

Abstract
Everolimus inhibits mammalian target of rapamycin complex 1 (mTORC1) and is known to cause induction of autophagy and G1 cell cycle arrest. However, it remains unknown whether everolimus-induced autophagy plays a critical role in its regulation of the cell cycle. We, for the first time, suggested that everolimus could stimulate autophagy-mediated cyclin D1 degradation in breast cancer cells. Everolimus-induced cyclin D1 degradation through the autophagy pathway was investigated in MCF-10DCIS.COM and MCF-7 cell lines upon autophagy inhibitor treatment using Western blot assay. Everolimus-stimulated autophagy and decrease in cyclin D1 were also tested in explant human breast tissue. Inhibiting mTORC1 with everolimus rapidly increased cyclin D1 degradation, whereas 3-methyladenine, chloroquine, and bafilomycin A1, the classic autophagy inhibitors, could attenuate everolimus-induced cyclin D1 degradation. Similarly, knockdown of autophagy-related 7 (Atg-7) also repressed everolimus-triggered cyclin D1 degradation. In addition, everolimus-induced autophagy occurred earlier than everolimus-induced G1 arrest, and blockade of autophagy attenuated everolimus-induced G1 arrest. We also found that everolimus stimulated autophagy and decreased cyclin D1 levels in explant human breast tissue. These data support the conclusion that the autophagy induced by everolimus in human mammary epithelial cells appears to cause cyclin D1 degradation resulting in G1 cell cycle arrest. Our findings contribute to our knowledge of the interplay between autophagy and cell cycle regulation mediated by mTORC1 signaling and cyclin D1 regulation.
Keywords: autophagy, cell cycle, cyclin D1, everolimus, mTORC1 inhibitor
INTRODUCTION
Rapamycin and its analogs (rapalogs) are the first-generation mammalian target of rapamycin complex 1 (mTORC1) inhibitors (11, 23). Compared with rapamycin, rapalogs have a more favorable pharmacokinetic profile (15). Everolimus (Afinitor; Novartis) is the second novel rapalog and approved for the treatment of advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib and for subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS). It is also approved for progressive neuroendocrine tumors of pancreatic origin (PNET), progressive gastrointestinal or lung neuroendocrine tumors, and advanced hormone receptor-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancer in combination with exemestane (https://www.us.afinitor.com/). Everolimus has therapeutic effects similar to those of rapamycin, which specifically inhibits the mTORC1 signaling pathway and stimulates autophagy (14) and late blockage of the G1 cell cycle (21). However, autophagy inhibition by mTORC1 usually is known to occur independently of the mTORC1 targets related to cell proliferation (6, 17).
Recently, a well-known cell cycle regulator, cyclin D1, has been shown to regulate autophagy (2, 4, 19, 25, 26). Normally, cyclin D1 dimerizes with cyclin-dependent kinases 4/6 (CDK4/6) to promote passage through the G1 phase by inhibiting the retinoblastoma protein. Cyclin D1 also has CDK-independent functions via binding to nuclear receptors [including estrogen receptor-α, thyroid hormone receptor, peroxisome proliferator-activated receptor-γ (PPARγ), and androgen receptor (AR)] to regulate cell proliferation, growth, and differentiation (18, 20). By interacting with oncogenes and autophagy-regulating proteins, cyclin D1 was also shown to inhibit autophagy (4, 19).
As a main downstream target of mTORC1, cyclin D1 expression can be regulated by mTORC1 inhibitors at both mRNA and protein levels. Rapalogs can decrease cyclin D1 RNA transcription via the hypoxia-inducible factor 1 (HIF-1) pathway (5, 9) and translation via the 70-kDa ribosomal protein S6 kinase 1 (P70S6K1, also known as S6K) and eukaryotic translation initiation factor 4E-binding protein-1 (4EBP1) pathway (7, 10). The drugs can also reduce the accumulation of cyclin D1 protein by promoting its ubiquitin-mediated proteolysis (8, 9). In this study, we investigated the role of everolimus-stimulated autophagy in regulating cyclin D1 level in breast cancer cells to further elucidate the interplay between everolimus-stimulated autophagy and cell cycle arrest.
MATERIALS AND METHODS
Compliance with ethical standards.
Human breast tissue specimens were obtained from Cooperative Human Tissue Network (CHTN) Western Division. The research with human tissue specimens was conducted with the appropriate approvals by the Institutional Review Board and Biosafety Committee at the University of Texas Health Science Center at San Antonio.
Cell culture and reagents.
The MCF-10DCIS.COM cell line was obtained from Wayne State University and maintained in DMEM-F-12 (1:1) culture medium (GIBCO) supplemented with 5% horse serum, 1.05 mM CaCl2, and 10 mM HEPES. The MCF-7 cell line was obtained from Wayne State University via Dr. Michael Brattain and maintained in McCoy’s 5A medium with 10% fetal bovine serum (FBS), pyruvate, vitamins, amino acids, and antibiotics (13). Cells were incubated in a humidified atmosphere of 95% air plus 5% CO2 at 37°C. Everolimus, 3-methyladenine (3-MA), and chlorhexidine (CHX) were obtained from Selleckchem (Houston, TX). Bafilomycin A1 (Baf A1) was from LC Laboratories (Woburn, MA). Chloroquine (CQ) was obtained from Sigma-Aldrich (St. Louis, MO).
Immunoblot analysis.
After different types of treatment, cells were harvested and resuspended in lysis buffer (50 mmol/l Tris·HCl, 150 mmol/l NaCl, 0.5% Nonidet P-40, 1 mmol/l Na3VO4, 5 mmol/l NaF, and 80 mmol/l B-glycerophosphate). The lysates were centrifuged at 10,000 g for 15 min at 4°C. Equivalent amounts of proteins (50 μg) were analyzed by SDS-PAGE. Appropriate antibodies to cyclin D1, cyclin E1, p27, p21, CDK4, and CDK6 (Santa Cruz Biotechnology, Santa Cruz, CA) and to phospho (p)-P70S6K1, P70S6K1, microtubule-associated protein 1A/1B-light chain 3 (LC3), autophagy-related 7 (ATG-7), and p62 (Cell Signaling Technology, Beverly, MA) were used. Proteins were visualized with peroxidase-coupled secondary antibody from Sigma-Aldrich, using ECL solution for detection.
Immunohistochemistry.
Tissue sections were rehydrated through xylene and graded concentrations of ethanol, incubated in sodium citrate (10 mM, pH 6.0) for 10 min, and then cooled down at room temperature, followed with blocking for endogenous peroxidase with 3% hydrogen peroxide (Thermo Fisher Scientific, Waltham, MA) for 30 min at room temperature. Sections were permeabilized with 0.1% Triton and blocked in 10% goat serum for 30 min. The primary antibodies were anti-cyclin D1 (1:400; Santa Cruz Biotechnology) and anti-LC3 (1:100; Cell Signaling Technology) diluted in PBS and incubated at 4°C overnight. Sections were then incubated with a biotinylated goat anti-rabbit antibody (1:200; BD PharMingen, San Diego, CA). For detection, streptavidin-horseradish peroxidase and DAB Substrate Kit (BD PharMingen) were used, and the counterstain was done with hematoxylin.
Immunofluorescence.
Cells were seeded on sterile coverslips and cultured for 48 h. After various treatments, cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.1% Triton X-100 for 10 min, followed by blocking with 3% BSA for 1 h. Immunofluorescence staining was conducted with antibodies to cyclin D1 (1:100; Santa Cruz Biotechnology) and to LC3 (1:50; Cell Signaling Technology) diluted in PBS and incubated at 4°C overnight. The cells were washed with PBS and incubated with Alexa Fluor 488 (Life Technologies) secondary antibody. Nuclei were stained using DAPI, and the cells were visualized using FV-1000 laser scanning confocal microscopes. Cells from 3 independent experiments in which at least 300 cells were counted were quantified. Images were captured using a fluorescence microscope (Olympus BX51).
Transfection of siRNA.
siRNA at 100 nM was transfected into MCF-10DCIS.COM cells using Lipofectamine 2000 reagent or RNAiMAX (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer. Mission siRNA duplexes (Sigma-Aldrich) were used for knockdown (ATG-7 siRNA-1, SASI_Hs01_00077648; ATG-7 siRNA-2, SASI_Hs01_00077649; cyclin D1 siRNA, SASI_Hs01_00213909). Scrambled siRNA was Mission Universal Negative Control siRNA (SIC001; Sigma-Aldrich).
Flow cytometry analysis.
Cell cycle distribution was measured using flow cytometry (LSR-II; Becton Dickinson) after fixation and propidium iodide staining (final concentration at 50 µg/ml) and analyzed with FACSDiva software (Becton Dickinson).
Treatment of tissue explants with everolimus.
Human breast tissue specimens were obtained from Cooperative Human Tissue Network (CHTN) Western Division. The research with human tissue specimens was conducted with the appropriate approvals by the Institutional Review Board and Biosafety Committee at the University of Texas Health Science Center at San Antonio
Under sterile conditions and 30 min before tissue dissection, gelatin sponges (no. 59-9863; Harvard Apparatus) were presoaked in an explant medium (RPMI 1640 supplemented with 10% FBS, 1% antibiotic-antimycotic, 0.01 mg/ml hydrocortisone, and 0.01% insulin) for 30 min. While sponges were hydrating, breast tumor and adjacent nontumor tissue specimens were cut into ~1–2-mm3 pieces and washed with the explant medium. Upon complete hydration of the gelatin sponges, the sponges were transferred to individual wells of a six-well plate containing 3 ml explant medium. Next, prewashed tumor pieces were placed on top of each gelatin sponge. Explant cultures were incubated at 37°C and 5% CO2. After 2 h of preincubation, an additional 3 ml of explant medium containing a 2X concentration of everolimus were added. After 48 h of culture at 37°C, tissue fragments were processed for histology.
RESULTS
Blockade of autophagy attenuated everolimus-induced G1 cell cycle arrest.
To understand the interplay between everolimus-stimulated autophagy and cell cycle arrest, we used the human breast cancer cell line MCF-10DCIS.COM, which was cloned from a cultured xenograft tumor formed by MCF10AT cells in a severe combined immunodeficient (SCID) mouse and can form ductal carcinoma in situ (DCIS) when injected into SCID mice (1, 16). The cells were treated with everolimus at 1 μM for 0.5, 1, 2, 4, and 6 h (for Western blot analysis), 48 h (for cell proliferation assay), or 2, 6, and 12 h (for cell cycle distribution assay with flow cytometry). Western blot analysis showed that everolimus decreased phospho-P70S6K1 level obviously, which is a readout for mTORC1 activity. Everolimus could also rapidly induce autophagy, as evidenced by decreased p62 and an increased LC3-II-to-LC3-I ratio, a higher level of LC3-II, and a decreased level of LC3-I in everolimus-treated cells compared with untreated cells (Fig. 1A). Interestingly, everolimus also induced G1 cell cycle arrest 6 h after the treatment (Fig. 1D), which was much longer than its effect on autophagy. Furthermore, treatment with 3-MA, a widely used autophagy inhibitor (Fig. 1B), could obviously attenuate everolimus-induced proliferative inhibition and G1 cell cycle arrest (Fig. 1, C and E). These data show that everolimus-stimulated autophagy might be involved in the process of its induction of cell cycle arrest.
Fig. 1.

Role of autophagy in everolimus-induced G1 cell cycle arrest. A: everolimus induced MCF-10DCIS.COM cell autophagy. Cells were exposed to 1 μM everolimus, harvested at the indicated time points, and then lysed to examine the relevant protein levels by Western blot analysis. B: 3-methyladenine (3-MA) at 5 mM blocked everolimus-induced autophagy. C: the autophagy inhibitor 3-MA attenuated everolimus-induced proliferative inhibition. Cells were treated with or without 3-MA at 5 mM for 48 h and then subjected to MTT assay. D: everolimus induced G1 cell cycle arrest in a time-dependent manner. E: the autophagy inhibitor 3-MA attenuated everolimus-induced G1 cell cycle arrest. Cells were treated with everolimus with or without 3-MA at 5 mM for the indicated number of hours and then subjected to flow cytometry analysis. Data shown are representative of at least 3 independent experiments. LC3, microtubule-associated protein 1A/1B-light chain 3; PI, propidium iodide; P70S6K1, 70-kDa ribosomal protein S6 kinase 1; p-P70S6K1, phospho-P70S6K1.
Everolimus arrested cells in G1 phase by decreasing cyclin D1 levels.
To elucidate the mechanism by which everolimus-stimulated autophagy regulates the process of its induction of cell cycle arrest, we first needed to confirm the downstream target of mTORC1 via which everolimus arrests the cell cycle at the G1 phase.
It has been reported that everolimus inhibits cancer growth through reduced synthesis of proteins involved in cell cycle progression, including cyclin D1, resulting in G1 cell cycle arrest. Indeed, among several cell cycle-related proteins, cyclin D1 was noticeably decreased upon everolimus treatment in a time-dependent manner in MCF-10DCIS.COM cells (Fig. 2A). Cyclin E and CDK6 were also decreased to some extent, whereas CDK4 level was stable. On the other hand, the CDK inhibitor p27 was slightly increased by everolimus treatment, which is consistent with a previous study (3), but another CDK inhibitor, p21, was reduced. Moreover, downregulation of cyclin D1 by siRNA rendered MCF-10DCIS.COM cells insensitive to everolimus-induced G1 cell cycle arrest. As shown in the Fig. 2B, cyclin D1 protein level was obviously reduced in MCF-10DCIS.COM cells either by treatment with everolimus or by knockdown of cyclin D1 with the transfection of a cyclin D1 siRNA. Consequently, treatment with everolimus or knockdown of cyclin D1 showed similar efficacy in causing cell accumulation in the G1 phase from ∼70 to ∼80%. Yet, the combination did not further increase the percentage of cells in the G1 phase. These results indicate that cyclin D1 is a target of everolimus in its induction of G1 arrest (Fig. 2C).
Fig. 2.

Everolimus arrested cells in G1 phase by decreasing cyclin D1 levels. A: everolimus decreased cyclin D1 protein levels. Cells were exposed to 1 μM everolimus, harvested at the indicated time points, and then lysed to examine the relevant protein levels by Western blot analysis. B and C: 30 h after transfection with cyclin D1 siRNA or mock siRNA, MCF-10DCIS.COM cells were treated with/without 1 μM everolimus for 6 h, harvested at the indicated time points, and then lysed to examine the relevant protein levels by Western blot analysis or subjected to flow cytometry. CDK, cyclin-dependent kinase; NC, negative control; PI, propidium iodide; P70S6K1, 70-kDa ribosomal protein S6 kinase 1; p-P70S6K1, phospho-P70S6K1.
Everolimus-stimulated autophagy mediated cyclin D1 degradation.
Previous studies have demonstrated that everolimus can inhibit cyclin D1 transcription and translation and promote its proteasome-mediated degradation (9). As one pathway of intracellular protein degradation (22), whether autophagy mediates rapalog-induced decrease of cyclin D1 protein has not been elucidated.
To investigate how everolimus regulates the degradation of cyclin D1 protein, we first determined whether degradation of newly synthesized cyclin D1 protein is accelerated by everolimus. As shown in Fig. 3A, we determined the half-life of the cyclin D1 protein by the introduction of cycloheximide, a well-known protein synthesis inhibitor. In everolimus-treated cells, the cyclin D1 protein had a shorter half-life (~15 min) than that of the control cells (~30 min), indicating that everolimus could impair the stability of the cyclin D1 protein, which is consistent with previous studies (9).
Fig. 3.

Everolimus stimulated autophagy-mediated cyclin D1 degradation in MCF-10DCIS.COM cells. A: everolimus treatment led to faster degradation of cyclin D1. Cells were pretreated with 1 μM everolimus for 1 h, followed by treatment with 100 μg/ml cycloheximide (CHX) to block protein synthesis for the indicated times, and then lysed to examine the protein levels by Western blot analysis (panels at bottom). Densitometric quantitation of cyclin D1 and GAPDH bands was used for calculation of the ratios of cyclin D1 to GAPDH, which are plotted as a function of time in the panel at top. B: the autophagy inhibitor attenuated everolimus-induced faster degradation of cyclin D1. Cells were pretreated with 1 μM everolimus combined with or without 3-methyladenine (3-MA, 10 mM) for 1 h, followed by treatment with 100 μg/ml CHX to block protein synthesis for the indicated times, and then lysed to examine the protein levels by Western blot analysis (panels at bottom). Densitometric quantitation of cyclin D1 and GAPDH bands was used for calculation of the ratios of cyclin D1 to GAPDH, which are plotted as a function of time in the panel at top. C: the autophagy inhibitors 3-MA (10 mM), bafilomycin A1 (Baf A1, 100 nM), and chloroquine (CQ, 10 μM) rescued the downregulation of cyclin D1 induced by everolimus. Cells were treated with 1 μM everolimus combined with or without autophagy inhibitor for 1 h. Data shown are representative or means ± SD of at least 3 independent experiments. LC3, microtubule-associated protein 1A/1B-light chain 3; P70S6K1, 70-kDa ribosomal protein S6 kinase 1; p-P70S6K1, phospho-P70S6K1. *P < 0.05.
To further examine whether the rapid decrease of cyclin D1 protein in everolimus-treated cells is associated with its autophagic degradation, we exposed the cells to the autophagy inhibitor 3-MA; as shown in Fig. 3B, in 3-MA-treated cells, the everolimus-stimulated cyclin D1 protein degradation was attenuated. Furthermore, we also exposed the cells to two additional autophagy inhibitors, CQ and Baf A1. All autophagy inhibitors rescued the decline of cyclin D1 levels induced by everolimus (Fig. 3C). Similar results were observed in the breast cancer MCF-7 cell line (Supplemental Fig. S1; Supplemental Material for this article is available at https://doi.org/10.6084/m9.figshare.8039024.v1).
ATG-7 knockdown disrupted everolimus-induced cyclin D1 degradation.
To further determine the role of autophagy in the degradation of cyclin D1 induced by everolimus, we next used a specific ATG-7 siRNA to knock down Atg-7 expression in the MCF-10DCIS.COM cell line. Knockdown of ATG-7 reduced the conversion of LC3-I to LC3-II (LC3-II-to-LC3-I ratio was decreased by ~33%) and increased cyclin D1 expression (Fig. 4, A and B). In control cells, the cyclin D1 protein level was decreased more by everolimus treatment than in the ATG-7-silenced cells (Fig. 4A).
Fig. 4.

Autophagy-related 7 (Atg-7) knockdown disrupted everolimus-induced cyclin D1 degradation. A: Atg-7 knockdown rescued the downregulation of cyclin D1 induced by everolimus. Forty-eight hours after control or ATG-7 siRNA-1 transfection, MCF-10DCIS.COM cells were treated with/without 1 μM everolimus for 1 h. B and C: ATG-7 knockdown attenuated everolimus-induced degradation of cyclin D1. Forty-eight hours after transfection with Atg-7 siRNA-1 (B) or control siRNA, MCF-10DCIS.COM cells were pretreated with 1 μM everolimus for 1 h, followed by treatment with 100 μg/ml cycloheximide (CHX) to block protein synthesis for the indicated times, and then lysed to examine the protein levels by Western blot analysis. Data shown are representative (A and B) or means ± SD (C) of at least 3 independent experiments. LC3, microtubule-associated protein 1A/1B-light chain 3; NC, negative control; P70S6K1, 70-kDa ribosomal protein S6 kinase 1; p-P70S6K1, phospho-P70S6K1. *P < 0.05.
Next, we compared the alteration half-lives of the cyclin D1 protein by the introduction of cycloheximide in MCF-10DCIS.COM cells transfected with control or ATG-7 siRNA-1 upon everolimus treatment. A slower reduction in cyclin D1 protein level was found in ATG-7-silenced cells (Fig. 4, B and C). Similar results were obtained by transfecting an additional siRNA targeting distinct sequences of ATG-7 (ATG-7 siRNA-2) into MCF-10DCIS.COM cells (Supplemental Fig. S2). Taken together, these data provide biochemical evidence that the faster reduction in cyclin D1 protein level in the control cells is in part due to autophagy-mediated degradation induced by everolimus.
Everolimus stimulated autophagy and decreased cyclin D1 protein levels in human breast tissue explants.
Because everolimus could stimulate autophagy-mediated cyclin D1 degradation in human breast cancer cells in vitro, we extended our investigation in clinical samples by treating human breast tumor and adjacent nontumor tissue samples with everolimus ex vivo. As shown in Fig. 5A, everolimus could also induce the autophagic marker LC-3 and decrease cyclin D1 protein levels in both human breast tumor and adjacent nontumor tissues. We found that cyclin D1 levels were higher and most cyclin D1 was in the nucleus in tumor tissues compared with adjacent nontumor tissues, whereas everolimus treatment resulted in redistribution of cyclin D1 from nucleus to cytoplasm in tumor tissues and obviously reduced total cyclin D1 in the adjacent nontumor tissues. This nuclear-to-cytoplasmic translocation was also observed in MCF-10DCIS.COM cells in vitro after everolimus treatment, which was blocked by the treatment with 3-MA (Fig. 5B).
Fig. 5.

Effects of everolimus on protein levels and subcellular localization of cyclin D1 and status of autophagy in explant human breast tissues. A: explant tissues were treated with vehicle or everolimus for 48 h and were processed for immunohistochemistry. Magnification, ×400. Scale bars represent 25 μm. B: subcellular localization of cyclin D1 in MCF-10DCIS.COM cells. Cells were treated with everolimus for 6 h. The immunostained cyclin D1 protein is shown in green. Magnification, ×400. Scale bars represent 20 μm. Data shown are representative of at least 3 independent experiments. LC3, microtubule-associated protein 1A/1B-light chain 3; 3-MA, 3-methyladenine; PN, patient adjacent nontumor sample; PT, patient tumor sample.
DISCUSSION
Previous reports demonstrated that everolimus can inhibit the G1-to-S transition by affecting cyclin D1 mRNA and protein ubiquitin-proteasomal degradation. In the present study, we showed that autophagy-mediated degradation of cyclin D1 stimulated by this rapalog also plays a critical role in its regulation of the cell cycle.
Recently published articles have shown the possibility of autophagy-cyclin D1 interaction. In 2012, Brown et al. recognized a new function of cyclin D1 in suppressing autophagy in the mammary epithelium (2). Casimiro et al. also reported in 2017 that cyclin D1 restrains autophagy by modulating the activation of AMPK in human breast cancer cell lines (4). On the other hand, Pirtoli et al. (19) observed colocalization of cyclin D1 and beclin-1 and a negative correlation between cyclin D1 and beclin-1 expression in primary glioblastoma tissue after radiotherapy-temozolomide therapy. These observations led the authors to postulate that therapy-induced degradation of cyclin D1 might be due to autophagy without further in vitro or in vivo experimentation. Here, we showed that pretreatment with 3-MA, CQ, or Baf A1 [the classic autophagy inhibitors (12)] or ATG-7 knockdown attenuated everolimus-induced cyclin D1 degradation. In this study, we first showed that blockage of autophagy via the inhibitors or ATG-7 knockdown increased cyclin D1 protein levels and attenuated everolimus-induced cyclin D1 degradation (Figs. 2–4 and Supplemental Figs. S1 and S2). These data support the notion that the autophagy-mediated pathway is involved in the process of rapalog-induced cyclin D1 protein destabilization.
The mammalian target of rapamycin (mTORC1) is the key regulator of G1 cell cycle progression and autophagy. Previous studies showed that mTORC1 increases protein synthesis and cell growth by promoting ribosome biogenesis via its targets S6K and 4EBP1 (11, 23), whereas it regulates autophagy directly in an S6K/4EBP1-independent manner. Therefore, G1 cycle arrest by everolimus is generally known to happen independently of its autophagy induction (6, 17). As discussed above, cyclin D1 may mediate cross talk between autophagy and cell cycle regulated by mTORC1. Indeed, we found that blockade of autophagy attenuated everolimus-induced G1 cell cycle arrest and everolimus-induced autophagy occurred earlier than its induction of G1 arrest. Furthermore, we demonstrated a correlation between everolimus-induced cyclin D1 level decrease and everolimus-stimulated autophagy in explant human breast cancer and adjacent nontumor tissues. We found increased levels and accumulation of cyclin D1 in the nucleus in human breast cancer tissues compared with adjacent nontumor tissues, whereas everolimus induced nuclear export of cyclin D1 and decreased cyclin D1 levels in human breast tissues ex vivo, which is consistent with previous data that mTORC1 inhibitors triggered cyclin D1 nuclear export, ubiquitination, and degradation by the 26S proteasome via activating GSK-3β kinase (9, 26). The fact that 3-MA pretreatment could block everolimus-induced decrease of cyclin D1 in cytoplasm in breast cancer cells indicates that in addition to the ubiquitin-proteasome pathway, everolimus can also stimulate cyclin D1 degradation via the autophagy pathway. Thus, everolimus-stimulated autophagy likely contributes to its induction of G1 arrest.
Autophagy-lysosomal proteolysis is one pathway of intracellular degradation of proteins that occurs in sequence through a phagophore of isolation membrane, autophagosome formation, fusion of autophagosome with lysosome, and, finally, lysosomal degradation of autophagosome materials (24). In the present study, we utilized several autophagy inhibitors by targeting different stages of autophagic flux (Fig. 3C and Supplemental Fig. S1C). 3-MA targets the early stage of autophagy and can reduce the conversion of LC3-I to LC3-II and block the decrease of cyclin D1 levels induced by everolimus. On the other hand, CQ and Baf A1 are lysosome inhibitors and target the end stage of autophagic flux. Upon pretreatment with CQ or Baf A1, the conversion of LC3-I to LC3-II still happens, and the autophagosome can still be formed; however, cyclin D1 level reduction induced by everolimus can still be blocked because of the inhibition of lysosomal degradation. Thus, our study demonstrates one more mechanism of cyclin D1 regulation by mTORC1 inhibition through the autophagy-lysosomal proteolysis pathway (Fig. 6).
Fig. 6.
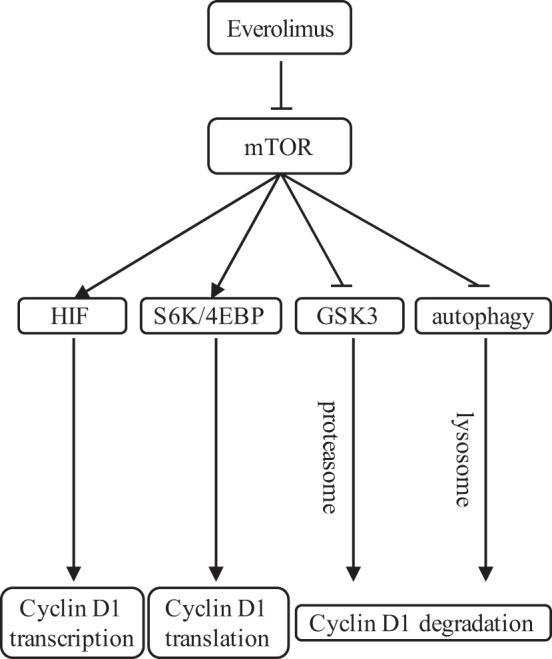
Regulation of cyclin D1 by rapalog. Currently, it is known that in various cell lines, everolimus downregulates the levels of cyclin D1 by inhibiting its transcription and translation and even inducing proteasome-mediated degradation. Our present study proposes a novel regulatory pathway of cyclin D1 via autophagy-mediated cyclin D1 degradation induced by everolimus in breast cancer cells. 4EBP, eukaryotic translation initiation factor 4E-binding protein; HIF, hypoxia-inducible factor; mTOR, mammalian target of rapamycin; S6K, 70-kDa ribosomal protein S6 kinase 1.
In conclusion, our data support the notion that the autophagy induced by everolimus in breast cancer cells appears to cause cyclin D1 protein degradation resulting in G1 cell cycle arrest. Thus, our findings contribute to our knowledge of the interplay between autophagy and cell cycle regulation mediated by mTORC1 signaling and cyclin D1 regulation.
GRANTS
This work was supported in part by National Institutes of Health Grant R01 CA192564 to L.-Z. Sun and National Natural Science Foundation of China Grant No. 81802657 to X.-F. Ding.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.C. and L.-Z.S. conceived and designed research; G.C., X.-F.D., H.B., and K.P. performed experiments; G.C., X.-F.D., H.B., and L.-Z.S. analyzed data; G.C., X.-F.D., H.B., and L.-Z.S. interpreted results of experiments; G.C. and L.-Z.S. prepared figures; G.C. and L.-Z.S. drafted manuscript; G.C. and L.-Z.S. edited and revised manuscript; G.C. and L.-Z.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the patients for donating samples for our research. We also thank CHTN for the collection of human breast tissue samples for our study.
Correspondence may also be addressed to G. Chen (gchen@tzc.edu.cn).
REFERENCES
- 1.Barnabas N, Cohen D. Phenotypic and molecular characterization of MCF10DCIS and SUM breast cancer cell lines. Int J Breast Cancer 2013: 872743, 2013. doi: 10.1155/2013/872743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown NE, Jeselsohn R, Bihani T, Hu MG, Foltopoulou P, Kuperwasser C, Hinds PW. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res 72: 6477–6489, 2012. doi: 10.1158/0008-5472.CAN-11-4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campos T, Ziehe J, Fuentes-Villalobos F, Riquelme O, Peña D, Troncoso R, Lavandero S, Morin V, Pincheira R, Castro AF. Rapamycin requires AMPK activity and p27 expression for promoting autophagy-dependent Tsc2-null cell survival. Biochim Biophys Acta 1863: 1200–1207, 2016. doi: 10.1016/j.bbamcr.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Casimiro MC, Di Sante G, Di Rocco A, Loro E, Pupo C, Pestell TG, Bisetto S, Velasco-Velázquez MA, Jiao X, Li Z, Kusminski CM, Seifert EL, Wang C, Ly D, Zheng B, Shen CH, Scherer PE, Pestell RG. Cyclin D1 restrains oncogene-induced autophagy by regulating the AMPK-LKB1 signaling axis. Cancer Res 77: 3391–3405, 2017. doi: 10.1158/0008-5472.CAN-16-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Bai M, Ning C, Xie B, Zhang J, Liao H, Xiong J, Tao X, Yan D, Xi X, Chen X, Yu Y, Bast RC, Zhang Z, Feng Y, Zheng W. Gankyrin facilitates follicle-stimulating hormone-driven ovarian cancer cell proliferation through the PI3K/AKT/HIF-1α/cyclin D1 pathway. Oncogene 35: 2506–2517, 2016. doi: 10.1038/onc.2015.316. [DOI] [PubMed] [Google Scholar]
- 6.Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M, D’Orazio M, Skobo T, Bordi M, Rohde M, Dalla Valle L, Helmer-Citterich M, Gretzmeier C, Dengjel J, Fimia GM, Piacentini M, Di Bartolomeo S, Velasco G, Cecconi F. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol 17: 20–30, 2015. [Erratum in Nat Cell Biol 17: 706, 2015.] 10.1038/ncb3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen JD, Gard JM, Nagle RB, Dietrich JD, Monks TJ, Lau SS. ERK crosstalks with 4EBP1 to activate cyclin D1 translation during quinol-thioether-induced tuberous sclerosis renal cell carcinoma. Toxicol Sci 124: 75–87, 2011. doi: 10.1093/toxsci/kfr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem 279: 2737–2746, 2004. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 9.Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivières S, Mercep L, Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem 273: 14424–14429, 1998. doi: 10.1074/jbc.273.23.14424. [DOI] [PubMed] [Google Scholar]
- 10.Holmes B, Lee J, Landon KA, Benavides-Serrato A, Bashir T, Jung ME, Lichtenstein A, Gera J. Mechanistic target of rapamycin (mTOR) inhibition synergizes with reduced internal ribosome entry site (IRES)-mediated translation of cyclin D1 and c-MYC mRNAs to treat glioblastoma. J Biol Chem 291: 14146–14159, 2016. doi: 10.1074/jbc.M116.726927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: from laboratory to patients. Cancer Treat Rev 59: 93–101, 2017. doi: 10.1016/j.ctrv.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, , et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222, 2016. [Erratum in Autophagy 12: 443, 2016.] 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei X, Yang J, Nichols RW, Sun LZ. Abrogation of TGFβ signaling induces apoptosis through the modulation of MAP kinase pathways in breast cancer cells. Exp Cell Res 313: 1687–1695, 2007. doi: 10.1016/j.yexcr.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lui A, New J, Ogony J, Thomas S, Lewis-Wambi J. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer 16: 487, 2016. doi: 10.1186/s12885-016-2490-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng LH, Zheng XF. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol Sin 36: 1163–1169, 2015. doi: 10.1038/aps.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst 92: 1185–1186, 2000. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- 17.Neufeld TP. Autophagy and cell growth: the yin and yang of nutrient responses. J Cell Sci 125: 2359–2368, 2012. doi: 10.1242/jcs.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pestell RG. New roles of cyclin D1. Am J Pathol 183: 3–9, 2013. doi: 10.1016/j.ajpath.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pirtoli L, Belmonte G, Toscano M, Tini P, Miracco C. Cyclin D1 co-localizes with beclin-1 in glioblastoma recurrences: a clue to a therapy-induced, autophagy-mediated degradative mechanism? Anticancer Res 36: 4057–4062, 2016. [PubMed] [Google Scholar]
- 20.Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl) 94: 1313–1326, 2016. doi: 10.1007/s00109-016-1475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saunders PO, Weiss J, Welschinger R, Baraz R, Bradstock KF, Bendall LJ. RAD001 (everolimus) induces dose-dependent changes to cell cycle regulation and modifies the cell cycle response to vincristine. Oncogene 32: 4789–4797, 2013. doi: 10.1038/onc.2012.498. [DOI] [PubMed] [Google Scholar]
- 22.Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem 86: 123–128, 2017. doi: 10.1146/annurev-biochem-061516-044859. [DOI] [PubMed] [Google Scholar]
- 23.Vicier C, Dieci MV, Arnedos M, Delaloge S, Viens P, Andre F. Clinical development of mTOR inhibitors in breast cancer. Breast Cancer Res 16: 203, 2014. doi: 10.1186/bcr3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang KC, Sathiyaseelan P, Ho C, Gorski SM. Evolution of tools and methods for monitoring autophagic flux in mammalian cells. Biochem Soc Trans 46: 97–110, 2018. doi: 10.1042/BST20170102. [DOI] [PubMed] [Google Scholar]
- 25.Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, Kondo S, Younes A. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 36: 443–450, 2008. doi: 10.1016/j.exphem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Yu XJ, Han QB, Wen ZS, Ma L, Gao J, Zhou GB. Gambogenic acid induces G1 arrest via GSK3β-dependent cyclin D1 degradation and triggers autophagy in lung cancer cells. Cancer Lett 322: 185–194, 2012. doi: 10.1016/j.canlet.2012.03.004. [DOI] [PubMed] [Google Scholar]