

Abstract
Glaucoma is the leading cause of blindness worldwide, resulting from degeneration of retinal ganglion cells (RGCs), which form the optic nerve. Prior to structural degeneration, RGCs exhibit physiological deficits. Müller glia provide homeostatic regulation of ions that supports RGC physiology through a process called K+ siphoning. Recent studies suggest that several retinal conditions, including glaucoma, involve changes in the expression of K+ channels in Müller glia. To clarify whether glaucoma-related stressors directly alter expression and function of K+ channels in Müller glia, we examined changes in the expression of inwardly rectifying K+ (Kir) channels and two-pore domain (K2P) channels in response to elevated intraocular pressure (IOP) in vivo and in vitro in primary cultures of Müller glia exposed to elevated hydrostatic pressure. We then measured outcomes of cell health, cation homeostasis, and cation flux in Müller glia cultures. Transcriptome analysis in a murine model of microbead-induced glaucoma revealed pressure-dependent downregulation of Kir and K2P channels in vivo. Changes in the expression and localization of Kir and K2P channels in response to elevated pressure were also found in Müller glia in vitro. Finally, we found that elevated pressure compromises the plasma membrane of Müller glia and induces cation dyshomeostasis that involves changes in ion flux through cation channels. Pressure-induced changes in cation flux precede both cation dyshomeostasis and membrane compromise. Our findings have implications for Müller glia responses to pressure-related conditions, i.e., glaucoma, and identify cation dyshomeostasis as a potential contributor to electrophysiological impairment observed in RGCs of glaucomatous retina.
Keywords: glaucoma, K2P, Kir, K+ siphoning, K+ channels, microbead, Müller glia, retina
INTRODUCTION
Glaucoma is one of the leading causes of irreversible blindness in the world (45). The two main risk factors for glaucoma are advanced age and elevated intraocular pressure (IOP). Lowering IOP is currently the only treatment, and while it slows, it does not halt, progression of the disease (4). Glaucomatous vision loss results from degeneration of retinal ganglion cells (RGCs) (4, 9, 16). Several studies show that maintaining activity is crucial for neurons, like RGCs, to fulfill their physiological function and to simply survive throughout adulthood. Support from surrounding glia plays a critical role in maintaining RGC physiology and may play an important role in the progression of pathology in glaucoma.
Müller glia are retina-specific glial cells and are the most abundant among glial subtypes in the retina (2, 27). A primary function of Müller glia is homeostatic regulation of the extracellular milieu, including a process referred to as K+ siphoning (38, 47). In response to action potentials generated by RGCs, Müller glia remove excess extracellular K+ through K+ channels with inward rectifying capabilities. This K+ siphoning is critical for RGCs to maintain proper physiology (2, 3, 33, 39). Studies suggest that inwardly rectifying Kir4.1 and Kir2.1 K+ channels in Müller glia are primarily responsible for K+ regulation in the retina (3, 13). However, when inwardly rectifying K+ (Kir) channels are blocked, the remaining K+ conductance in Müller glia is mediated by two-pore domain (K2P) K+ channels (12). Accordingly, many K2P channels, including TWIK-1, TREK-2, TRESK, TASK-1, and TRAAK channels, display weak inwardly rectifying properties (17).
Recent studies suggest that the expression and function of K+ channels in Müller glia is altered in glaucoma and other retinal disorders, including retinitis pigmentosa and diabetic retinopathy (42, 61, 62). In particular, chronic ocular hypertension in rats reduces Kir4.1, Kir2.1, and TASK-1 protein expression in Müller glia (in vivo) (61). Decreased expression of Kir4.1 in Müller glia is also noted in rat models of retinitis pigmentosa and diabetic retinopathy (42, 62). This decreased expression leads to decreased K+ currents and altered electrophysiological properties in both conditions (42, 62). However, in the DBA/2J mouse model of glaucoma, reactive Müller glia do not exhibit changes in membrane current or membrane potential, as compared with Müller glia from control mice (25). Rather, Müller glia in aged DBA/2 mice exhibit increased membrane capacitance, which is characteristic of hypertrophied, reactive glial cells (1).
Here, we sought to clarify whether glaucoma-related stressors directly alter expression and function of K+ channels in Müller glia. Using the microbead occlusion model (MOM) of murine glaucoma (54), we examined changes in the expression of K+ channels with inward-rectifying capabilities (Kir and K2P) in the retina. We then assessed whether similar changes in K+ channel expression accompany exposure to elevated pressure in vitro, in primary cultures of purified Müller glia exposed to elevated hydrostatic pressure (34, 52, 53, 55, 56). We also measured outcomes of K+ homeostasis and cell health in Müller glia cultures.
We found that transcriptome analysis, as well as immunohistochemistry, in the microbead occlusion model indicated pressure-induced changes in Kir and K2P channel expression. Long-term pressure elevation in Müller glia cultures produced changes in expression and localization of K+ channels with inward-rectifying capabilities (Kir and K2P). Long-term pressure elevation also resulted in significant lactate dehydrogenase (LDH) release, increased extracellular concentrations of K+ and Na+, and reduced cation flux. In contrast, short-term pressure elevation only resulted in reduced cation flux. Finally, the pressure-induced decrease in cation influx can be recapitulated by treatment with a cation channel inhibitor. Our findings have implications for Müller glia responses to pressure-related conditions, i.e., glaucoma, as well as electrophysiological impairment noted in RGCs of glaucomatous retina.
METHODS
Microbead occlusion model.
Mice were housed in accordance with NIH guidelines and maintained on a 12:12-h light-dark cycle with free access to food and water. All experiments were approved by the Institutional Animal Care and Use Committee of Vanderbilt University Medical Center. Male C57Bl/6 mice were obtained from Charles River Laboratories (Wilmington, MA). IOP elevation was induced in 1-mo-old C57Bl/6 mice, using the microbead occlusion model, as previously described (10, 14, 15, 54, 59). Briefly, anesthetized animals received bilateral injections of 1.5 μl sterile 15-µm polystyrene beads (1 × 106 microbeads/ml; cat. no. F8844; Life Technologies). Control mice received bilateral injections of an equal volume of saline. IOP elevation lasted 4 wk, at which point, the animals were euthanized. IOP was measured in awake, behaving mice, using a TonoLab tonometer (TonoLab; Reichert, Depew, NY) (14, 21). IOP was determined as the mean of 10 individual measurements. Prior to initial microbead or saline injections, baseline IOP was recorded for 3 consecutive days. Following injections, IOP was recorded three times a week throughout the 4-wk experiment.
RNA sequencing.
Whole, intact retina was dissected from microbead occlusion model mice following euthanization. Immediately following dissection, RNA was extracted from retina using TRIzol (cat. no. 15596026; Invitrogen, Carlsbad, CA) and treated with deoxyribonuclease (DNase) I (cat. no. LS006333; Worthington). Experiments were performed through the Vanderbilt Technologies for Advanced Genomics core at Vanderbilt University Medical Center. DNase-treated total RNA quality was assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Samples with integrity values greater than 6 were used to generate polyA (mRNA)-enriched libraries, using stranded mRNA sample kits with indexed adaptors (New England BioLabs, Ipswich, MA). Library quality was assessed using the 2100 Bioanalyzer (Agilent Technologies), and libraries were quantitated using KAPA Library quantification kits (KAPA Biosystems, Wilmington, MA). Pooled libraries were subjected to 100 bp paired-end sequencing, according to the manufacturer’s protocol (Illumina NovaSeq6000). Bcl2fastq2 Conversion Software (Illumina, San Diego, CA) was used to generate demultiplexed Fastq files. Analysis of RNAseq results was performed through the Vanderbilt Technologies for Advanced Genomics Analysis and Research Design core at Vanderbilt University. Reads were aligned to the GENCODE GRCm38.p5 genome using STAR v2.5.3a. GENCODE vM12 gene annotations were provided to STAR to improve the accuracy of mapping. Quality control on raw reads was performed using FastQC. FeatureCounts v1.15.2 was used to count the number of mapped reads to each gene. Significantly, differential-expressed genes with adjusted P value <0.05 and absolute fold change >2 were detected by DESeq2 v1.14. Data are reported as fold change (log scale) and percent change in RNA transcript levels between saline and microbead retina. Data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE116915 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE116915).
Immunohistochemistry.
We assessed expression and localization patterns for Kir and K2P channels using immunocytochemistry (34, 52, 53, 55, 56). Immunohistochemistry experiments were done on longitudinal paraffin-embedded retina sections of whole eyes from saline- and microbead-injected mice. A separate set of experiments was also done in Müller glia cultures exposed to ambient or elevated pressure for 48 h. Cells were fixed in 4% paraformaldehyde (cat. no. 15714-S; Electron Microscopy Sciences, Hatfield, PA) for 15 min at room temperature and washed with 1× PBS. Samples were incubated in blocking solution containing 5% normal horse serum (NHS; Life Technologies) and 0.1% Triton-X (Fisher Scientific) in 1× PBS. Samples were then incubated overnight at 4°C in primary antibody solution (3% NHS and 0.1% Triton X-100 diluted in 1× PBS) containing: rabbit anti-Kir2.1 (5 µg/µl, cat. no. APC-026, Alomone Labs, Jerusalem, Israel), rabbit anti-Kir4.1 (2 µg/ml, cat. no. APC-035; Alomone Labs), rabbit anti-TRAAK (5 µg/ml, cat. no. APC-108; Alomone Labs), rabbit anti-TRESK (5 µg/ml, cat. no. APC-122; Alomone Labs), rabbit anti-TREK2 (5 µg/ml, cat. no. APC-055; Alomone Labs), rabbit anti-TASK1 (5 µg/ml, cat. no. APC-024; Alomone Labs), or rabbit anti-TWIK1 (5 µg/ml, cat. no. APC-110; Alomone Labs); and rat anti-CD44 (6.7 µg/ml, cat. no. 14-0441; Invitrogen). Following 1× PBS washes, samples were incubated for 2 h at room temperature in a secondary antibody solution containing: 1% NHS, 0.1% Triton X-100, and either 647-donkey anti-rat (7.5 µg/ml; cat. no. 712-605-153; Jackson ImmunoResearch, West Grove, PA) or 488-donkey anti-rabbit (7.5 µg/ml; cat. no. 711-546-152; Jackson ImmunoResearch) in 1× PBS. Cells were counterstained with the nuclear stain DAPI (50 µg/ml; cat. no. D1306; Life Technologies) and coverslipped in aqueous mounting media (Southern Biotech, Birmingham, AL). Immunolabeling was imaged at 20× for intensity analysis and 60× for representative images using an Olympus FV-1000 inverted confocal microscope (Olympus, Tokyo, Japan). Images compared between control and experimental groups were collected at the same time, under the same imaging parameters. Mean fluorescence intensity was calculated using the FIJI Color Histogram tool (ImageJ, NIH, Bethesda, MD).
Purified Müller glia primary cultures.
Primary cultures of purified Müller glia were prepared as previously described (34). Briefly, Müller glia were purified by immunomagnetic separation, using mouse anti-CD44 IgG antibody (16 µg/ml, cat. no. MA5-16909; Invitrogen, Carlsbad, CA) and metallic microbeads conjugated to anti-mouse IgG secondary antibody (cat. no. 130-047-102; Miltenyi Biotec, Auburn, CA). Müller glia were plated in eight-well chamber slides and grown in DMEM-F12 (cat. no. 11330-032; Gibco, Carlsbad, CA) containing 1% G5 supplement (cat. no. 17503012; Gibco), 0.1% gentamycin (cat. no. 15710-064; Gibco), and 10% fetal bovine serum (cat. no. 16140071, Gibco). Experiments were performed ~1–2 wk after plating. The drug fluoxetine, used for thallium flux experiments, was dissolved in the culture media (100 µM, cat. no. F132; Sigma-Aldrich, St. Louis, MO).
Elevated hydrostatic pressure.
Primary cultures of purified Müller glia were maintained at ambient or at +70 mmHg hydrostatic pressure, for 4 or 48 h, as previously described (34, 52, 53, 55, 56). Briefly, a humidified pressure chamber equipped with a regulator and a gauge was placed in a 37°C incubator; a mixture of 95% air and 5% CO2 was pumped into the chamber to obtain a pressure of +70 mmHg (9% increase above atmospheric pressure) that was maintained by the regulator. For ambient pressure experiments, cells were kept in a standard incubator.
Quantitative RT-PCR.
RNA was isolated from primary, purified cultures of Müller glia exposed to 4 or 48 h of ambient or elevated pressure, using RNeasy micro kit (cat. no. 74004; Qiagen, Valencia, CA), according to the manufacturer’s instructions. cDNA synthesis was performed with iScript cDNA synthesis kit (cat. no. 170889; Bio-Rad, Hercules, CA), according to the manufacturer’s instructions. PCR was performed with a 7300 real-time PCR system (Applied Biosystems, Foster City, CA) using FastStart Universal SYBR Green Master (ROX) kit (cat. no. 04913850001; Roche, Basel, Switzerland) together with 25-ng template DNA and the following primer concentrations: Kcnj6 = 500 nM, Kcnj12 = 500 nM, Kcnj14 = 700 nM, Kcnk1 = 500 nM, Kcnk3 = 800 nM, Kcnk12 = 300 nM, and Gapdh = 500 nM, in 50 µl final volume. Initial denaturation for 10 min at 95°C was followed by 40 cycles of amplification (95°C for 15 s and 60°C for 60 s) and melting curve analysis for 15 s at 95°C, 30 s at 60°C, and 15 s at 95°C. We used primer sequences found in existing literature for the following genes: Kcnj6 (29), Kcnj12 (37), Kcnj14 (44), Kcnk1 (43), Kcnk3 (43), Kcnk12 (36), and Gapdh (36). All products reported correspond to well-defined single peaks in the melting curve. No such peaks were observed in the absence of the template. The results were analyzed using the 7300 System SDS software (Applied Biosystems). Ct (threshold cycle) values were calculated by setting the threshold in the linear phase of the amplification curve. All samples were run in triplicate and Ct values shown are the means of the three values, normalized to the control gene, Gapdh.
TUNEL reactivity.
In Müller glia cultures exposed to ambient or elevated pressure for 4 or 48 h, we measured apoptosis with TdT-mediated dUTP-X nick-end labeling (TUNEL; cat. no. 12156792910; Roche, Basel, Switzerland). Labeling was performed according to manufacturer’s specifications, as previously described (22, 23, 34, 55). Cells were counterstained with CD44 (rat anti-CD44, 6.7 µg/ml, cat. no. 14-0441; Invitrogen) to label Müller glia for confirmation of cell type and DAPI for quantification of total cell density. To quantify % TUNEL+ cells, 20× images were taken using a Roper Scientific black and white camera (Photometrics, Tucson, AZ) mounted to a Nikon Ti microscope (Nikon Instruments, Melville, NY). Images compared between control and experimental groups were collected at the same time, under the same imaging parameters. Five images were taken per well, across four wells per condition. Total number of CD44+/DAPI+ and TUNEL+/ CD44+/DAPI+ were counted for each image and summed for each well. Data are shown as % TUNEL+ cells relative to the total number of CD44+/DAPI+ cells.
Lactate dehydrogenase assay.
In Müller glia cultures maintained at ambient or elevated pressure for 4 or 48 h, we measured cell toxicity, which is related to necrotic cell death, using a lactate dehydrogenase (LDH) assay (cat. no. G1780; Promega, Madison, WI). The assay was performed according to manufacturer’s specifications and as previously described (55). Briefly, culture supernatant was collected following pressure elevation and immediately frozen at −80°C. The concentration of LDH in culture supernatants was determined by enzymatic reaction and measurement of optical density (OD) at 490 nm. All samples were run in triplicate, and OD levels were averaged. Background OD levels were obtained from blank media samples and subtracted from OD levels of experimental samples. Data are shown as average of OD at 490 nm, with background subtracted.
Inductively coupled plasma mass spectrometry.
In supernatants from Müller glia cultures exposed to 4 or 48 h ambient or elevated pressure, we measured the extracellular concentration of K+ and Na+, using inductively coupled plasma mass spectrometry (ICP-MS). Experiments were performed through the Mass Spectrometry Research Center at Vanderbilt University. Culture supernatants were diluted 1000× with Milli-Q water (Millipore, Milli-Q synthesis advantage A-10; Millipore, Burlington, MA) and prepared alongside a calibration curve made using the same water. Na+ and K+ standards (Fluka, Sigma-Aldrich, L'Isle-d'Abeau Chesnes, Saint-Quentin Fallavier, France) were serially diluted for the calibration curve, which ranged from 10 ppm to 1 ppb. Samples and standards were immediately analyzed by ICP-MS on an Agilent model 7700X (Agilent Technologies). Each sample was introduced to the instrument manually, without use of an autosampler, and the probe was rinsed in 2% nitric acid (Optima grade, Fisher Scientific, Pittsburgh, PA) between each sample. Three blank samples of pure Milli-Q water between introduction of standards and samples confirmed the absence of background Na+ and K+. The data were analyzed by the offline data analysis package (Agilent Technologies), where the calibration curve was plotted linearly and both R2 values, as well as detection limits, were calculated. The calculated concentrations of each sample were multiplied by 1,000 to account for the initial dilution factor. Data are shown as average K+ and Na+ concentration in parts per million (ppm).
Thallium flux imaging.
We assessed ion channel activity in live Müller glia cultures exposed to 4 or 48 h of ambient or elevated pressure by thallium flux imaging, as previously described (20). Thallium flux indirectly measures inward ion flux, through the fluorescence intensity of Thallos dye. Briefly, cells were loaded with Thallos-AM dye (0.5 μg/μl, cat. no. 0902, TEFlabs, Austin, TX), generously provided by Dave Weaver of Vanderbilt University, by incubating with the dye for 30 min at 37°C. Following dye loading, cells were washed with fresh media. Using live cell fluorescence microscopy, we recorded baseline fluorescence of the Thallos dye continuously for 6–8 s (images taken every 1 s). 1 mM thallium (TI+) solution was then added to cell culture media, and live imaging was performed continuously for 45 s (images taken every 1 s). Fluorescence intensity was calculated for each image using Nikon NIS-Elements software. Data were plotted as normalized fluorescence intensity at each time point and as the average fluorescence intensity after the addition of thallium.
Statistical analysis.
All statistical tests were conducted with SigmaPlot (Systat Software, San Jose, CA). Experimental groups were compared within time points by Student’s t-test. Normality (Shapiro-Wilk) and equal variance was also assessed for each comparison. Comparisons between time points within groups were assessed by one-way ANOVA followed by pairwise comparison by either the Tukey or Dunn’s method. All data are presented as means ± SD, with significant comparisons marked by brackets and asterisks. For all analyses, P ≤ 0.05 was considered statistically significant.
RESULTS
Ocular hypertension alters expression of Kir and K2P channels in vivo.
Kir and K2P channels, particularly those with inward-rectifying capabilities, are implicated in K+ homeostasis in Müller glia (17, 28, 32, 61). To determine whether ocular hypertension alters expression of Kir and K2P channels, we examined transcription of these channels after 4 wk of elevated IOP in the Microbead Occlusion Model (MOM) of murine glaucoma (54). Transcriptional regulation of Kir and K2P channel gene families (Kcnj and Kcnk, respectively) was quantified by RNA sequencing in whole retina of C57Bl/6 mice with either bilateral saline injection (IOP = 15 mmHg ± 0.43 mmHg) or bilateral microbead injection (IOP = 21 mmHg ± 0.53mmHg). Transcriptome analysis in whole retina represents the entire complement of retinal neurons, glia, and vascular elements. While this analysis is not Müller glia-specific, it can establish whether elevated IOP in glaucoma globally alters K+ homeostatic processes.
Transcriptome analysis revealed IOP-dependent decreases in the transcription of genes encoding several Kir channels (Fig. 1, A and B, left). In order of magnitude, microbead-induced IOP elevation decreased transcription of Kir3.2 (Kcnj6; 56%), Kir2.2 (Kcnj12; 51%), and Kir2.4 (Kcnj14; 42%), as compared with saline-injected retina (P < 0.05 for all; Fig. 1B). There was no significant difference observed in transcription of genes encoding Kir1.1 (Kcnj1), Kir2.1 (Kcnj2), Kir2.3 (Kcnj4), Kir3.4 (Kcnj5), Kir4.1 (Kcnj10), Kir6.1 (Kcnj8), Kir5.1 (Kcnj16), Kir3.1 (Kcnj3), Kir3.3 (Kcnj9), Kir6.2 (Kcnj11), Kir7.1 (Kcnj13), and Kir4.2 (Kcnj15) in microbead- versus saline-injected eyes (P > 0.05; Fig. 1, A and B, left).
Fig. 1.

Ocular hypertension alters RNA expression of K+ channels in vivo. RNA sequencing was performed on whole retina from saline- and microbead-injected eyes. A: box plots of fold change (log scale) of expression change between saline and microbead retinas from the Kcnj (left) and Kcnk (right) gene families. B: box plots of percentage of expression change between saline and microbead retinas from the Kcnj (left) and Kcnk (right) gene families. n(PBS) = 5 retinas; n(MOM) = 5 retinas. C: quantitative RT-PCR was performed on RNAs isolated from primary, purified cultures of Müller glia exposed to ambient or elevated pressure for 48 h. Bar graphs of mean threshold cycle (Ct) values normalized to Gapdh, compared between ambient and elevated pressure for Kcnk1 (left) and Kcnk3 (right). n = 3 for all. *P < 0.05. MOM, microbead occlusion model.
For K2P channels, elevated IOP in vivo reduced transcription of THIK2 (Kcnk12; 60%), TASK-1 (Kcnk3; 34%), and TWIK-1 (Kcnk1; 26%), as compared with saline-injected eyes (P < 0.05 for all; Fig. 1B, right). Like Kir channel expression, there were no statistically significant increases in K2P channel transcription (Fig. 1, A and B, right). There was no significant difference in transcription of genes encoding TRAAK (Kcnk4), TREK-1 (Kcnk2), TREK-2 (Kcnk10), TRESK (Kcnk18), TASK-2 (Kcnk5), TWIK-2 (Kcnk6), TASK-3 (Kcnk9), THIK-1 (Kcnk13), TASK-5 (Kcnk15), or TWIK-3 (Kcnk7) in microbead- versus saline-injected eyes (P > 0.05 for all; Fig. 1, A and B). Together, these data suggest that elevated IOP in vivo decreases transcription of a small subset of Kir and Kir and K2P channels with inward-rectifying capabilities. Interestingly, elevated IOP does not increase transcription of any Kir or K2P channels at the whole retina level.
Elevated pressure does not alter expression of Kcnk1 and Kcnk3 in Müller glia in vitro.
To determine whether downregulation of Kir and K2P channels noted in whole retina are attributable to Müller glia, we performed qPCR for Kcnj6, Kcnj12, Kcnj14, Kcnk1, Kcnk3, and Kcnk12 in primary cultures of Müller glia exposed to ambient or elevated hydrostatic pressure for 48 h. Expression levels for Kcnj6, Kcnj12, Kcnj14, and Kcnk12 were below the detectable range. This is consistent with previous literature, indicating that these channels are not expressed in Müller glia (7, 26, 33, 46). Kcnk1 and Kcnk3 were expressed in Müller glia; however, exposure to elevated pressure did not alter the level of gene expression, compared with ambient pressure controls (Fig. 1C).
Our transcriptome analysis indicates that elevated IOP induces downregulation of some Kir and K2P channels. As indicated above, this analysis is useful for identifying global shifts in transcriptional regulation of K+ channels. However, changes in a single population of cells (i.e., Müller glia) may not be sufficient for detection in the context of whole retina. This is supported by our qPCR analysis in purified, primary Müller cells, which did not reflect the same expression pattern as our whole retina analysis. Furthermore, ion channel activity is often regulated at the protein level, i.e., by changes in their representation on the membrane. Thus, we elected to examine protein localization and relative level of protein expression for individual Kir and K2P channels in both ocular hypertensive retina and in purified, primary cultures of Müller glia.
Ocular hypertension alters expression of Kir2.1 and Kir4.1 in retina.
Kir2.1 and Kir4.1 are thought to be primarily responsible for K+ siphoning in Müller glia (3, 13). To determine how ocular hypertension may alter protein expression patterns for these channels, we performed immunolabeling for Kir2.1 and Kir4.1 in retinal sections from saline-injected (IOP = 15 mmHg ± 0.9mmHg) and microbead-injected (IOP = 21 mmHg ± 1.1mmHg) mice. Qualitatively, immunolabeling for Kir2.1 was increased, while immunolabeling for Kir4.1 was reduced in retina from microbead-injected mice, as compared with saline-injected controls (Fig. 2A). In addition to Müller glia, this immunolabeling was also strongly associated with neuronal processes in the plexiform layers of the retina (Fig. 2A). Quantification of immunolabeling intensity confirmed that elevated IOP increased intensity of Kir2.1 by 25% and decreased intensity of Kir4.1 by 32%, as compared with saline-injected controls (Fig. 2B). These data suggest that elevated IOP increases the protein representation of Kir2.1, while decreasing that of Kir4.1.
Fig. 2.
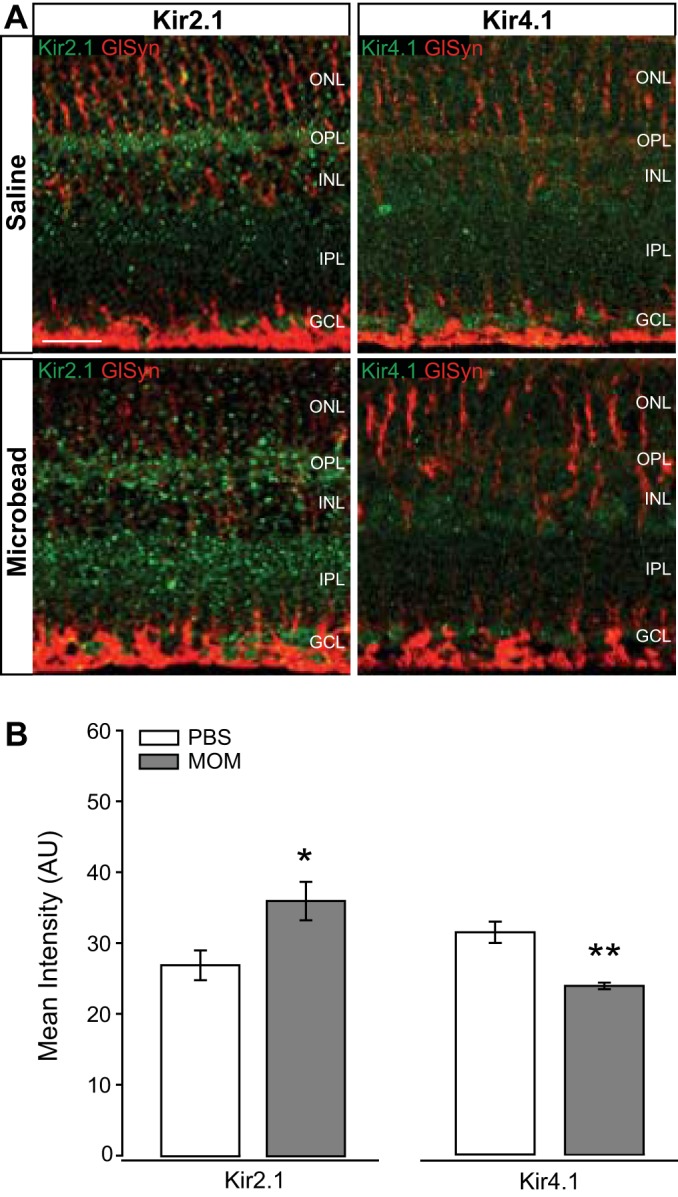
Intensity of inwardly rectifying K+ (Kir) channel staining in glaucomatous retinal sections. A: representative fluorescent micrographs of retinal sections from saline- or microbead-injected eyes. Immunolabeling of Kir2.1 (green), Kir4.1 (green), and CD44 (red) was performed. B: bar graphs of the mean intensity of Kir2.1 and Kir4.1 staining compared between saline and microbead retina. Kir2.1: n(PBS)= 7 retina sections; n(MOM)= 7; Kir4.1: n(PBS)= 6; n(MOM) = 7. Images taken at ×40; scale bar = 20 µm. Student’s t-test was used to analyze statistical significance. *P < 0.05, **P < 0.005. MOM, microbead occlusion model.
Elevated pressure alters expression and localization of Kir2.1 and Kir4.1 in Müller glia in vitro.
To examine Müller cell-specific changes in Kir protein, we exposed Müller glia cultures to ambient or +70 mmHg hydrostatic pressure for 48 h and immunolabeled for Kir2.1 and Kir4.1. Both channels exhibited a similar localization pattern in Müller glia exposed to ambient pressure (Fig. 3A). For both, Kir immunolabeling appeared to be ubiquitous with some more concentrated staining within intracellular vesicles (Fig. 3A). These localization patterns are evident in the z-plane view of the images directly below the fluoromicrographs, as well as in the associated plots of fluorescent intensity below the z-plane view. This is consistent with previous studies indicating the presence of sequence motifs in Kir channels that signal export from the ER and the Golgi via adaptor proteins that initiate vesicle formation (35, 57). Ubiquitous labeling is likely attributable, in part, to plasma membrane localization, as Kir channels exist both in the plasma membrane and in vesicles trafficking to the plasma membrane (57). Qualitatively, elevated pressure appeared to increase Kir2.1 immunolabeling, but decrease Kir4.1 immunolabeling, as compared with ambient pressure (Fig. 3A). For both Kir2.1 and Kir4.1, changes in the intensity of immunolabeling were most evident in staining not associated with vesicles, suggesting that elevated pressure likely alters the cell surface representation for these channels (Fig. 3A). Quantification of immunolabeling intensity confirmed pressure-induced changes in Kir channel representation. Elevated pressure increased the labeling intensity for Kir2.1 by 28% (P < 0.001), but decreased the intensity of Kir4.1 labeling by 29% (P < 0.005), as compared with ambient pressure (Fig. 3B). These data reflect the changes in protein representation observed in retina sections and thus, support reciprocal regulation of Kir2.1 and Kir4.1 protein levels.
Fig. 3.
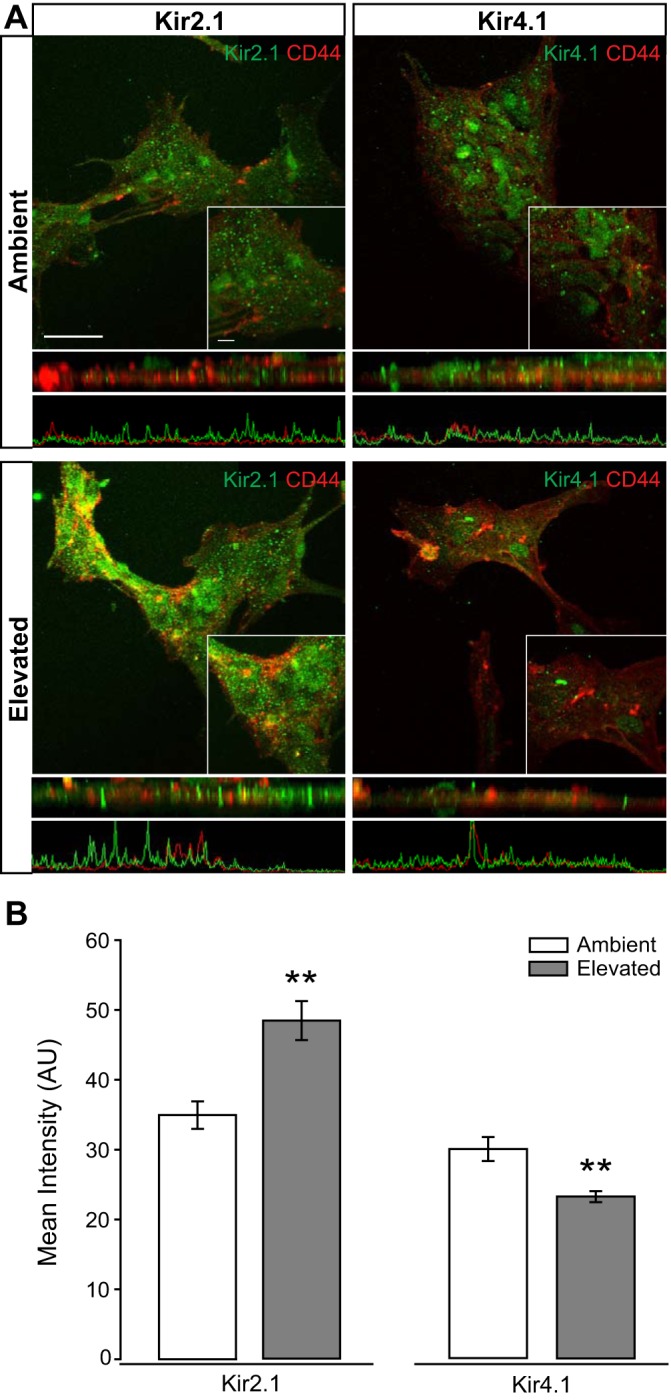
Intensity of inwardly rectifying K+ (Kir) channel staining in Müller glia cultures following pressure elevation. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated (Elev) pressure for 48 h. A: representative fluorescent micrographs of Müller glia from each condition. Immunolabeling of Kir2.1 (green), Kir4.1 (green), and CD44 (red) was performed. Panels directly below the fluoromicrographs are a z-plane view of the image, with associated plot of fluorescent intensity. B: bar graphs of the mean intensity of Kir2.1 and Kir4.1 staining compared between ambient and elevated pressure. Kir2.1: n(Amb) = 29 cells; n(Elev) = 18; Kir4.1: n(Amb) = 19; n(Elev) = 20. Images taken at ×60; scale bar = 40 µm. Inset taken at ×60 + ×2.5 zoom; scale bar = 10 µm. Student’s t-test was used to analyze statistical significance. **P < 0.005.
Ocular hypertension alters expression of TASK-1, but not TWIK-1, in retina.
K2P channels with inwardly rectifying properties (17), such as TWIK-1 and TASK-1, are responsible for some of the K+ conductance in Müller glia and contribute to K+ siphoning along with Kir channels (12). We performed immunolabeling for TWIK-1 and TASK-1 in retinal sections from saline-injected and microbead-injected mice. Qualitatively, immunolabeling for TWIK-1 did not appear to be significantly altered in retina from microbead-injected mice, as compared with saline-injected controls (Fig. 4A). However, it appeared that immunolabeling for TASK-1 was reduced in retina from microbead-injected mice, as compared with saline-injected controls (Fig. 4A). TASK-1 expression was also more restricted to Müller glia than TWIK-1 (Fig. 4A). Quantification of immunolabeling intensity confirmed that elevated IOP did not significantly alter the intensity of TWIK-1, but did decrease intensity of TASK-1 by 40%, as compared with saline-injected controls (Fig. 4B). These data suggest that TASK-1 regulation is impacted more by ocular hypertension than TWIK-1.
Fig. 4.
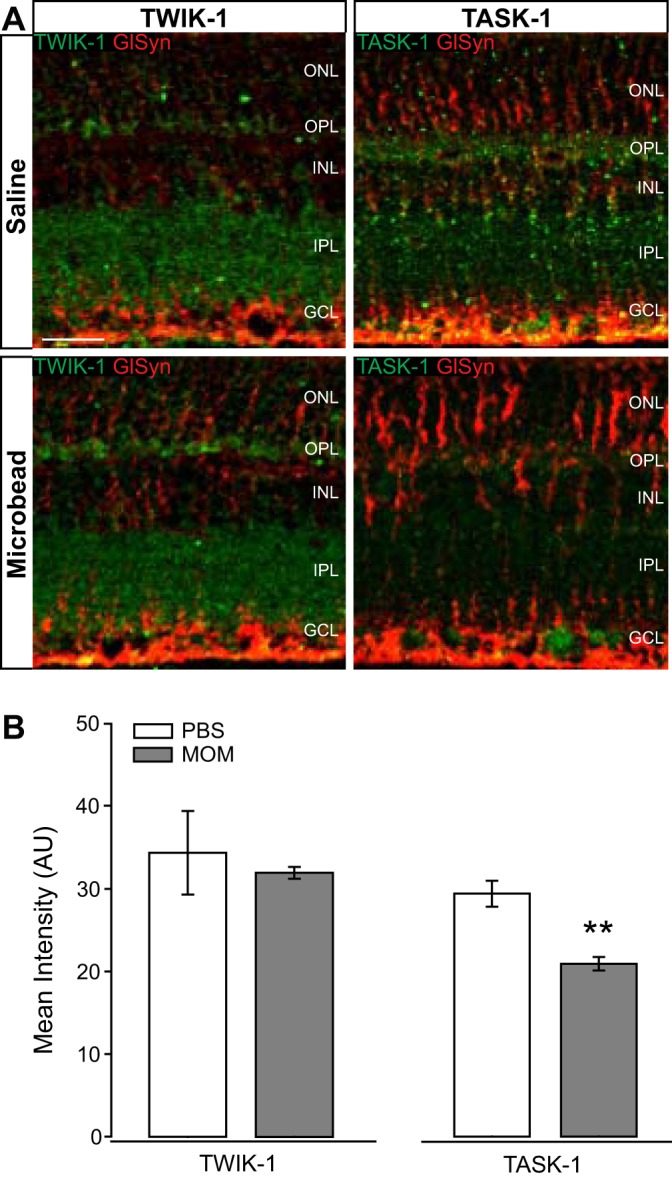
Intensity of TWIK-1 and TASK-1 channel staining in glaucomatous retinal sections. A: representative fluorescent micrographs of retinal sections from saline- or microbead-injected eyes. Immunolabeling of TWIK-1 (green), TASK-1 (green), and CD44 (red) was performed. B: bar graphs of the mean intensity of TWIK-1 and TASK-1 staining compared between saline and microbead retina. TWIK-1: n(PBS) = 6 retina sections; n(MOM) = 7; TASK-1: n(PBS) = 7; n(MOM) = 7. Images taken at ×40; scale bar = 20 µm. Student’s t-test was used to analyze statistical significance. **P < 0.005. MOM, microbead occlusion model.
Elevated pressure alters expression and localization of TASK-1, but not TWIK-1, in Müller glia in vitro.
We examined Müller cell-specific TWIK-1 and TASK-1 protein in Müller glia cultures exposed to 48 h of ambient or elevated pressure. The TWIK-1 immunolabeling in Müller glia maintained at ambient pressure appeared to associate with circular structures (Fig. 5A). These circular structures are also represented in the z-plane view of the images directly below the fluoromicrographs, as well as in the associated plots of fluorescent intensity below the z-plane view. This is consistent with previous findings indicating endosomal localization of TWIK-1 and its interaction with EFA6 on the inner surface of endosomal membranes (5, 18). In contrast, TASK-1 immunolabeling appeared more ubiquitous, with possibly some staining concentrated to vesicles (Fig. 5A). This is consistent with previous findings that TASK-1 interacts with both p11 and syntaxin-8, which are associated with cell surface trafficking and endocytosis (18, 48, 49). Elevated pressure did not appear to alter the localization pattern or labeling intensity of TWIK-1 (Fig. 5A). However, the labeling intensity of TASK-1 appeared to decrease, particularly that with a more diffuse labeling pattern (Fig. 5A). Quantification of immunolabeling intensity revealed that elevated pressure decreased TASK-1 by 35%, as compared with ambient pressure (P < 0.001, Fig. 5B). In contrast, there was no change in the intensity of TWIK-1 immunolabeling between cultures maintained at ambient or elevated pressure (Fig. 5B). These data reflect the protein profiles of TWIK-1 and TASK-1 in retina from our ocular hypertension model and indicate preferential regulation of the TASK-1 K2P channels in Müller glia by elevated pressure.
Fig. 5.
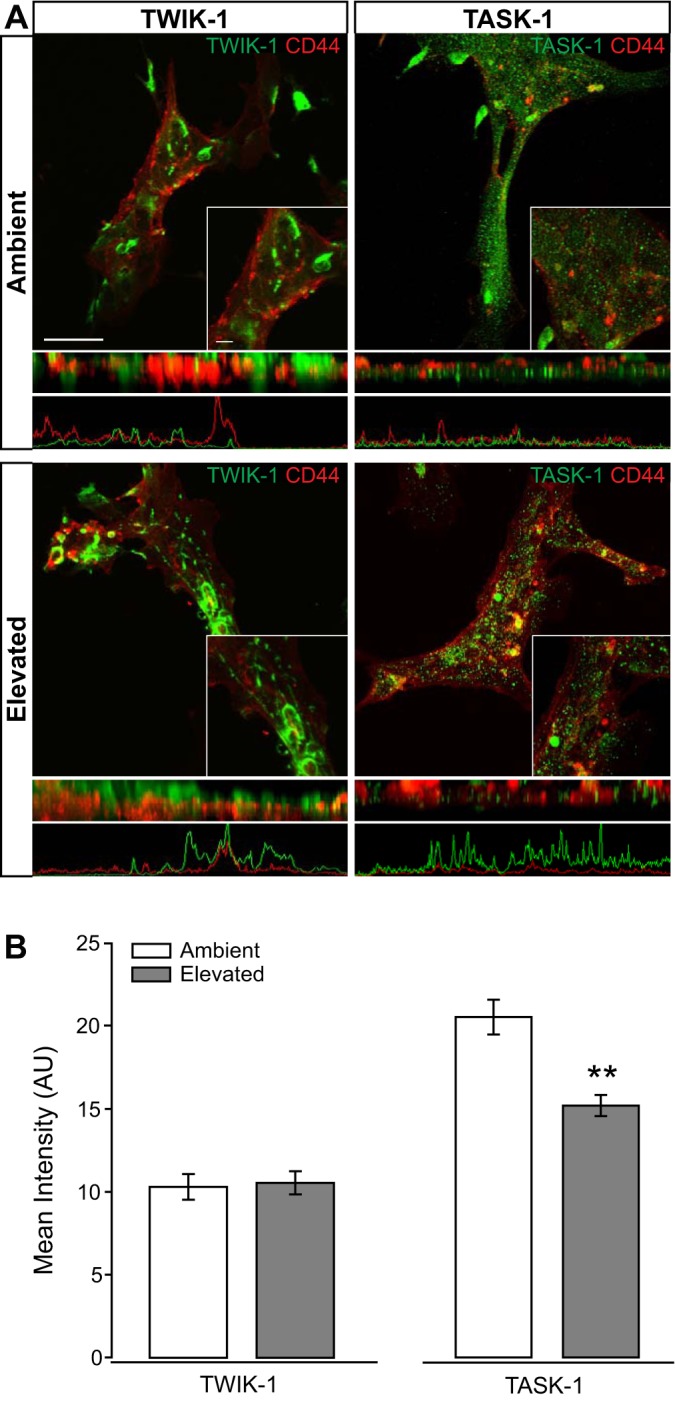
Intensity of TWIK-1 and TASK-1 channel staining in Müller glia cultures following pressure elevation. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated (Elev) pressure for 48 h. A: representative fluorescent micrographs of Müller glia from each condition. Immunolabeling of TWIK-1 (green), TASK-1 (green), and CD44 (red) was performed. Panels directly below the fluoromicrographs are a z-plane view of the image, with associated plot of fluorescent intensity. B: bar graphs of the mean intensity of TWIK-1 and TASK-1 staining compared between ambient and elevated pressure. TWIK-1: n(Amb) = 22 cells; n(Elev) = 13; TASK-1: n(Amb) = 21; n(Elev) = 22. Images taken at ×60; scale bar = 40 µm. Inset taken at ×60 + ×2.5 zoom; scale bar = 10 µm. Student’s t-test was used to analyze statistical significance. **P < 0.005.
Ocular hypertension alters expression of TRESK, but not TREK-2 and TRAAK, in retina.
Next, we profiled protein expression of the K2P channels, TREK-2, TRAAK, and TRESK. These channels also possess inwardly rectifying capabilities, but they are not necessarily linked to Müller glia physiology. We performed immunolabeling for TREK-2, TRAAK and TRESK in retinal sections from saline-injected and microbead-injected mice. Qualitatively, immunolabeling for TREK-2 and TRAAK did not appear to be significantly altered in retina from microbead-injected mice, as compared with saline-injected controls (Fig. 6A). However, immunolabeling for TRESK appeared to increase in the retina from microbead-injected mice, as compared with saline-injected controls (Fig. 6A). As expected, the pattern of immunolabeling did not indicate preferential expression in Müller glia for TREK-2, TRESK, or TRAAK (Fig. 6A). Quantification of immunolabeling intensity confirmed that elevated IOP did not significantly alter the intensity of TREK-2 and TRAAK, but did decrease intensity of TRESK by 25%, as compared with saline-injected controls (Fig. 6B). The lack of IOP-dependent changes in TREK-2 and TRAAK reflect transcriptome data and, thus, indicate that these channels are likely not impacted by ocular hypertension in vivo. In contrast, the protein representation of TRESK appears to decrease in response to elevated IOP.
Fig. 6.

Intensity of TREK-2, TRAAK, and TRESK channel staining in glaucomatous retinal sections. A: representative fluorescent micrographs of retinal sections from saline- or microbead-injected eyes. Immunolabeling of TREK-2 (green), TRAAK (green), TRESK (green), and CD44 (red) was performed. B: bar graphs of the mean intensity of TREK-2, TRAAK, and TRESK staining compared between saline and microbead retina. TREK-2: n(PBS) = 7 retina sections; n(MOM) = 7; TRAAK: n(PBS)= 6; n(MOM)= 7; TRESK: n(PBS) = 6; n(MOM) = 6. Images taken at ×40; scale bar = 20 µm. Student’s t-test was used to analyze statistical significance. *P < 0.05. MOM, microbead occlusion model.
Elevated pressure alters expression and localization of TRAAK and TRESK, but not TREK-2, in Müller glia in vitro.
To assess Müller glia-specific expression of TREK-2, TRAAK, and TRESK, we performed immunolabeling in Müller glia cultures exposed to 48 h of ambient or elevated pressure. In cultures maintained at ambient pressure, TREK-2 and TRESK immunolabeling exhibited similar patterns of localization that appears to be mostly ubiquitous, with some staining concentrated to vesicles (Fig. 7A, top). The differences in localization patterns between channels are clearly evident in the z-plane view of the images directly below the fluoromicrographs, as well as in the associated plots of fluorescent intensity below the z-plane view. This pattern of localization is consistent with the known association between microtubules and TREK-2- and TRESK-containing vesicles (18, 51). Despite similar localization patterns, the intensity of TRESK immunolabeling appeared qualitatively lower than that of TREK-2 (Fig. 7A, top). TRAAK immunoreactivity was the lowest of the three and appeared to be concentrated primarily in vesicles (Fig. 7A, top). Qualitatively, elevated pressure appeared to increase immunoreactivity for TRAAK and TRESK (Fig. 7A, bottom). For TRAAK, this increase in immunoreactivity was also accompanied by a change in localization pattern from primarily vesicular to a combination of vesicular and plasma membrane (Fig. 7A, bottom). For TRESK, the increased immunoreactivity appeared mostly associated with microtubule-like structures, which may indicate an increase in the number of TRESK-containing vesicles and/or increased trafficking of this channel (Fig. 7A, bottom). Elevated pressure did not appear to drastically alter the intensity or localization of TREK-2 (Fig. 7A, bottom). Quantification of immunolabeling intensity confirmed pressure-induced changes in TRAAK and TRESK representation, as well as the pressure insensitivity of TREK-2 representation. Elevated pressure increased the intensity of TRAAK immunolabeling by 24% (P < 0.05) and of TRESK by 57% (P < 0.001), as compared with ambient pressure (Fig. 7B). As qualitatively observed, elevated pressure did not significantly alter the intensity of TREK-2 immunolabeling (P > 0.05; Fig. 7B). These data do not reflect that of protein representations in retina sections, suggesting that the retinal milieu may be important for the regulation of these K2P channels in ocular hypertension.
Fig. 7.

Intensity of TREK-2, TRAAK, and TRESK channel staining in Müller glia cultures following pressure elevation. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated (Elev) pressure for 48 h. A: representative fluorescent micrographs of Müller glia from each condition. Immunolabeling of TREK-2 (green), TRAAK (green), TRESK (green), and CD44 (red) was performed. Panels directly below the fluoromicrographs are a z-plane view of the image, with associated plot of fluorescent intensity. B: bar graphs of the mean intensity of TREK-2, TRAAK, and TRESK staining compared between ambient and elevated pressure. TREK-2: n(Amb) = 24 cells; n(Elev) =21; TRAAK, n(Amb) = 19; n(Elev) = 21; TRESK: n(Amb) = 12; n(Elev) = 22. Images taken at ×60; scale bar = 40 µm. Inset taken at ×60 + ×2.5 zoom; scale bar = 10 µm. Student’s t-test was used to analyze statistical significance. *P < 0.05, **P < 0.005.
Long-term pressure elevation induces cytotoxicity, but not apoptosis in Müller glia.
To determine the effect of elevated pressure on the overall health of Müller glia, we exposed primary, purified cultures of Müller glia to ambient or elevated pressure for either 4 or 48 h. We measured apoptotic death with TUNEL labeling and cytotoxicity with an LDH assay. As demonstrated previously, 48 h of +70 mmHg hydrostatic pressure induces RGC apoptosis and leads to production of inflammatory and trophic factors by retinal microglia, astrocytes and Müller glia (34, 52, 53, 55, 56). Consistent with our previous findings (34), DIC imaging to analyze cell morphology revealed no major changes indicative of cell death, i.e., retraction of processes and swelling or shrinkage of soma, following exposure to either 4 or 48 h of elevated pressure (Fig. 8A). Accordingly, the percentage of TUNEL+ cells did not change in Müller glia cultures exposed to either 4 or 48 h of elevated pressure, as compared with those maintained at ambient pressure (P > 0.05; Fig. 8B). However, the presence of LDH in culture media increased twofold in Müller glia cultures exposed to 48 h of elevated pressure, as compared with ambient pressure (P < 0.001; Fig. 8C). This pressure-induced increase in membrane permeability was not detected after 4 h of elevated pressure (P > 0.05; Fig. 8C). Thus, exposure to elevated pressure for 48 h or less does not induce apoptosis in Müller glia cultures. However, increased membrane permeability, an indicator of cytotoxicity, is detectable by 48 h of pressure exposure.
Fig. 8.

Effect of short- and long-term pressure elevation on Müller glia cell health. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated Elev) pressure for 48 h. A: representative DIC micrographs of Müller glia from each condition. B: bar graphs of the percentage of TUNEL-positive Müller glia compared between each condition. n = 4 wells per condition (combined 5 images at ×20/well). C: bar graphs of the amount of lactate dehydrogenase (LDH) released in the Müller glia culture media compared between each condition. n(Amb 4 h) = 5 wells; n(Elev 4 h) = 5; n(Amb 48 h) = 15; n(Elev 48 h) = 17. Student’s t-test was used to analyze statistical significance. **P < 0.001.
Elevated pressure alters ion homeostasis in Müller glia.
To determine whether elevated pressure alters cation homeostasis in Müller glia, we measured the extracellular concentration of K+ ([K+]E) and Na+ ([Na+]E) in the culture media of Müller glia exposed to ambient or elevated pressure for either 4 or 48 h, using ICP-MS. Exposure to 4 h of elevated pressure did not alter [K+]E or [Na+]E, as compared with ambient pressure (Fig. 9, A and B). In contrast, 48 h of elevated pressure significantly increased [K+]E by 12% (P < 0.001; Fig. 9A) and [Na+]E by 11% (P < 0.001, Fig. 9B), as compared with ambient pressure. At elevated pressure, [K+]E increased by 31% and [Na+]E increased by 12% between 4 and 48 h (P < 0.001 both; Fig. 9B). These data suggest that exposure to 48 h, but not 4 h or less, of elevated pressure induces both K+ and Na+ dyshomeostasis in Müller glia. However, it is important to note that the 48-h exposure time was also associated with increased LDH release, and thus, decreased integrity of the plasma membrane could contribute to this pressure-induced increase in [K+]E and [Na+]E.
Fig. 9.

Elevated pressure alters Na+ and K+ homeostasis in Müller glia. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated (Elev) pressure for 4 or 48 h. A: box plots of the concentration of K+ ions in the culture media of Müller glia at each condition. One-way ANOVA with pairwise comparison by Tukey’s method was used to analyze statistical significance. B: box plots of the concentration of Na+ ions in the culture media of Müller glia compared between each condition. One-way ANOVA with pairwise comparison by Dunn’s method was used to analyze statistical significance. n(Amb 4 h) = 5 wells; n(Elev 4 h) = 4; n(Amb 48 h) = 12; n(Elev 48 h) = 12. **P < 0.001.
Short- and long-term elevated pressure alters K+ flux in Müller glia.
To determine whether the pressure-induced increases in [K+]E and [Na+]E arose from reduced integrity of the plasma membrane or from altered flux through cation channels, we quantified inward flux of cations, using real-time thallium flux imaging (20, 60). Thallium acts as a surrogate for monovalent cations and a fluorescent signal is generated by thallium binding to a cell-permeable Thallos dye (60). In this assay, increased fluorescence signal indicates opening of cation channels, which are promiscuously permeable to thallium (60). Müller glia cultures were loaded with Thallos dye following either 4 or 48 h exposure to ambient or elevated pressure. After the addition of thallium, fluorescence was imaged in live cells at 1-s intervals for 45 s. Representative heat maps of the resultant fluorescent signal are depicted in Fig. 10A. For quantification, we measured the fluorescent intensity/cell at each 1-s time point between ambient and elevated pressure (Fig. 10B), as well as the overall mean fluorescence intensity across cells (Fig. 10C). At ambient pressure, mean thallium flux decreased by 13% between 4- and 48-h time points (P < 0.05; Fig. 10C). Exposure to 4 h of elevated pressure decreased thallium flux by 30%, as compared with 4 h of ambient pressure (P < 0.05; Fig. 10, B and C). By 48 h of exposure to elevated pressure, thallium flux decreased further to 59% of that measured in the respective ambient pressure condition (P < 0.05; Fig. 10, B and C). For cultures maintained at elevated pressure, thallium flux decreased by ~50% between 4 and 48 h of exposure (P < 0.05; Fig. 10C). Together, these data indicate that time in culture alters cation homeostasis by reducing inward cation flux. Exposure to both 4 and 48 h of elevated pressure further reduced cation flux, where the magnitude of this decrease positively correlated with exposure time.
Fig. 10.

Cation channel activity in Müller glia exposed to elevated pressure. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated (Elev) pressure for 4 or 48 h. A: representative heat map showing the fluorescent signal of Thallos dye in Müller glia from each condition. Images were taken after the addition of thallium. B: line graphs displaying the normalized fluorescence intensity of Thallos dyes over time for each condition. Student’s t-test was used to analyze statistical significance. C: box plots of the average fluorescence intensity following addition of thallium compared between each condition. One-way ANOVA with pairwise comparison by Dunn’s method was used to analyze statistical significance. n(Amb 4 h) = 5 cells; n(Elev 4 h) = 12; n(Amb 48 h) = 5; n(Elev 48 h) = 18. *P < 0.05.
Inhibition of cation channels alters pressure-induces changes in K+ flux in Müller glia.
To confirm whether the reduced inward cation flux seen in Müller glia exposed to elevated pressure results from altered flux through cation channels, rather than reduced integrity of the plasma membrane, we treated Müller glia cultures with the cation channel inhibitor, fluoxetine (100 μM), while exposing them to either ambient or elevated pressure for 4 h. Fluoxetine is a broad, nonspecific inhibitor of cation channels (8, 11, 24, 31, 58), which allows for testing of changes in overall membrane conductance versus membrane leakiness. Fluoxetine also specifically has been shown to inhibit several channels involved in K+ siphoning, including Kir4.1 (40) and TREK (30), which makes it a relevant tool to examine changes in cation influx for these studies. Consistent with Fig. 10, 4 h of elevated pressure decreased thallium influx by 30% in vehicle-treated cultures, as compared with those maintained at ambient pressure (Fig. 11, A–C). At ambient pressure, treatment with fluoxetine decreased thallium influx by 50%, as compared with vehicle treatment at ambient pressure (P < 0.05; Fig. 11, B and C). Treatment with fluoxetine at ambient pressure reduced thallium influx to levels comparable to vehicle treatment at elevated pressure. Treatment with fluoxetine at elevated pressure further decreased thallium influx by 39%, as compared with vehicle treatment at elevated pressure (P < 0.05; Fig. 11, B and C). These data indicate that inhibition of cation channels reduces cation influx in Müller glia cultures to an extent that is similar to pressure elevation. This further suggests that pressure-induced changes in ion homeostasis and K+ flux in Müller glia are due to altered cation channel flux, rather than reduced integrity of the plasma membrane.
Fig. 11.

Inhibition of cation channel activity in Müller glia by fluoxetine (Fluox) treatment. Primary, purified cultures of Müller glia were exposed to ambient (Amb) or elevated (Elev) pressure for 4 h, in the presence of vehicle or 100 µM fluoxetine. A: representative heat map showing the fluorescent signal of Thallos dye in Müller glia from each condition. Images were taken after the addition of thallium. B: line graphs displaying the normalized fluorescence intensity of Thallos dyes over time for each condition. Student’s t-test was used to analyze statistical significance. C: box plots of the average fluorescence intensity following the addition of thallium compared between each condition. One-way ANOVA with pairwise comparison by Dunn’s method was used to analyze statistical significance. n(Amb) = 5 cells; n(Elev) = 12; n(Amb+Fluox) = 28; n(Elev+Fluox) = 22. *P < 0.05.
DISCUSSION
Here, we evaluated the effect of elevated pressure, both in vivo and in vitro, on expression and localization of K+ channels in the retina generally and Müller glia specifically. Transcriptome analysis revealed that pressure-induced decreases in the expression of a subset of Kir and K2P channels are a phenotype of ocular hypertension in the microbead-occlusion model (Fig. 1). Assessment of Müller glia-specific changes in gene expression revealed that these decreases do not likely arise from Müller glia alone (Fig. 1). Altered representation of K+ channels with inward rectifying capabilities (Kir and K2P) was observed through immunohistochemical staining in both retinal sections from mice with ocular hypertension and primary cultures of purified Müller glia (Figs. 2–7).
Next, we examined the impact of elevated pressure on health and ion homeostasis in primary cultures of purified Müller glia cells. We evaluated early and sustained responses to pressure in vitro with two exposure paradigms: 4 h and 48 h. We found that at 48 h of elevated pressure, a stimulus known to induce apoptotic death in RGCs (55, 56), Müller glia cultures exhibited: significant LDH release (Fig. 8), increased extracellular concentrations of K+ and Na+ (Fig. 9), and reduced inward cation flux (Fig. 10). Of these pressure-induced changes, only the reduction in inward cation flux was also noted at the 4-h time point (Figs. 8–10). Furthermore, inhibition of cation channels at ambient pressure reduced inward cation flux to levels comparable to elevated pressure (Fig. 11).
Our transcriptome analysis and immunohistochemical studies revealed pressure-induced changes in the expression and localization of both Kir and K2P K+ channels. Elevated pressure decreased the representation of the Kir channel Kir4.1 and the K2P channel TASK-1, while increasing representation of Kir2.1, TRAAK, and TRESK. Elevated pressured also reduced transcription of TWIK-1 (Kcnk1). TWIK-1 and TASK-1 are acid-sensitive K2P channels and exhibit dynamic ion selectivity (18, 35). Under acidic extracellular conditions, TWIK-1 and TASK-1 become permeable to extracellular Na+ (35). Thus, these pressure-induced reductions of these channels on the plasma membrane could contribute to the increase in both K+ and Na+ extracellular concentrations. Interestingly, TRAAK activation is induced by mechanical stretch or strain to the cell (18). This mechanical gating could be relevant for pressure-sensitivity in our study and an increase in the representation of this channel could have significant implications for Müller glia-mediated ion homeostasis in glaucoma.
RNA sequencing of whole retina in the Microbead Occlusion Model revealed only downregulation of Kir and K2P channels after 4 wk of elevated IOP in vivo. The Kir channels Kir3.2, Kir2.2, and Kir2.4 and the K2P channels THIK-2, TASK-1, and TWIK-1 were significantly downregulated (42–60%). Kir3.2 is strongly expressed in RGCs, where it localizes to the plasma membrane, dendrites, and axons (7). K+ currents mediated by the Kir3 channels in RGCs appear to be G-protein-coupled and associated with GABAB receptors (7). Thus, it is likely that downregulation of Kir3.2 in our glaucoma model arises from modulation in RGCs rather than Müller glia, as expression was undetectable through quantitative RT-PCR (qRT-PCR) in Müller glia cultures. Expression and localization of Kir2.2 and Kir2.4 in the retina are less clear. A study of glial cells from guinea pig retina identified expression of mRNA encoding both Kir2.2 and Kir2.4 (46). However, Kir2.2 protein expression was undetectable by immunohistochemistry in murine retina in a later study (33). Our transcriptome analysis indicates that both Kir2.2 and Kir2.4 channels are transcribed in murine retina constitutively and that this expression is sensitive to elevated IOP. Further studies are required to determine the cell type-specific localization and IOP-dependent modulation of these channels. However, expression of both of these channels was undetectable through qRT-PCR in Müller glia cultures. K2P channel expression is predominantly localized to Müller glia and RGCs (26). Based on a recent study examining circadian and developmental modulation of K2P channels in murine retina, TWIK-1 is predominantly expressed by Müller glia, while TASK-1 is predominantly expressed by RGCs (26). Our in vitro data for TWIK-1 are consistent with this study and suggest that downregulation of TWIK-1 in the microbead occlusion model is likely attributable to modulation in Müller cells. While we detected pressure-sensitive expression of TASK-1 protein in purified Müller cells, it is possible that IOP-dependent modulation in vivo also involves modulation of expression by RGCs. Our transcriptome analysis identified constitutive THIK-2 expression in retina that was substantially downregulated by elevated IOP. In the aforementioned study of K2P channel expression in retina (26), THIK-2 was not detectable by qPCR profiling, consistent with our studies. These conflicting results are likely attributable to methodological differences in detection and require further investigation. It is important to note that TASK-1, TWIK-1, and THIK-2 channels are all known to heterodimerize with other channels from their subfamilies (18). Thus, the functional outcomes of changes in expression are likely diverse and dependent on cell type-specific expression. Despite differences in the identity of K2P channels modulated by elevated pressure in vitro and in vivo, both model approaches revealed that K2P channels with dynamic ion selectivity are modulated by elevated pressure (18). Overall, our in vivo findings suggest that ocular hypertension shifts the expression profile of K+ channels with inward-rectifying capabilities, including those with dynamic ion selectivity. Thus, both K+ and Na+ homeostasis may be chronically altered in glaucoma.
Pressure-induced increases in membrane permeability, a hallmark of cytotoxicity, in Müller glia is a novel finding with important implications for studies investigating the role of glia in glaucoma pathology. Elevated pressure in vitro and in vivo leads to apoptosis of RGCs (55, 56). The containment of intracellular contents and removal by phagocytes during apoptotic cell death avoid eliciting inflammation in the neighboring cells (19). However, increased permeability of the membrane, like that observed with cytotoxicity, can release intracellular contents that have inflammatory and potentially, detrimental consequences to surrounding tissue (19). This cytotoxic response could indirectly compromise the health of nearby cells in the retina and perpetuate inflammatory and pathological processes associated with glaucoma. Further investigations should determine the nature, source, and significance of Müller glia cytotoxicity in response to glaucoma-related stressors.
We discovered that elevated pressure induces cation dyshomeostasis in Müller glia cultures by 48 h of exposure. Increases in the extracellular concentration of K+ and Na+ after 48, but not 4, h of elevated pressure could arise from either 1) release of ions through a compromised plasma membrane, like that indicated by LDH release, or 2) reduced ion influx that results from a process with selectivity. Our live cell, thallium flux imaging revealed reduced inward cation flux at both 4- and 48-h time points. While time in culture slightly reduces cation channel activity (ambient pressure 4 versus 48 h), exposure to elevated pressure over time dramatically reduces channel activity (elevated pressure 4 versus 48 h). That changes in cation flux are detectable after only 4 h of exposure indicates that modulation of cation channel activity precedes plasma membrane compromise and elevations in [K+]E and [Na+]E noted at 48 h. Together, our findings indicate that reduced inward flux of cations is an early component of the Müller glia response to elevated pressure.
Maintenance of ion concentration gradients is critical for neuronal function. In the retina, Müller glia siphon K+ to assist in the establishment of proper ion gradients for RGCs (2, 3, 33, 38, 39, 47). Our findings indicate that elevated pressure alters cation homeostasis and cation channel flux in Müller glia in vitro. This is accompanied by changes in the expression and localization profile of Kir and K2P channels with inward-rectifying capabilities. Downregulation of Kir and K2P channels in the microbead occlusion model of glaucoma further supports that disrupting ion homeostasis could impact proper establishment of K+ and Na+ concentration gradients. Prolonged disruption of these gradients could contribute to recently reported alterations in the electrophysiological properties of RGCs in glaucomatous retina (6, 41, 50). Future studies should further evaluate the contribution of Müller glia and ion homeostasis to glaucoma-related changes in RGC physiology.
GRANTS
These studies were supported by the National Eye Institute awards R21EY026176 (D. Li, Y. Xu, and R. M. Sappington), R01EY027729 (Y. Xu D. Li, and R. M. Sappington), T32EY007135 (R. A. Fischer; Vanderbilt Vision Research Center), and P30EY08126 (Vanderbilt Vision Research Center), and an Unrestricted Departmental Award (Vanderbilt Eye Institute) from Research to Prevent Blindness, Inc.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.A.F. and R.M.S. conceived and designed research; R.A.F., A.L.R., and L.K.W. performed experiments; R.A.F., A.L.R., and R.M.S. analyzed data; R.A.F. and R.M.S. interpreted results of experiments; R.A.F., A.L.R., and R.M.S. prepared figures; R.A.F. drafted manuscript; R.A.F and R.M.S. edited and revised manuscript; R.A.F., A.L.R., L.K.W., and R.M.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors would like to thank the Cell Imaging Shared Resource, the Vanderbilt Technologies for Advanced Genomics and the Vanderbilt Technologies for Advanced Genomics Analysis and Research Design core facilities at Vanderbilt University Medical Center for assistance in confocal imaging, RNA sequencing and transcriptome analysis, respectively.
REFERENCES
- 1.Bolz S, Schuettauf F, Fries JE, Thaler S, Reichenbach A, Pannicke T. K+ currents fail to change in reactive retinal glial cells in a mouse model of glaucoma. Graefes Arch Clin Exp Ophthalmol 246: 1249–1254, 2008. doi: 10.1007/s00417-008-0872-x. [DOI] [PubMed] [Google Scholar]
- 2.Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, Reichelt W, Reichenbach A. Role of glial K+ channels in ontogeny and gliosis: a hypothesis based upon studies on Müller cells. Glia 29: 35–44, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 3.Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med 10: 33–44, 2006. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res 31: 702–719, 2012. doi: 10.1016/j.preteyeres.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatelain FC, Bichet D, Douguet D, Feliciangeli S, Bendahhou S, Reichold M, Warth R, Barhanin J, Lesage F. TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci USA 109: 5499–5504, 2012. doi: 10.1073/pnas.1201132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H, Zhao Y, Liu M, Feng L, Puyang Z, Yi J, Liang P, Zhang HF, Cang J, Troy JB, Liu X. Progressive degeneration of retinal and superior collicular functions in mice with sustained ocular hypertension. Invest Ophthalmol Vis Sci 56: 1971–1984, 2015. doi: 10.1167/iovs.14-15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Yu YC, Zhao JW, Yang XL. Inwardly rectifying potassium channels in rat retinal ganglion cells. Eur J Neurosci 20: 956–964, 2004. doi: 10.1111/j.1460-9568.2004.03553.x. [DOI] [PubMed] [Google Scholar]
- 8.Choi JS, Hahn SJ, Rhie DJ, Yoon SH, Jo YH, Kim MS. Mechanism of fluoxetine block of cloned voltage-activated potassium channel Kv1.3. J Pharmacol Exp Ther 291: 1–6, 1999. [PubMed] [Google Scholar]
- 9.Crish SD, Calkins DJ. Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience 176: 1–11, 2011. doi: 10.1016/j.neuroscience.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci USA 107: 5196–5201, 2010. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deák F, Lasztóczi B, Pacher P, Petheö GL, Valéria Kecskeméti, Spät A. Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacology 39: 1029–1036, 2000. doi: 10.1016/S0028-3908(99)00206-3. [DOI] [PubMed] [Google Scholar]
- 12.Eaton MJ, Veh RW, Makarov F, Shuba YM, Reichenbach A, Skatchkov SN. Tandem-pore K+ channels display an uneven distribution in amphibian retina. Neuroreport 15: 321–324, 2004. doi: 10.1097/00001756-200402090-00022. [DOI] [PubMed] [Google Scholar]
- 13.Eberhardt C, Amann B, Feuchtinger A, Hauck SM, Deeg CA. Differential expression of inwardly rectifying K+ channels and aquaporins 4 and 5 in autoimmune uveitis indicates misbalance in Müller glial cell-dependent ion and water homeostasis. Glia 59: 697–707, 2011. doi: 10.1002/glia.21139. [DOI] [PubMed] [Google Scholar]
- 14.Echevarria FD, Formichella CR, Sappington RM. Interleukin-6 deficiency attenuates retinal ganglion cell axonopathy and glaucoma-related vision loss. Front Neurosci 11: 318, 2017. doi: 10.3389/fnins.2017.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Echevarria FD, Rickman AE, Sappington RM. Interleukin-6: a constitutive modulator of glycoprotein 130, neuroinflammatory and cell survival signaling in retina. J Clin Cell Immunol 7: 439, 2016. doi: 10.4172/2155-9899.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Danaf RN, Huberman AD. Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J Neurosci 35: 2329–2343, 2015. doi: 10.1523/JNEUROSCI.1419-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559–605, 2010. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 18.Feliciangeli S, Chatelain FC, Bichet D, Lesage F. The family of K2P channels: salient structural and functional properties. J Physiol 593: 2587–2603, 2015. doi: 10.1113/jphysiol.2014.287268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907–1916, 2005. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer RA, Zhang Y, Risner ML, Li D, Xu Y, Sappington RM. Impact of graphene on the efficacy of neuron culture substrates. Adv Healthc Mater 7: e1701290, 2018. doi: 10.1002/adhm.201701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Formichella CR, Abella SK, Sims SM, Cathcart HM, Sappington RM. Astrocyte reactivity: a biomarker for retinal ganglion cell health in retinal neurodegeneration. J Clin Cell Immunol 5: 188, 2014. doi: 10.4172/2155-9899.1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493–501, 1992. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorczyca W, Gong J, Darzynkiewicz Z. Detection of DNA strand breaks in individual apoptotic cells by the in situ terminal deoxynucleotidyl transferase and nick translation assays. Cancer Res 53: 1945–1951, 1993. [PubMed] [Google Scholar]
- 24.Hahn SJ, Choi JS, Rhie DJ, Oh CS, Jo YH, Kim MS. Inhibition by fluoxetine of voltage-activated ion channels in rat PC12 cells. Eur J Pharmacol 367: 113–118, 1999. doi: 10.1016/S0014-2999(98)00955-8. [DOI] [PubMed] [Google Scholar]
- 25.Heiduschka P, Julien S, Schuettauf F, Schnichels S. Loss of retinal function in aged DBA/2J mice—new insights into retinal neurodegeneration. Exp Eye Res 91: 779–783, 2010. doi: 10.1016/j.exer.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Hughes S, Foster RG, Peirson SN, Hankins MW. Expression and localisation of two-pore domain (K2P) background leak potassium ion channels in the mouse retina. Sci Rep 7: 46085, 2017. doi: 10.1038/srep46085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon C-J, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci 18: 8936–8946, 1998. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji M, Miao Y, Dong L-D, Chen J, Mo X-F, Jiang S-X, Sun X-H, Yang X-L, Wang Z. Group I mGluR-mediated inhibition of Kir channels contributes to retinal Müller cell gliosis in a rat chronic ocular hypertension model. J Neurosci 32: 12744–12755, 2012. doi: 10.1523/JNEUROSCI.1291-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawano T, Zhao P, Nakajima S, Nakajima Y. Single-cell RT-PCR analysis of GIRK channels expressed in rat locus coeruleus and nucleus basalis neurons. Neurosci Lett 358: 63–67, 2004. doi: 10.1016/j.neulet.2003.12.104. [DOI] [PubMed] [Google Scholar]
- 30.Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A. Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol 144: 821–829, 2005. doi: 10.1038/sj.bjp.0706068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirsch GE, Trepakova ES, Brimecombe JC, Sidach SS, Erickson HD, Kochan MC, Shyjka LM, Lacerda AE, Brown AM. Variability in the measurement of hERG potassium channel inhibition: effects of temperature and stimulus pattern. J Pharmacol Toxicol Methods 50: 93–101, 2004. doi: 10.1016/j.vascn.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A. Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia 39: 292–303, 2002. doi: 10.1002/glia.10112. [DOI] [PubMed] [Google Scholar]
- 33.Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience 129: 1045–1056, 2004. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SJ, Duncan DS, Echevarria FD, McLaughlin WM, Hatcher JB, Sappington RM. Pressure-induced alterations in PEDF and PEDF-R expression: implications for neuroprotective signaling in glaucoma. J Clin Exp Ophthalmol 6: 491, 2015. doi: 10.4172/2155-9570.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma L, Zhang X, Zhou M, Chen H. Acid-sensitive TWIK and TASK two-pore domain potassium channels change ion selectivity and become permeable to sodium in extracellular acidification. J Biol Chem 287: 37145–37153, 2012. doi: 10.1074/jbc.M112.398164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsh B, Acosta C, Djouhri L, Lawson SN. Leak K+ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol Cell Neurosci 49: 375–386, 2012. doi: 10.1016/j.mcn.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Millar ID, Wang S, Brown PD, Barrand MA, Hladky SB. Kv1 and Kir2 potassium channels are expressed in rat brain endothelial cells. Pflugers Arch 456: 379–391, 2008. doi: 10.1007/s00424-007-0377-1. [DOI] [PubMed] [Google Scholar]
- 38.Newman EA. Distribution of potassium conductance in mammalian Müller (glial) cells: a comparative study. J Neurosci 7: 2423–2432, 1987. [PMC free article] [PubMed] [Google Scholar]
- 39.Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 225: 1174–1175, 1984. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51, 2007. doi: 10.1016/j.brainres.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 41.Pang JJ, Frankfort BJ, Gross RL, Wu SM. Elevated intraocular pressure decreases response sensitivity of inner retinal neurons in experimental glaucoma mice. Proc Natl Acad Sci USA 112: 2593–2598, 2015. doi: 10.1073/pnas.1419921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pannicke T, Iandiev I, Wurm A, Uckermann O, vom Hagen F, Reichenbach A, Wiedemann P, Hammes HP, Bringmann A. Diabetes alters osmotic swelling characteristics and membrane conductance of glial cells in rat retina. Diabetes 55: 633–639, 2006. doi: 10.2337/diabetes.55.03.06.db05-1349. [DOI] [PubMed] [Google Scholar]
- 43.Pollema-Mays SL, Centeno MV, Ashford CJ, Apkarian AV, Martina M. Expression of background potassium channels in rat DRG is cell-specific and down-regulated in a neuropathic pain model. Mol Cell Neurosci 57: 1–9, 2013. doi: 10.1016/j.mcn.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pondugula SR, Raveendran NN, Ergonul Z, Deng Y, Chen J, Sanneman JD, Palmer LG, Marcus DC. Glucocorticoid regulation of genes in the amiloride-sensitive sodium transport pathway by semicircular canal duct epithelium of neonatal rat. Physiol Genomics 24: 114–123, 2006. doi: 10.1152/physiolgenomics.00006.2005. [DOI] [PubMed] [Google Scholar]
- 45.Quigley HA, Vitale S. Models of open-angle glaucoma prevalence and incidence in the United States. Invest Ophthalmol Vis Sci 38: 83–91, 1997. [PubMed] [Google Scholar]
- 46.Raap M, Biedermann B, Braun P, Milenkovic I, Skatchkov SN, Bringmann A, Reichenbach A. Diversity of Kir channel subunit mRNA expressed by retinal glial cells of the guinea-pig. Neuroreport 13: 1037–1040, 2002. doi: 10.1097/00001756-200206120-00012. [DOI] [PubMed] [Google Scholar]
- 47.Reichenbach A, Bringmann A. New functions of Müller cells. Glia 61: 651–678, 2013. doi: 10.1002/glia.22477. [DOI] [PubMed] [Google Scholar]
- 48.Renigunta V, Fischer T, Zuzarte M, Kling S, Zou X, Siebert K, Limberg MM, Rinné S, Decher N, Schlichthörl G, Daut J. Cooperative endocytosis of the endosomal SNARE protein syntaxin-8 and the potassium channel TASK-1. Mol Biol Cell 25: 1877–1891, 2014. doi: 10.1091/mbc.e13-10-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rescher U, Gerke V. S100A10/p11: family, friends and functions. Pflugers Arch 455: 575–582, 2008. doi: 10.1007/s00424-007-0313-4. [DOI] [PubMed] [Google Scholar]
- 50.Risner ML, Pasini S, Cooper ML, Lambert WS, Calkins DJ. Axogenic mechanism enhances retinal ganglion cell excitability during early progression in glaucoma. Proc Natl Acad Sci USA 115: E2393–E2402, 2018. doi: 10.1073/pnas.1714888115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandoz G, Tardy MP, Thümmler S, Feliciangeli S, Lazdunski M, Lesage F. Mtap2 is a constituent of the protein network that regulates twik-related K+ channel expression and trafficking. J Neurosci 28: 8545–8552, 2008. doi: 10.1523/JNEUROSCI.1962-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sappington RM, Calkins DJ. Contribution of TRPV1 to microglia-derived IL-6 and NF-κB translocation with elevated hydrostatic pressure. Invest Ophthalmol Vis Sci 49: 3004–3017, 2008. doi: 10.1167/iovs.07-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sappington RM, Calkins DJ. Pressure-induced regulation of IL-6 in retinal glial cells: involvement of the ubiquitin/proteasome pathway and NF-κB. Invest Ophthalmol Vis Sci 47: 3860–3869, 2006. doi: 10.1167/iovs.05-1408. [DOI] [PubMed] [Google Scholar]
- 54.Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci 51: 207–216, 2010. doi: 10.1167/iovs.09-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sappington RM, Chan M, Calkins DJ. Interleukin-6 protects retinal ganglion cells from pressure-induced death. Invest Ophthalmol Vis Sci 47: 2932–2942, 2006. doi: 10.1167/iovs.05-1407. [DOI] [PubMed] [Google Scholar]
- 56.Sappington RM, Sidorova T, Long DJ, Calkins DJ. TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci 50: 717–728, 2009. doi: 10.1167/iovs.08-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stockklausner C, Klocker N. Surface expression of inward rectifier potassium channels is controlled by selective Golgi export. J Biol Chem 278: 17000–17005, 2003. doi: 10.1074/jbc.M212243200. [DOI] [PubMed] [Google Scholar]
- 58.Traboulsie A, Chemin J, Kupfer E, Nargeot J, Lory P. T-type calcium channels are inhibited by fluoxetine and its metabolite norfluoxetine. Mol Pharmacol 69: 1963–1968, 2006. doi: 10.1124/mol.105.020842. [DOI] [PubMed] [Google Scholar]
- 59.Wareham LK, Dordea AC, Schleifer G, Yao V, Batten A, Fei F, Mertz J, Gregory-Ksander M, Pasquale LR, Buys ES, Sappington RM. Increased bioavailability of cyclic guanylate monophosphate prevents retinal ganglion cell degeneration. Neurobiol Dis 121: 65–75, 2019. doi: 10.1016/j.nbd.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen 9: 671–677, 2004. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z, Huang P, Liu X, Huang S, Deng L, Jin Z, Xu S, Shen X, Luo X, Zhong Y. Effect of adenosine and adenosine receptor antagonist on Müller cell potassium channel in rat chronic ocular hypertension models. Sci Rep 5: 11294, 2015. doi: 10.1038/srep11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao T, Li Y, Weng C, Yin Z. The changes of potassium currents in RCS rat Müller cell during retinal degeneration. Brain Res 1427: 78–87, 2012. doi: 10.1016/j.brainres.2011.10.011. [DOI] [PubMed] [Google Scholar]