V. , Ghiani, M. , Selicorni, A. , … Franco, B. (2001). Identification of the gene for oral‐facial‐digital type I syndrome. American Journal of Human Genetics, 68(3), 569–576. 10.1086/318802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante, M. I. , Romio, L. , Castro, S. , Collins, J. E. , Goulding, D. A. , Stemple, D. L. , … Wilson, S.W., (2009). Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral‐facial‐digital type 1 syndrome gene. Human Molecular Genetics, 18(2), 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante, M. I. , Zullo, A. , Barra, A. , Bimonte, S. , Messaddeq, N. , Studer, M. , … Franco, B., (2006). Oral‐facial‐digital type I protein is required for primary cilia formation and left‐right axis specification. Nature Genetics, 38(1), 112–117. [DOI] [PubMed] [Google Scholar]
- Field, M. , Scheffer, I. E. , Gill, D. , Wilson, M. , Christie, L. , Shaw, M. , … Gecz, J. (2012). Expanding the molecular basis and phenotypic spectrum of X‐linked Joubert syndrome associated with OFD1 mutations. European Journal of Human Genetics, 20(7), 806–809. 10.1038/ejhg.2012.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodship, J. , Platt, J. , Smith, R. , & Burn, J. (1991). A male with type I orofaciodigital syndrome. Journal of Medical Genetics, 28(10), 691–694. 10.1136/jmg.28.10.691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin, R. J. , Anderson, V. E. , & Scott, C. R. (1961). Hypertrophied frenuli, oligophrenia, familial trembling and anomalies of the hand—Report of four cases in one family and a forme fruste in another. New England Journal of Medicine, 264, 486–489. 10.1056/NEJM196103092641004 [DOI] [PubMed] [Google Scholar]
- Jones, K. L. , Jones, M. C. , & Del Campo, M. (2013). Smith’s recognizable patterns of human malformation, 7th ed Philadelphia, PA: Elsevier Saunders. [Google Scholar]
- Knowles, M. R. , Daniels, L. A. , Davis, S. D. , Zariwala, M. A. , & Leigh, M. W. (2013). Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. American Journal of Respiratory and Critical Care Medicine, 188(8), 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro, N. C. , Currie, D. C. , Lindsay, K. S. , Ryder, T. A. , Rutman, A. , Dewar, A. , … Cole, P. J. (1994). Fertility in men with primary ciliary dyskinesia presenting with respiratory infection. Thorax, 49(7), 684–687. 10.1136/thx.49.7.684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon‐Léage, M. , & Psaume, J. (1954). Dysmorphie des freins buccaux: Huit observations. Actual Odontostomatol, 8(25), 7–26. [PubMed] [Google Scholar]
- Parisi, M. A. (2009). Clinical and molecular features of Joubert syndrome and related disorders. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 151C(4), 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prattichizzo, C. , Macca, M. , Novelli, V. , Giorgio, G. , Barra, A. , Franco, B. ; the Oral‐Facial‐Digital Type I (OFDI) Collaborative Group (2008). Mutational spectrum of the oral‐facial‐digital type I syndrome: A study on a large collection of patients. Human Mutation, 29(10), 1237–1246. 10.1002/humu.20792 [DOI] [PubMed] [Google Scholar]
- Shapiro, A. J. , Zariwala, M. A. , Ferkol, T. , Davis, S. D. , Sagel, S. D. , Dell, S. D. , … Leigh, M. W. (2016). Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatric Pulmonology, 51(2), 115–132. 10.1002/ppul.23304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z. , Lin, M. G. , Stowe, T. R. , Chen, S. , Zhu, M. , Stearns, T. , … Zhong, Q., (2013). Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature, 502(7470), 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin‐Robinet, C. , Cossée, M. , Cormier‐Daire, V. , van Maldergem, L. , Toutain, A. , Alembik, Y. , … Faivre, L., (2006). Clinical, molecular, and genotype‐phenotype correlation studies from 25 cases of oral‐facial‐digital syndrome type 1: A French and Belgian collaborative study. Journal of Medical Genetics, 43(1), 54–61. 10.1136/jmg.2004.027672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin‐Robinet, C. , Lee, J. S. , Lopez, E. , Herranz‐Pérez, V. , Shida, T. , Franco, B. , … Nachury, M. V., (2014). The oral‐facial‐digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nature Genetics, 46(8), 905–911. 10.1038/ng.3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin‐Robinet, C. , Thomas, S. , Sinico, M. , Aral, B. , Burglen, L. , Gigot, N. , … Attié‐Bitach, T., (2013). OFD1 mutations in males: Phenotypic spectrum and ciliary basal body docking impairment. Clinical Genetics, 84(1), 86–90. 10.1111/cge.12013 [DOI] [PubMed] [Google Scholar]
- Tsurusaki, Y. , Kosho, T. , Hatasaki, K. , Narumi, Y. , Wakui, K. , Fukushima, Y. , … Matsumoto, N., (2013). Exome sequencing in a family with an X‐linked lethal malformation syndrome: Clinical consequences of hemizygous truncating OFD1 mutations in male patients. Clinical Genetics, 83(2), 135–144. 10.1111/j.1399-0004.2012.01885.x [DOI] [PubMed] [Google Scholar]
- Webb, T. R. , Parfitt, D. A. , Gardner, J. C. , Martinez, A. , Bevilacqua, D. , Davidson, A. E. , … Hardcastle, A. J. (2012). Deep intronic mutation in OFD1, identified by targeted genomic next‐generation sequencing, causes a severe form of X‐linked retinitis pigmentosa (RP23). Human Molecular Genetics, 21(16), 3647–3654. 10.1093/hmg/dds194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen, I. M. , Johnston, J. J. , Patton, J. H. , Graham, J. M. , Sapp, J. C. , & Biesecker, L. G. (2016). Exome sequencing identifies a mutation in OFD1 in a male with Joubert syndrome, orofaciodigital spectrum anomalies and complex polydactyly. Hum Genome Var, 3, 15069 10.1038/hgv.2015.69 [DOI] [PMC free article] [PubMed] [Google Scholar]