

Abstract
Background
Hearing loss or hearing impairment is a clinically and genetically heterogeneous disorder. More than 117 genes were discovered to date in hereditary, nonsyndromic hearing loss (NSHL). Identifying novel gene variants and their frequency in specific populations is valuable for public health and potentially for genetic screening of NSHL.
Aims
To identify the gene variants underlying NSHL in a Pakistani cohort.
Methods and Results
A cohort of 40 school‐aged children with NSHL was initially screened for variants in GJB2, the gene with the highest incidence of variants in other populations with NSHL. We found known homozygous as well as compound heterozygous GJB variants in 15 individuals. Next, we used targeted next generation sequencing (TNGS) for the remaining 25 individuals and identified 20 different variants in 14 genes (SLC26A4, KCNQ4, MYO7A, MYO15A, TMPRSS3, ESPN, TMC1, GIPC3, LHFPL5, WFS1, DFNB59, GRXCR1, ESRRB, and LRTOMT).
Conclusions
We described common and novel variants in 15 genes in a Pakistani cohort of NSHL.
Keywords: GJB2, nonsyndromic hearing loss (NSHL), Pakistani Cohort, Targeted Next Generation Sequencing
To the Editor:
Hearing loss is a clinically and genetically heterogeneous disorder and is the most common human disability (Shrivastava, Shrivastava, & Ramasamy, 2016). Both environmental and genetic factors were shown in the pathogenesis of hearing loss such as iodine deficient diet, infections, ototoxic drug treatment, or gene variants (http://hereditaryhearingloss.org/) (Morton & Nance, 2016; Shrivastava et al., 2016; Wonkam et al., 2013). Nonsyndromic hearing loss (NSHL) can be inherited as an autosomal recessive (AR), autosomal dominant (AD), X‐linked (dominant or recessive), or mitochondrial trait (Liu et al., 2005; Snoeckx et al., 2005; Wang, Han, Khan, & Zhang, 2017). Identifying novel sequence variants in populations provide valuable information for public health, particularly for genetic screening of the NSHL.
In our study, we recruited a total of 40 unrelated individuals affected by NSHL from Bannu and Kohat districts in Khyber Pakhtunkhwa (KPK) province, Pakistan. A clinical questionnaire was used to collect medical history and rule out the history of other diseases potentially affecting hearing and environmental factors such as antibiotic use, excessive noise exposure, and infection that could cause hearing loss. Detailed physical examination did not reveal other abnormal findings than hearing loss. Palpable goiters and other dysmorphic features were not observed in any of the participants with hearing loss; their hearing was ranging from severely impaired to deaf, listed in Figure 1 and Table 1. All of the probands had bilateral, prelingual hearing loss consistent with a congenital origin. There was no previous family history of hearing loss in the participants. The study design and protocol were approved by the Institutional Review Board of (IRB) Quaid‐i‐Azam University Islamabad Pakistan, the Ethical Review Committee (ERC) of Peking Union Medical College (Beijing, China), and China Medical University (Shenyang, China). Written informed consents to conduct and publish the study were obtained from all participating individuals and their parents. All 40 individuals were screened by polymerase chain reaction (PCR) and Sanger sequencing for variants in GJB2 (gap junction protein beta 2; NM_004004.5) gene which is the most commonly mutated gene in recessive NSHL (Liu et al., 2005; Snoeckx et al., 2005). We found known homozygous as well as compound heterozygous variants in 15 individuals. Four homozygous patients had GJB2; c. 370C > T; p.Gln124*, six had GJB2; c.35delG; p.Gly12Valfs*2, three compound heterozygotes had GJB2; c.[71G > A];c.[231G > A]; p.[p.Trp24*];p.[Trp77*], and two had GJB2; c.[−23 + 1G>A]; c.[231G > A]; p.[N/A]; p.[Trp77*] variants respectively (Table 1). The 25 individuals, who were negative for GJB2 gene variants, were screened by targeted next generation sequencing (TNGS) technology using the Ion Torrent platform for a panel of 63 genes implicated in hearing loss as described previously (Wang et al., 2017). We identified 20 variants in 14 genes (SLC26A4, KCNQ4, MYO15A, TMPRSS3, ESPN, TMC1, GIPC3, LHFPL5, WFS1, DFNB59, GRXCR1, ESRRB, and LRTOMT). Ten were novel such as two homozygous missense variant in MYO15A c.[9518‐2A > G];p.[N/A], c.3944G > A;p.Gly1315Glu, one novel variant c.199T > G; p.Tyr67Asp in LHFPL5 in two unrelated individuals, one novel variant c.2519G > A; p.Trp840* in ESPN, one in KCNQ4 c.1288G > A; p.Glu430Lys, one in ESRRB c521G > A; p.Arg174His, one in DFNB59 c.147T > A; p.Tyr49*, one in GIPC3 c.759C > G; p.Ser253Arg, one in TMC1 c.662A > G; p.Tyr221Cys, and one novel variant in LRTOMT c.154C > T; p.Arg52Trp (Figure 1, Table 1). The functional effect of the variants was predicted using the in silico tools including Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant from Tolerant (SIFT) (http://sift.jcvi.org/), PROVEAN (http://www.provean.jcvi.org) Mutation Taster (http://www.mutationtaster.org/), Human Splicing Finder (HSF) (http://www.umd.be/HSF) and CADD (https://cadd.gs.washington.edu) (Table 1).
Figure 1.
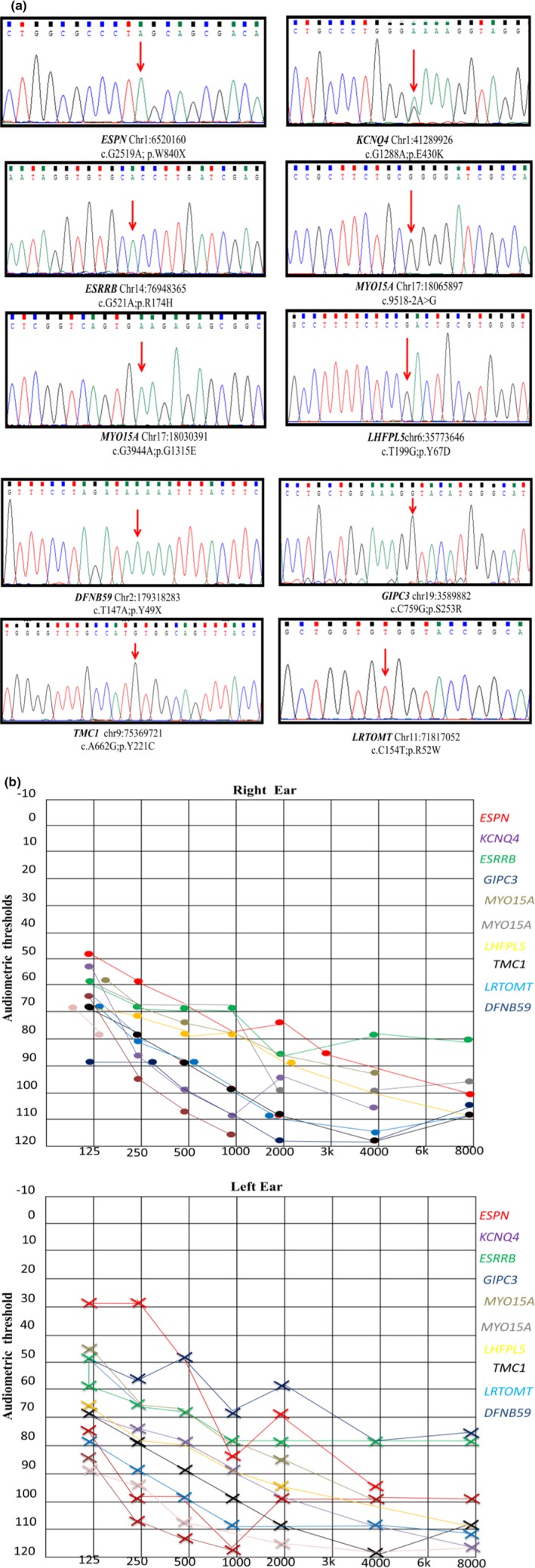
(a) Novel sequence variants identified in ESPN, KCNQ4, ESRRB, MYO15A, LHFPL5, DFNB59, GIPC3, TMC1, and LRTOMT in our cohort from Pakistan. (b) Audiometry of individuals in our cohort with hearing loss
Table 1.
Clinical features and genotyping in hearing loss individuals
| Subjects | Sex | Age | Gene | Transcript ID | Variant | Allele | Effect on protein | Mutation type | Known/Novel | Variant interpretation prediction scores | ACMG classification | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polyphen2 | PROVEAN | SIFT | Mutation taster | CADD | |||||||||||
| 1 | M | 7 | GJB2 | NM_004004.5 | c.[71G > A]; [71G > A] | Hom | p.[Trp24*];[Trp24*] | Nonsense | Known | – | – | – | 1 | 36 | PVS1 |
| 2 | M | 7 | GJB2 | NM_004004.5 | c.[71G > A];[71G > A] | Hom | p.[Trp24*];[Trp24*] | Nonsense | Known | – | – | – | 1 | 36 | PVS1 |
| 3 | M | 9 | GJB2 | NM_004004.5 | c.[71G > A];c.[231G > A] | Comp Het | p.[Trp24*];p.[Trp77*] | Nonsense | Known | – | – | – | 1 | 36/38 | PVS1 |
| 4 | F | 6 | GJB2 | NM_004004.5 | c.[370C > T];[370C > T] | Hom | p.[Gln124*];[Gln124*] | Nonsense | Known | – | – | 1 | 34 | PVS1 | |
| 5 | F | 5 | GJB2 | NM_004004.5 | c.[370C > T];[370C > T] | Hom | p.[Gln124*];[Gln124*] | Nonsense | Known | – | – | – | 1 | 34 | PVS1 |
| 6 | M | 11 | GJB2 | NM_004004.5 | c.[370C > T];[370C > T] | Hom | p.[Gln124*];[Gln124*] | Nonsense | Known | – | – | – | 1 | 34 | PVS1 |
| 7 | F | 13 | GJB2 | NM_004004.5 | c.[370C > T];[370C > T] | Hom | p.[Gln124*];[Gln124*] | Nonsense | Known | – | – | – | 1 | 34 | PVS1 |
| 8 | M | 9 | GJB2 | NM_004004.5 | c.[35delG];[35delG] | Hom | p.[(Gly12Valfs*2)];[(Gly12Valfs*2)] | Frameshift | Known | – | – | – | – | – | PVS1 |
| 9 | F | 8 | GJB2 | NM_004004.5 | c.[35delG];[35delG] | Hom | p.[(Gly12Valfs*2)];[(Gly12Valfs*2)] | Frameshift | Known | – | – | – | – | – | PVS1 |
| 10 | F | 10 | GJB2 | NM_004004.5 | c.[35delG];[35delG] | Hom | p.[(Gly12Valfs*2)];[(Gly12Valfs*2)] | Frameshift | Known | – | – | – | – | – | PVS1 |
| 11 | F | 7 | GJB2 | NM_004004.5 | c.[35delG];[35delG] | Hom | p.[(Gly12Valfs*2)];[(Gly12Valfs*2)] | Frameshift | Known | – | – | – | – | – | PVS1 |
| 12 | M | 5 | GJB2 | NM_004004.5 | c.[35delG];[35delG] | Hom | p.[(Gly12Valfs*2)];[(Gly12Valfs*2)] | Frameshift | Known | – | – | – | – | – | PVS1 |
| 13 | M | 8 | GJB2 | NM_004004.5 | c.[35delG];[35delG] | Hom | p.[(Gly12Valfs*2)];[(Gly12Valfs*2)] | Frameshift | Known | – | – | – | 15 | – | PVS1 |
| 14 | M | 9 | GJB2 | NM_004004.5 | c.[−23 + 1G>A];c.[231G > A] | Comp Het | p.[N/A];p.[Trp77*] | Splice site; Frameshift | Known | – | – | – | – | −/38 | PVS1 |
| 15 | M | 10 | GJB2 | NM_004004.5 | c.[−23 + 1G>A];[−23 + 1G>A] | Hom | p.[N/A];[N/A] | Splice site | Known | – | – | – | – | – | PVS1 |
| 16 | M | 8 | SLC26A4 | NM_000441.1 | c.[679G > C];[679G > C] | Hom | p.[Ala227Pro];[Ala227Pro] | Missense | Known | 1 | −4.94 | 0.001 | 1 | 30 | PM2 |
| 17 | F | 7 | SLC26A4 | NM_000441.1 | c.[679G > C];[679G > C] | Hom | p.[Ala227Pro];[Ala227Pro] | Missense | Known | 1 | −4.94 | 0.001 | 1 | 30 | PM2 |
| 18 | F | 7 | SLC26A4 | NM_000441.1 | c.[679G > C];[679G > C] | Hom | p.[Ala227Pro];[Ala227Pro] | Missense | Known | 1 | −4.94 | 0.001 | 1 | 30 | PM2 |
| 19 | M | 6 | SLC26A4 | NM_000441.1 | c.[716T > A];[716T > A] | Hom | p.[Val239Asp];[Val239Asp] | Missense | Known | 0.845 | −5.67 | 0.001 | 1 | 29.5 | PP2 |
| 20 | M | 7 | SLC26A4 | NM_000441.1 | c.[716T > A];[716T > A] | Hom | p.[Val239Asp];[Val239Asp] | Missense | Known | 0.845 | −5.67. | 0.001 | 1 | 29.5 | PP2 |
| 21 | M | 8 | MYO7A | NM_000260.3 | c.[1258A > T];[1258A > T] | Hom | p.[Lys420*];[Lys420*] | Nonsense | Known | – | – | – | 1 | 41 | PVS1 |
| 22 | M | 9 | MYO7A | NM_000260.3 | c.[1258A > T];[1258A > T] | Hom | p.[Lys420*];[Lys420*] | Nonsense | Known | – | – | – | 1 | 41 | PVS1 |
| 23 | F | 14 | MYO7A | NM_000260.3 | c.[4838delA];[4838delA] | Hom | p.[Asp1613Valfs*32];[Asp1613Valfs*32] | Frameshift | Known | – | – | – | – | – | PVS1 |
| 24 | M | 13 | MYO15A | NM_016239.3 | c.[9518−2A > G]; [9518−2A > G] | Hom | p.[N/A];[N/A] | Splice site | Novel | – | – | – | 1 | 19 | PVS1 |
| 25 | F | 15 | MYO15A | NM_016239.3 | c.[3944G > A]; [3944G > A] | Hom | p.[Gly1315Glu]; [Gly1315Glu] | Missense | Known | 1 | −7.7 | 0.0 | 1 | 24.6 | PM2 |
| 26 | M | 7 | GRXCR1 | NM_001080479.2 | c.[784C > T];[784C > T] | Hom | p.[Arg262*];[Arg262*]; | Nonsense | Known | – | – | – | 1 | 50 | PVS1 |
| 27 | M | 12 | GRXCR1 | NM_001080479.2 | c.[784C > T];[784C > T] | Hom | p.[Arg262*];[Arg262*]; | Nonsense | Known | – | – | – | 1 | 50 | PVS1 |
| 28 | M | 10 | LHFPL5 | NM_182548.3 | c.[199T > G];[199T > G] | Hom | p.[Tyr67Asp];[Tyr67Asp] | Missense | Novel | 1 | −6.12 | 0.032 | 1 | 27.8 | PM2 |
| 29 | M | 8 | LHFPL5 | NM_182548.3 | c.[199T > G];[199T > G] | Hom | p.[Tyr67Asp];[Tyr67Asp] | Missense | Novel | 1 | −6.12 | 0.032 | 1 | 27.8 | PM2 |
| 30 | M | 7 | TMPRSS3 | NM_024022.2 | c.[727G > A];[727G > A] | Hom | p.[Gly243Arg];[Gly243Arg] | Missense | Known | 1 | −7.53 | 0.01 | 1 | 35 | PM2 |
| 31 | F | 9 | TMPRSS3 | NM_024022.2 | c.[1219T > C];[1219T > C] | Hom | p.[Cys407Arg];[Cys407Arg] | Missense | Known | 0.997 | −3.98 | 0.122 | 1 | 23.3 | PM2 |
| 32 | M | 6 | WFS1 | NM_006005.3 | c.[2338G > A];[1219T > C] | Hom | p.[Gly780Ser];[Gly780Ser] | Missense | Known | 0.896 | −1.34 | 0.060 | 1 | 23.7 | PM2 |
| 33 | M | 16 | WFS1 | NM_006005.3 | c.[2590G > A];[2590G > A] | Hom | p.[Glu864Lys];[Glu864Lys] | Missense | Known | 1 | −1.68 | 0.045 | 1 | 28.6 | PM2 |
| 34 | M | 22 | ESPN | NM_031475.2 | c.[2519G > A];[2519G > A] | Hom | p.[Trp840*];[Trp840*] | Nonsense | Known | – | – | – | 1 | 42 | PVS1 |
| 35 | M | 13 | KCNQ4 | NM_004700.3 | c.[1288G > A];[1288G > A] | Hom | p.[Glu430Lys];[Glu430Lys] | Missense | Novel | 0.956 | −0.72 | 0.376 | 1 | 18.19 | PP2 |
| 36 | M | 16 | ESRRB | NM_004452.3 | c.[521G > A];[521G > A] | Hom | p.[Arg174His];[Arg174His] | Missense | Novel | 0.860 | −4.27 | 0.046 | 1 | 27.5 | PM2 |
| 37 | M | 10 | DFNB59 | NM_001042702.4 | c.[147T > A];[521G > A] | Hom | p.[Tyr49*];[Tyr49*] | Nonsense | Novel | – | – | – | 1 | 35 | PVS1 |
| 38 | M | 8 | GIPC3 | NM_133261.2 | c.[759C > G];[759C > G] | Hom | p.[Ser253Arg];[Ser253Arg] | Missense | Novel | 0.995 | −3.87 | 0.043 | 0.9994 | 23.2 | PM2 |
| 39 | F | 9 | TMC1 | NM_138691.2 | c.[662A > G];[662A > G] | Hom | p.[Tyr221Cys];[Tyr221Cys] | Missense | Novel | 1 | −6.34 | 0.045 | 1 | 27.3 | PM2 |
| 40 | F | 11 | LRTOMT | NM_001145308.4 | c.[154C > T];[154C > T] | Hom | p.[Arg52Trp];[Arg52Trp] | Missense | Known | 0.988 | −1.79 | 0.001 | 1 | 26.5 | PVS1 |
Abbreviations: Comp. Het, Compound Heterozygous; F, Female; Hom, Homozygous; M, Male.
Furthermore, these novel variants was neither present in the dbSNP (http://www.ncbi.nlm.nih.gov/SNP/),Exome Variant Server (http://evs.gs.washington.edu/EVS/), GnomAD (https://gnomad.broadinstitute.org), Human gene mutation database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), 1,000 Genomes (http://www.1000genomes.org/), and Exome aggregation consortium (ExAC) (http://www.exac.broadinstitute.org). Finally, for the interpretation of variants, the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines were used (Table 1) (Richards et al., 2015).
None of the novel variants were present in 200 ethnically matched control individuals confirmed by PCR and Sanger sequencing. The '15 of 40 patients had known variants in GJB2 (described above), 5 of 40 patients had known variants in SLC26A4 c.679G > C; p.Ala227Pro, 3 of 40 had known variants in MYO7A c.1258A > T; p.Lys420*, c.4838delA; p.Asp1613Valfs*32, 2 of 40 had known variants in GRXCR1 c.784C > T; p.Arg262*, 2 of 40 had known variants in TMPRSS3 c.727G > A; p.Gly243Arg, c.1219T > C; p.Cys407Arg, 2 of 40 had known variants in WFS1 c.2338G > A; p.Gly780Ser, c.2590G > A; p.Glu864Lys, 1 of 40 had known variant in ESPN c.2519G > A; p.Trp840*,1 of 40 had known variant in LRTMOT c.154C > T; p.Arg52Trp and were already known to be associated with syndromic or NSHL (Salman et al., 2015; Wang et al., 2017). The GJB2 variants were the most common variants in our patient cohort. Our study showed that GJB2 is the most common gene found mutated in our Pakistani cohort which is similar to other cohorts around the world (Salman et al., 2015; Snoeckx et al., 2005; Wang et al., 2017). We found the GJB2 variants c.35delG; p.Gly12Valfs*2, c.71G > A; p.Trp24* and c. [71G > A]; c. [231G > A]; p. [p.Trp24*]; p. [Trp77*] as the most common, 15 of 40 patients in our cohort however their frequency is variable worldwide (Salman et al., 2015).
In our patient cohort we found 15/40 patients had GJB2 variants, (5/40) had SLC26A4, and (3/40) had MYO7A variants responsible for NSHL. We found the incidence of GJB2 variants is higher compared to other populations. While this would argue for screening for GJB2 variants in the first place, we found a comparably high incidence for SLC26A4 and MYO7A variants together. Future research could specifically take advantage of using TNGS in the Pakistani cohort to capture a larger proportion of NSHL cases of genetic origin. We expect our study will enhance public awareness toward this hearing loss trouble and genetic counseling significance for the affected families and their members. This study expanded a spectrum of disease causing variants in genes involved in causing hearing loss.
WEB RESOURCES
The URLs for data presented herein are as follows:
1,000 Genomes: http://www.1000genomes.org/
Exome Variant Server: http://evs.gs.washington.edu/EVS/
ExAC: http://exac.broadinstitue.org/
dbSNP: http://www.ncbi.nlm.nih.gov/SNP/
OMIM: http://www.omim.org/
HGMD: http://www.biobase-international.com/products/hgmd
SIFT: http://sift.jcvi.org/
Polyphen2: http://genetics.bwh.harvard.edu/pph2/
IGV: http://www.broadinstitute.org/igv/
ANNOVAR: http://annovar.openbioinformatics.org/en/latest/
Mutation Taster: http://www.mutationtaster.org/
OMIM: http://www.omim.org/
UCSC Genome Browser: http://genome.ucsc.edu
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
We thank all the individuals for their participation in this study. This work was financially supported by the National Key Research and Development Program of China (grant number 2016YFC0905100), the CAMS Innovation Fund for Medical Sciences (CIFMS) (grant numbers 2017‐I2M‐B & R‐05 and 2016‐I2M‐1‐002], the National Natural Science Foundation of China (NSFC) (grant number 81230015), the Beijing Municipal Science and Technology Commission (grant number Z151100003915078), and the Central Research Institutes of Basic Research and Public Service Special Operations (grant number 2018PT32024). We thank Dr. Peter Gergics, the University of Michigan for helpful comments on the manuscript.
REFERENCES
- Liu, X. Z. , Pandya, A. , Angeli, S. , Telischi, F. F. , Arnos, K. S. , Nance, W. E. , & Balkany, T. (2005). Audiological features of GJB2 (connexin 26) deafness. Ear and Hearing, 26(3), 361–369. 10.1097/00003446-200506000-00011 [DOI] [PubMed] [Google Scholar]
- Morton, C. C. , & Nance, W. E. (2016). Newborn hearing screening–a silent revolution. New England Journal of Medicine, 354, 2151–2164. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Laboratory Quality Assurance Committee, A. C. M. G. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman, M. , Bashir, R. , Imtiaz, A. , Maqsood, A. , Mujtaba, G. , Iqbal, M. , & Naz, S. (2015). Mutations of GJB2 Encoding Connexin 26 Contribute to Nonsyndromic Moderate and Severe Hearing Loss in Pakistan”. European archives of oto‐rhino‐laryngology : Official journal of the European Federation of Oto‐Rhino‐Laryngological Societies (EUFOS): Affiliated with the German Society for Oto‐Rhino‐Laryngology ‐. Head and Neck Surgery, 272(8), 2071–2075. 10.1007/s00405-015-3523-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, S. R. , Shrivastava, P. S. , & Ramasamy, J. (2016). Supporting the global initiative of preventing childhood hearing loss: Act now, here's how!. Noise Health, 18, 280–281. 10.4103/1463-1741.192478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeckx, R. L. , Huygen, P. L. , Feldmann, D. , Marlin, S. , Denoyelle, F. , Waligora, J. , … Van Camp, G. (2005). GJB2 mutations and degree of hearing loss: A multicenter study. American Journal of Human Genetics, 77(6), 945–957. 10.1086/497996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Han, S. , Khan, A. , & Zhang, X. (2017). Molecular Analysis of Twelve Pakistani Families with Nonsyndromic or Syndromic Hearing Loss. Genet Test Mol Biomarkers., 316–321 10.1089/gtmb.2016.0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonkam, A. , Noubiap, J. J. , Djomou, F. , Fieggen, K. , Njock, R. , & Toure, G. B. (2013). Aetiology of childhood hearing loss in Cameroon (sub‐Saharan Africa). European Journal of Medical Genetics, 56, 20–25. 10.1016/j.ejmg.2012.09.010 [DOI] [PubMed] [Google Scholar]