

Abstract
Acid-sensing ion channels (ASICs) are voltage-insensitive cation channels that contribute to cellular excitability. We previously reported that ASIC1 in pulmonary artery smooth muscle cells (PASMC) contribute to pulmonary vasoreactivity and vascular remodeling during the development of chronic hypoxia (CH)-induced pulmonary hypertension. However, the roles of ASIC2 and ASIC3 in regulation of pulmonary vasoreactivity and the development of CH-induced pulmonary hypertension are unknown. We tested the hypothesis that ASIC2 and ASIC3 contribute to increased pulmonary vasoreactivity and development of CH-induced pulmonary hypertension using ASIC2- and ASIC3-knockout (−/−) mice. In contrast to this hypothesis, we found that ASIC2−/− mice exhibit enhanced CH-induced pulmonary hypertension compared with WT and ASIC3−/− mice. This response was not associated with a change in ventilatory sensitivity or systemic cardiovascular function but was instead associated with direct changes in pulmonary vascular reactivity and pulmonary arterial morphology in ASIC2−/− mice. This increase in reactivity correlated with enhanced pulmonary arterial basal tone, elevated basal PASMC [Ca2+] and store-operated calcium entry (SOCE) in PASMC from ASIC2−/− mice. This increase in PASMC [Ca2+] and vasoreactivity was dependent on ASIC1-mediated Ca2+ influx but was not contingent upon an increase in ASIC1 mRNA or protein expression in PASMC from ASIC2−/− mice. Together, the results from this study demonstrate an important role for ASIC2 to regulate pulmonary vascular reactivity and for ASIC2 to modulate the development of CH-induced pulmonary hypertension. These data further suggest that loss of ASIC2 enhances the contribution of ASIC1 to overall pulmonary vascular reactivity.
NEW & NOTEWORTHY This study demonstrates that loss of ASIC2 leads to increased baseline pulmonary vascular resistance, enhanced responses to a variety of vasoconstrictor stimuli, and greater development of hypoxic pulmonary hypertension. Furthermore, these results suggest that loss of ASIC2 enhances the contribution of ASIC1 to pulmonary vascular reactivity.
Keywords: acute hypoxia, blood pressure, plethysmography, pulmonary vascular resistance, store-operated Ca2+ entry
INTRODUCTION
Acid-sensing ion channels (ASICs) belong to the degenerin/epithelial sodium channel (DEG/ENaC) superfamily, which includes several amiloride-sensitive cation channels. Five ASIC genes (ACCN1-5) encoding at least seven distinct ASIC subunits have been identified: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, ASIC4, and ASIC5 (11, 13, 21, 32, 41, 42, 50). Three individual subunits combine to form either a homotrimeric or a heterotrimeric channel that is pH sensitive and voltage independent. The subunit composition of ASICs largely determines the pH sensitivity and gating characteristics of the individual channel (2, 3, 6, 24, 43). The classical activation of ASICs is via extracellular acidosis; however, various nonproton ligands, effector proteins, and signaling molecules have been shown to regulate the function of ASICs (56, 58). ASICs are permeable primarily to Na+, and activation of ASICs in neurons of the central and peripheral nervous system induces membrane depolarization and generation of action potentials (15, 38, 49). In addition to Na+, ASIC1a can additionally conduct Ca2+ (51, 54, 55). The physiological and pathological roles of ASICs have been characterized largely in the nervous system, where they were first identified (9). ASICs have been shown to contribute to cardiovascular homeostasis via sensory signaling from arterial baroreceptors and peripheral chemoreceptor afferent sensory neurons (1). However, recent studies provide evidence that ASICs also exist in several nonneural tissues, including vascular smooth muscle cells (12, 17, 20, 28), although the functional roles of ASICs within the vasculature remain largely unknown.
We have previously shown that ASIC1, ASIC2, and ASIC3 are expressed in the pulmonary circulation and, in particular, pulmonary arterial smooth muscle cells (PASMC) (28). Our previous studies have focused on the contribution of ASIC1a to G-protein coupled receptor (GPCR)-induced vasoconstriction (27, 36). Activation of GPCRs causes depletion of Ca2+ from the sarcoplasmic reticulum, which induces Ca2+ influx through store-operated channels (SOC), leading to pulmonary arterial constriction. ASIC1a appears to be a necessary component of this store-operated Ca2+ entry (SOCE) in PASMC (28). In addition, activation of ASIC1a contributes to hypoxic pulmonary vasoconstriction (36). These data suggest ASIC1a is an important regulator of pulmonary vascular resistance. The goal of this study is to determine whether the presence of ASIC2 and ASIC3 in PASMC additionally contributes to the regulation of pulmonary vascular reactivity.
Chronic vasoconstriction is an important mechanism of increased pulmonary vascular resistance in the pathogenesis of pulmonary hypertension that accompanies many long-term lung diseases associated with chronic hypoxia (CH). This vasoconstrictor hyperresponsiveness is multifaceted and includes increases in both basal- and agonist-induced tone attributable to persistent membrane depolarization of PASMC (35, 45, 46, 48, 57), increased PASMC basal and stimulated Ca2+ levels (8, 31, 44), and enhanced Ca2+ sensitivity of the myofilament contractile apparatus in PASMC (4, 10, 29, 34, 53). Our previous data show that ASIC1-mediated Ca2+ entry in PASMC is an important mediator of both the active vasoconstrictor and vascular remodeling components of pulmonary hypertension. Although ASIC2 and ASIC3 do not conduct Ca2+, they do conduct Na+, and their activation could induce membrane depolarization and subsequent activation of L-type voltage-gated Ca2+ channels (LTCC). Therefore, in this study we used both ASIC2- and ASIC3-null mice to test the working hypothesis that ASIC2 and ASIC3 contribute to increased pulmonary vasoreactivity and development of CH-induced pulmonary hypertension.
METHODS
Animals
All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine and abide by the National Institutes of Health guidelines for animal use. ASIC2- (B6.129-Asic2tm1Wsh/J, stock no. 013126; The Jackson Laboratory; also see Ref. 39) and ASIC3-knockout (−/−) mice (B6.129-Asic3tm1Wsh/J, stock no. 013127; The Jackson Laboratory; also see Ref. 40) were bred on a C57BL/6 background and compared with age-matched C57BL/6 wild-type (WT) controls. Disruption of the relevant ASIC was confirmed by PCR and agarose gel electrophoresis to detect both WT and disrupted alleles (Table 1). Animals were housed one to five per cage in a specific, pathogen-free (SPF) animal care facility and maintained on a 12:12-h light-dark cycle. Standard chow (Teklad soy protein-free diet no. 2920; Envigo) and water were available and accessible at all times. Animals were randomly allocated to experimental groups and when possible, and treatment assignments were blinded to investigator. To obtain sufficient numbers of knockout mice and reduce the number of animals being bred, this study used both male and female mice (14–20 wk old). Potential differences in responses based on sex were determined for key experiments, and P values were reported in figure legends. Our previous experiments showed no significant interaction between sex and development of pulmonary hypertension (36).
Table 1.
Primers and bp product size used for genotyping, RT-PCR, and qPCR for ASIC1, ASIC2, ASIC3, β-actin, and RNA18S
| Gene Product Size | Primer Pair Sequence (Sense/Antisense; 5′–3′) |
|---|---|
| For Genotyping | |
| ASIC2+/+ (365 bp) | AGTCCTGCACGGTGGGAGCTTCTA/GAAGAGGAAGGGAGCCATGATGAG |
| ASIC2−/− (300 bp) | ATGGTTTCGGAGTGGTTTGGCATTGTG/TGGATGTGGAATGTGTGCGA |
| ASIC3+/+ (400 bp) | GAACCTGGAAAACAGAGGCAGGAAGGAT/CAGGGAGTAAGATCTTATGTAGCCTGGC |
| ASIC3−/− (650 bp) | TGGATGTGGAATGTGTGCGA/CCCTGGGCACCAGAGTTGAAGGTGTAGC |
| For RT-PCR | |
| ASIC2 (240 bp) | TCCGAGAACATTCTTGTTCTGGAT/GTTCTCATCATGGCTCCCTTCCTC |
| ASIC3 (432 bp) | TGAGAGCCACCAGCTTACC/GGCAGATACTCCTCCTGCT |
| β-Actin (294 bp) | Quantum RNA internal standards (Invitrogen cat. no. AM1720) |
| For qPCR | |
| ASIC1 (91 bp) | CTGGCCCTGCTCAACAAC/GGAAGTTGGCCTTGTCCTG |
| RNA18S (151 bp) | GTAACCCGTTGAACCCCATT/CCATCCAATCGGTAGTAGCG |
ASIC, acid-sensing ion channel; bp, base pairs; qPCR, quantitative PCR; RNA18S, 18s ribosomal RNA.
Exposure to chronic hypoxia.
Pulmonary hypertension was induced by chronic hypoxia (CH). Animals designated for exposure to CH were housed in a clear plexiglass hypobaric chamber (∼18 ft3), with barometric pressure maintained at ∼380 mmHg for 4 wk. The hypobaric chamber was partially evacuated with a vacuum pump, allowing for continuous air flow of 30 l/min through the chamber. The chamber was opened three times/wk to change bedding and provide fresh water and food. Age-matched control mice were housed at ambient barometric pressure (∼630 mmHg in Albuquerque, NM). We have previously demonstrated that this mouse model mimics cardiopulmonary changes observed in human pulmonary hypertension, including right ventricular hypertrophy, enhanced vasoconstriction, and arterial remodeling (36).
Assessment of Systemic Mean Arterial Blood Pressure
Blood pressure and heart rate were recorded in male mice using radiotelemetry devices (PA-C10 implant; Data Systems International). Telemetry transmitters were surgically implanted under sterile conditions with inhaled isoflurane anesthesia (2% isoflurane and 98% O2 gas mixture). The analgesic Buprenex (buprenorphine; 0.1 mg/kg im) and lidocaine (8 mg/kg sc) were administered before the start of surgery to provide effective recovery and preemptive pain management. Using sterile techniques, an incision was made midline and the carotid artery exposed by blunt dissection. A small incision was made in the carotid artery between two silk sutures and the end of the catheter of a PA-C10 small implantable telemetry probe inserted and advanced toward the heart. The tip was tied in place and the body of the telemeter secured subcutaneously in the mid-flank region of carotid artery. The wound was closed with sterile suture and the mouse recovered 5 days before blood pressure measurements.
Collection of Intrapulmonary Arteries and Generation of Primary PASMC Culture
WT, ASIC2−/−, and ASIC3−/− mice were anesthetized with pentobarbital sodium (200 mg/kg body wt ip), and the heart and lungs were removed by midline thoracotomy. Intrapulmonary arteries (∼2nd to 5th order) were dissected from surrounding lung parenchyma and either snap-frozen for Western blot analysis or enzymatically digested for primary culture. For primary culture, isolated arteries were incubated in reduced-Ca2+ Hank’s Balanced Salt Solution (HBSS) containing papain (9.5 U/ml), type I collagenase (750 CDU/ml), dithiothreitol (1 mg/ml), and BSA (2 mg/ml) at 37°C for 15 min. Single smooth muscle cells were dispersed by gentle trituration with a fire-polished pipette in Ca2+-free HBSS. The cell suspension was plated in smooth muscle cell growth media (Cell Biologics) and 1% penicillin-streptomycin for 48 h in a humidified atmosphere of 5% CO2-95% air at 37°C. Then, cells were switched to 1% FBS for 72 h. Cellular purity was assessed by morphological appearance under phase contrast microscopy and the presence of immunofluorescence staining for rabbit anti-myosin (smooth muscle) heavy chain BT-562 (1:200, no. J64817; Alfa Aesar), goat anti-smooth muscle 22 α-transgelin (TAGLN) (1:200, ab10135; Abcam), and goat anti-smooth muscle α-actin (1:200; no. ab21027; Abcam), as shown in Fig. 1.
Fig. 1.

Representative immunofluorescence images in primary pulmonary arterial smooth muscle cells (PASMC) showing expression of smooth muscle 22α (SM 22α)/transgelin (TAGLN) and smooth muscle myosin heavy chain (SM-MHC) (A) and smooth muscle α-actin (SMA) and SM-MHC (B). Merged images show nuclear counterstain (TO-PRO-3; blue).
ASIC Expression
RT-PCR.
Total RNA was prepared from PASMC using TRIzol extraction, and 1 µg of total RNA was reverse transcribed to cDNA using the Transcriptor First-Strand cDNA Synthesis kit (Roche). Specific primers were used to detect transcripts for ASIC2, ASIC3, and β-actin (Table 1). PCR was performed with the iCycler PCR system (Bio-Rad) using Taq polymerase. First-strand cDNA (4 µl) was amplified by annealing at 59°C for 30 s, extending at 68°C for 1 min, and denaturing at 94°C for 30 s. PCR products were electrophoresed through a 3% agarose gel and stained with ethidium bromide for visualization under UV light.
ASIC1 qPCR.
Total RNA was prepared from PASMC using TRIzol extraction, and 900 ng of total RNA was reverse transcribed to cDNA using the Transcriptor First-Strand cDNA Synthesis kit (Roche). Specific primers were used to detect transcripts for ASIC1 and 18s ribosomal RNA as an internal control (Integrated DNA Technologies) (Table 1) by real-time quantitative PCR in PASMC from WT, ASIC2−/−, and ASIC3−/− mice. SYBR Green (Applied Biosystems) was used with the StepOne Plus Real-Time PCR system (Applied Biosystems), and relative quantification of gene expression was determined by the comparative CT method ().
Western blot analysis.
Intrapulmonary arteries (< 500 µm) from control and CH WT mice or PASMC lysates (25 μg) from WT, ASIC2−/−, and ASIC3−/− mice were homogenized in 10 mM Tris·HCl homogenization buffer (containing 255 mM sucrose, 2 mM EDTA, 12 μM leupeptin, 1 μM pepstatin A, 0.3 μM aprotinin) and centrifuged at 10,000 g for 10 min at 4°C to remove insoluble debris (27). Sample protein concentrations were determined by the Qubit Protein Assay (Life Technologies). Pulmonary artery lysates (25 µg) were separated by SDS-PAGE (7.5% TGX) and transferred to polyvinylidene difluoride membranes. Blots were blocked for 1 h with 5% milk then incubated for 48 h at 4°C with rabbit anti-ASIC1 (1:500, no. ab5674P; Millipore), rabbit anti-ASIC2 (1:500, no. ab5460; Millipore), or rabbit anti-ASIC3 (1:400, no. ab49333; Abcam). For immunochemical labeling, blots were incubated with goat anti-rabbit IgG-horseradish peroxidase (1:3,000 for 1 h; Bio-Rad). Blots were exposed to chemiluminescence-sensitive film (GeneMate), and quantification of ASIC bands was accomplished by densitometric analysis of scanned images using ImageJ gel analysis (National Institutes of Health). We normalized total ASIC expression to total protein per lane, because we have consistently observed that CH increases both mRNA and protein expression of typical “housekeeping” genes/proteins in pulmonary arterial homogenates and isolated PASMC from rats and mice. To determine total protein levels, PVDF membranes were stained with Coomassie brilliant blue R-250 (1610400; Bio-Rad). Densitometric analysis of total lane protein from scanned images was performed using ImageJ gel analysis.
Assessment of Pulmonary Hypertension
Following 4 wk of CH, mice were anesthetized (2% isoflurane and 98% O2 gas mixture), and right ventricular systolic pressure (RVSP) and heart rate were measured via transdiaphragmatic direct cardiac puncture, as previously described (36). An upper transverse laparotomy was performed to expose the diaphragm. A 25-gauge needle, connected to a pressure transducer (model APT300; Harvard Apparatus) through a saline-filled catheter, was inserted into the RV via a closed-chest transdiaphragmatic approach, and the output was amplified using a TAM-A bridge amplifier (Hugo Saks Electronik; Harvard Apparatus) and recorded using Powerlab data acquisition and LabChart software (ADInstruments). The change in RV pressure over time (dP/dt) also provides an indication of RV contractility. However, dP/dt is influenced by preload (Cyon-Frank-Starling law), afterload (Anrep effect), HR (Bowditch effect), and myocardial hypertrophy. To account for the increases in RV hypertrophy, afterload, and to some degree preload following the development of pulmonary hypertension, we assessed RV contractility using the contractility index: dP/dtmax divided by the pressure (P) at the time of dP/dtmax (1/s; LabChart Software). Right ventricular hypertrophy in response to CH was assessed by measuring the mass ratio of the right ventricle to left ventricle plus septum (Fulton’s index). Hematocrit was measured using the iStat hand-held blood analyzer (Abbott Laboratories).
Assessment of Arterial Remodeling
To determine the degree of pulmonary arterial remodeling, lung sections were prepared for immunofluorescence from paraffin-embedded lung tissue. Mice were anesthetized with pentobarbital sodium (200 mg/kg ip). After a median sternotomy, heparin (100 U/20 g body wt) was injected directly into the RV, and the pulmonary artery was cannulated with a 22-gauge feeding needle. The preparation was immediately perfused with 25 ml of 0.1 M PBS containing 10−4 M papaverine to maximally dilate the vasculature and flush the circulation of blood. The lungs were then perfused with 25 ml of fixative (0.1 M PBS containing 4% sucrose, 4% paraformaldehyde, and 10−4 M papaverine) at a pressure of 50 cm H2O above the hilum, and the trachea was inflated to a pressure of 25 cm H2O causing a transmural distending pressure of 25 cm H2O during fixation. The trachea was ligated with 4-0 silk, and the lungs were immersed in fixative overnight, dehydrated, and then mounted in paraffin.
Sections were cut (5 μm thick) and mounted onto Superfrost Plus slides (Fisher Scientific). Antibody/antigen binding was enhanced by an antigen retrieval method in which sections were held just below boiling for 15 min with 10 mM Tris (pH 9.0) buffer (containing 1 mM EDTA + 0.05% Tween 20). Sections were incubated with rabbit anti-smooth muscle α-actin (1:200; Abcam) overnight at 4°C. Smooth muscle α-actin was detected by incubating the slides with DyLight 549-donkey anti-rabbit (1:400, 3.5 h; Jackson ImmunoResearch). Cross-section images of ∼30–50 pulmonary arterioles (<100 µm inner diameter) were randomly collected per animal using a ×63 objective on a confocal microscope (TCS SP5, Leica). Images were thresholded using ImageJ software (National Institutes of Health). Regions of interest (ROIs) were drawn around each fully muscularized artery. The percent thresholded area to total ROI area was calculated for each artery as the percent muscularization. Arterial diameter was calculated based on the circumference of the ROI. Fluorescence images were digitally inverted to provide better contrast and visibility of immunofluorescence.
Assessment of Ventilatory Parameters Using Whole Body Plethysmography
Respiratory rate and tidal volume were measured using whole body plethysmography, as first described by Drorbaugh and Fenn (16) and later adapted by our group in rodents (14, 52). Conscious, unrestrained CH mice were placed into a ∼50 cubic centimeter plexiglass chamber fitted with a Validyne DP45-16 differential pressure transducer. Each breath corresponds to a pressure deflection, the magnitude of which can be used to calculate tidal volume, using the following equation: VT = PT/PK × VK × (TR[PB-PC])/(TR[PB-PC] − TC[PB − PR]), where VT = tidal volume VK = the volume of air injected into the animal chamber for calibration, PT = the pressure deflection associated with each tidal volume, PK = the pressure deflection associated with injection of the calibrating volume, VK TR = body temperature, which is assumed to be 37°C for all animals, TC = the air temperature in the chamber, which varied from 20 to 23°C, PB = barometric pressure, 630 mmHg in Albuquerque, NM, PR = vapor pressure of water at body temperature, and PC = vapor pressure of water in the chamber, which was derived from TC assuming 100% humidity, which was confirmed in pilot experiments (gas mixtures were humidified by bubbling through 3consecutive flasks of water before entry into the plethysmography chamber).
Tidal volume and minute ventilation were normalized to body mass. After a baseline measurement was taken, the chamber was flushed with gas mixture to achieve hypoxia (7% O2, 0% CO2, balance N2) or hypercapnia (21% O2, 6% CO2, balance N2). To specifically assess the roles of ASICs in the ventilatory response to hypoxia without the confounding influence of hypocapnia due to hypoxic-induced hyperventilation, we additionally assessed responses to isocapnic hypoxia (7% O2, 3.2% CO2, balance N2) by supplementing the hypoxic gas mixture with 3.2% CO2 to offset hypocapnia, as previously determined (14).
Femoral Artery Catheterization and Blood Gas Measurement
Mice were implanted with a femoral artery catheter (tapered PE-10 filled with a solution of 0.9% saline containing 100 U/ml heparin) under anesthesia (2% isoflurane and 98% O2 gas mixture) for arterial blood sampling to determine arterial Po2, Pco2, and pH. Blood gas measurements were performed 2 days after the implantation surgery. Mice were placed in a polycarbonate chamber, and catheters were routed out through the top of the cage through spring tethers to enable blood sampling in conscious, unrestrained mice. Mice were exposed to different inspired gas mixtures using a PEGAS 4000MF gas mixer (Columbus Instruments), and gas levels within the chamber were confirmed using an O2Cap Oxygen/CO2 Analyzer (OxiGraf). To measure blood gases, ∼100 μl of blood was allowed to flow directly from the femoral artery catheter onto the iStat handheld blood gas analyzer cartridges (EG6+ or G3+; Abbott Laboratories). Each measurement was taken after 5 min of exposure to the respective inspired gas mixture, with ≥20 min between subsequent exposures. The catheter was flushed with a volume of saline (containing 100 U/ml heparin) equivalent to 1.5 times the dead space of the catheter between each measurement. For each individual mouse, all blood gas measurements were performed on a single day.
Pulmonary Vasoconstrictor Responses in the Isolated Perfused Mouse Lung
Baseline pulmonary vascular resistance (PVR) and changes in PVR were assessed in isolated, saline-perfused lungs from control WT, ASIC2−/−, and ASIC3−/− mice, as described previously (36). Mice were anesthetized with pentobarbital sodium (200 mg/kg ip). The trachea was cannulated with a 18-gauge blunt needle stub, and the lungs were ventilated with a positive-pressure mouse ventilator (Model 845; Hugo Sachs Elektronik) at a frequency of 150 breaths/min and a tidal volume of 8 μl/g body wt with a humidified normoxic gas mixture (21% O2, 6% CO2, balance N2), with a peak end-expiratory pressure of 2 cm H2O. After a median sternotomy, heparin (5 U/g body weight) was injected directly into the right ventricle, and the pulmonary artery was cannulated with a 22-gauge feeding needle. The preparation was immediately perfused with NaHCO3-buffered PSS [containing (in mM) 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 19 NaHCO3, 1.8 CaCl2, 0.5 NaH2PO4, and 5.5 glucose] containing 4% (wt/vol) albumin (Sigma), 300 µM NG-nitro-l-arginine (Sigma), and 30 µM meclofenamate (Sigma) at 0.2 ml/min with a peristaltic pump (Ismatec). NG-nitro-l-arginine (nitric oxide synthase inhibitor) and meclofenamate (cyclooxygenase inhibitor) were added to minimize potential complicating influences of endogenous nitric oxide and cyclooxygenase products on vascular reactivity. The perfusion rate was gradually increased to 30 µl·min−1·g body wt−1 over the course of 5 min and maintained at this rate for the duration of the experiment. Pulmonary arterial and venous pressures were measured with P75 transducers linked to PLUGSYS TAM-A amplifiers (Harvard Apparatus). Pulmonary venous pressure was maintained at 1 mmHg by adjusting the height of the venous outflow tubing in the perfusate reservoir. After a 20-min stabilization period, changes in PVR were assessed in response to acute hypoxic ventilation (6% CO2, balance N2), KCl (30 mM depolarizing stimulus; achieved by replacing the equivalent concentration of NaCl), or the stable thromboxane A2 analog U-46619 (10−9 to 10−6 M). Data were stored and processed with a computer-based data acquisition analysis system (AT-CODAS; DATAQ Instruments).
Cannulation and Fura-2 Loading of Small Pulmonary Arteries
To determine changes in pulmonary vasoreactivity, small intrapulmonary arteries were cannulated and pressurized for simultaneous dimensional and [Ca2+]i analysis, as previously described (36). Briefly, mice were anesthetized with pentobarbital sodium (200 mg/kg ip). The left lung was removed, and small intrapulmonary arteries (4th to 5th order, ∼150 µm passive inner diameter) were dissected free of bronchioles and lung parenchyma, transferred to a vessel chamber (Living Systems), and secured to tapered glass pipettes with a single strand of silk ligature. After cannulation, the artery was pressurized with a servo-controlled peristaltic pump (Living Systems) to 12 mmHg. Any artery that failed to maintain pressure was discarded. The vessel chamber was superfused (5 ml/min at 37°C) with HEPES-based PSS (in mM: 130 NaCl, 4 KCl, 1.2 MgSO4, 4 NaHCO3, 10 HEPES, 1.18 KH2PO4, 1.8 CaCl2, 6 glucose, and 3 EGTA, pH adjusted to 7.4 with NaOH). Arteries were incubated abluminally with Fura-2 AM (2 μM and 0.05% pluronic acid in PSS; Molecular Probes) for 45 min at room temperature, as previously described (36). Fura-2-loaded small pulmonary arteries were alternately excited at 340 and 380 nm at a frequency of 1 Hz with a dual-excitation light source (HyperSwitch; IonOptix), and the respective 510-nm emissions were detected with a photomultiplier tube. Red-wavelength, bright-field images were obtained simultaneously using an Eclipse TS100 microscope (Nikon) and IonOptix CCD100M camera to measure inner diameter, and dimensional analysis was performed by IonOptix Ion Wizard software (IonOptix).
Experiments were conducted in WT and ASIC2−/− mice in the presence or absence of the specific ASIC1a inhibitor psalmotoxin 1 (PcTX1, 20 nM; Phoenix Peptides). Basal tone was determined by calculating the percent change in baseline diameter compared with passive diameter taken during Ca2+-free, HEPES-based PSS (in mM: 130 NaCl, 4 KC1, 1.2 MgSO4, 4 NaHCO3, 10 HEPES, 1.18 KH2PO4, 6 glucose, and 3 EGTA, pH adjusted to 7.4 with NaOH). Depolarization-mediated vasoconstriction was determined using depolarizing concentrations of equal molar KCl (30 mM; Sigma-Aldrich). Receptor-mediated vasoconstrictor reactivity was assessed by superfusion (5 ml/min at 37°C) of cumulative concentrations of the thromboxane analog U-46619 (10−10 to 10−6 M; Cayman Chemicals). For U-46619 concentration-response curves, statistical analysis was performed on the two main pharmacodynamics properties: maximum effect (Emax) and the concentration producing 50% of the maximum effect (log EC50).
Assessment of Basal PASMC Intracellular Ca2+ and Store-Operated Ca2+ Entry
PASMC were incubated with fura-2 AM (3 μM) and 0.05% pluronic acid in HEPES-based PSS for 30 min at 32°C. PASMC were superfused (5 ml/min at 37°C) with HEPES-based PSS for 15 min, and then basal PASMC [Ca2+]i was assessed in fura-2-loaded PASMC and alternately excited at 340 and 380 nm at a frequency of 1 Hz with a dual-excitation light source (HyperSwitch, IonOptix), and the respective 510-nm emissions were collected with a photomultiplier tube.
Store-operated Ca2+ entry.
The effect of ASIC2 and ASIC3 deletion on SOCE was determined as described previously (19, 23, 36). After basal fura-2 ratios were determined, PASMC were superfused (5 ml/min at 37°C) with Ca2+-free HEPES-based PSS containing 50 μM diltiazem to prevent Ca2+ entry through LTCC. Previously, we demonstrated that this concentration of diltiazem blocks the increase in [Ca2+]i and vasoconstrictor response to a depolarizing concentration of KCl (50 mM) (26). In addition, arteries/cells were incubated with the sarco/endoplasmic reticulum Ca2+-ATPase inhibitor cyclopiazonic acid (10 μM) to deplete intracellular Ca2+ stores and prevent Ca2+ reuptake. Changes in [Ca2+]i were determined upon repletion of HEPES-based PSS containing 1.8 mM CaCl2 in the continued presence of diltiazem and cyclopiazonic acid. Area under the curve was calculated as previously described (37). Some experiments were additionally conducted in the presence or absence of the ASIC1a inhibitor PcTX1 (20 nM) by incubation of fura-2-loaded PASMC with PcTX1 for 30 min before and during the SOCE response.
Antibody Validation
The ASIC antibodies used in our present study have been validated previously by our laboratory using genetically deficient animals and/or lysates from genetically silenced cells (28, 36). Full-length gel images have been included for all antibodies. SM-MHC, SM22α, and SMA immunofluorescence antibodies were validated by lack of labeling in non-smooth muscle cells.
Statistics
Based on previous studies from our laboratory, eight to 10 animals per group will be needed to achieve ≥80% power (α = 0.05) for detecting physiologically significant differences (P < 0.05). For experiments involving measurements from multiple cells from an individual animal, data will be averaged for each animal and considered as a single observation and indicated in the graphs as single data points. Statistical tests are specified in the figure legends and were made using Prism 7.02 (GraphPad Software).
RESULTS
ASIC2−/− and ASIC3−/− Mice Characterization
ASIC2 and ASIC3 mRNA are expressed in PASMC from WT mice (Fig. 2). The lack of expression in the corresponding null mice verifies genetic knockout in PASMC. To determine the effect of ASIC2−/− and ASIC3−/− on baseline cardiovascular hemodynamics, we measured systemic MABP and heart rate in conscious control mice by radiotelemetry. ASIC2−/− and ASIC3−/− mice had similar MABP and heart rate compared with WT mice (Table 2), indicating a lack of significant differences in normal cardiovascular hemodynamics between WT, ASIC2−/−, ASIC3−/− conscious mice.
Fig. 2.
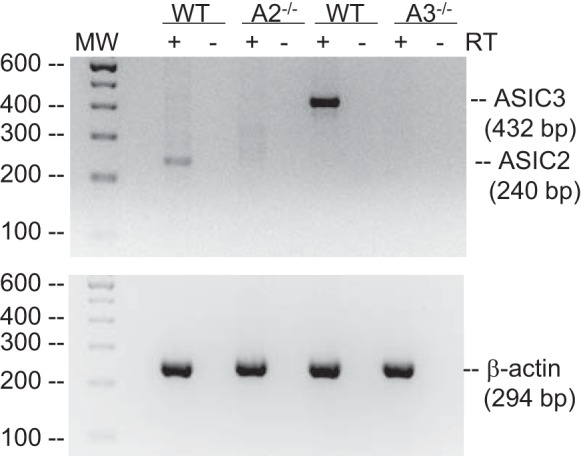
Acid-sensing ion channel (ASIC)2 and ASIC3 are expressed in pulmonary arterial smooth muscle cells (PASMC). Representative electrophoresis gel showing reverse transcriptase (RT)-PCR for ASIC2 (240 bp), ASIC3 (432 bp), and β-actin (294 bp) in PASMC from wild-type (WT), ASIC2−/−, and ASIC3−/− mice.
Table 2.
Average systemic MABP (mm Hg) and HR in beats/min in conscious WT, ASIC2−/−, and ASIC3−/− male mice by radiotelemetry recorded over 72 h
| MABP, mm Hg | P Value vs. WT | HR, beats/min | P Value vs. WT | |
|---|---|---|---|---|
| WT | 114 ± 4 | 546 ± 22 | ||
| ASIC2−/− | 116 ± 7 | 0.9 | 565 ± 11 | 0.73 |
| ASIC3−/− | 116 ± 6 | 0.9 | 529 ± 18 | 0.8 |
Values are means ± SE; n = 5/group, analyzed by 1-way ANOVA. ASIC, acid-sensing ion channel; HR, heart rate; MABP, mean arterial blood pressure; WT, wild type.
ASIC2−/− Mice Exhibit an Enhanced CH-Induced Pulmonary Hypertensive Response
Exposure of animals to CH for 4 wk did not significantly alter ASIC2 (P = 0.30) or ASIC3 (P = 0.55) protein expression in isolated intrapulmonary arteries (Fig. 3). RVSP was similar between WT, ASIC2−/−, and ASIC3−/− control mice (Fig. 4A). After 4 wk of exposure to CH, all mice exhibited increased RVSP compared with their respective control; however, RVSP was significantly greater in ASIC2−/− CH-mice (44 ± 1 mmHg) compared with WT CH-mice (38 ± 1 mmHg; Fig. 4A). In contrast, we observed no such effect of ASIC3−/− on RVSP. RV diastolic pressure (RVDP) was similar between all groups and close to zero. Heart rate was not different between groups or genotypes (Table 3).
Fig. 3.
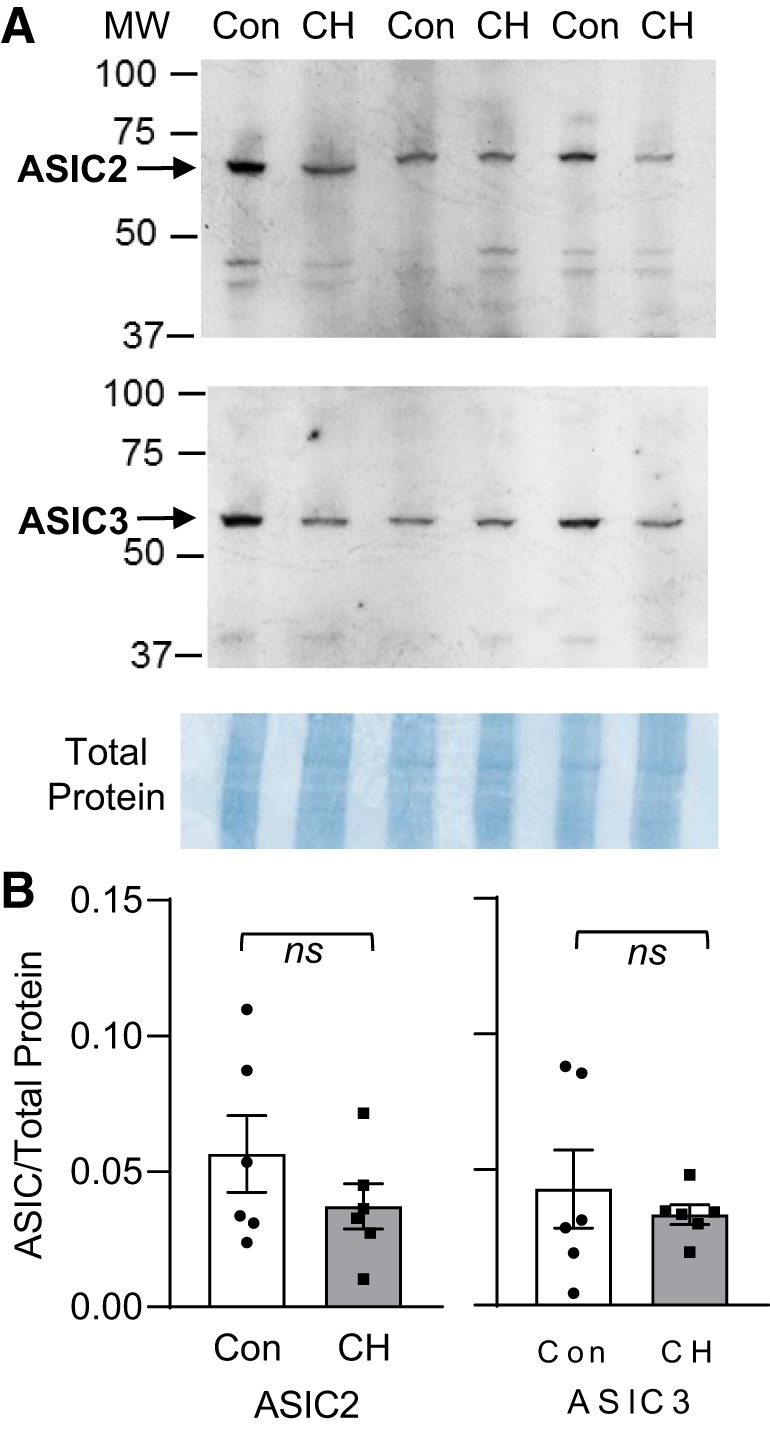
Chronic hypoxia (CH) does not significantly alter acid-sensing ion channel (ASIC)2 or ASIC3 protein levels in intrapulmonary arteries. A: representative Western blots of ASIC2 (predicted ∼69 kDa), ASIC3 (predicted ∼59 kDa), and corresponding Coomassie-stained blot. All samples were collected at the same time and processed in parallel on 2 gels for each antibody to accommodate 12 samples [6 control (Con) and 6 CH] from male wild-type (WT) mice. B: summary data detecting ASIC2/3 expression (normalized to entire lane on Coomassie-stained blot) in isolated intrapulmonary arteries from Con and CH WT mice. Values are means ± SE; dots indicate n = 6/group, analyzed by unpaired t-test.
Fig. 4.

Right ventricular systolic pressure (RVSP) is augmented in ASIC2−/− mice. Summary data for RVSP (A) and ratio of right ventricular (RV) to left ventricular plus septum (LV + S) heart weight (B) in anesthetized wild-type (WT), acid-sensing ion channel (ASIC)2−/−, and ASIC3−/− mice following exposure control or chronic hypoxia (CH) conditions. Values are means ± SE; n/group are indicated at the bottom of the bars as no. of males/females and as color-coded dots: black circle, control male; red circle, control female; black square, CH male; red square, CH female. There is no significant difference between males and females (P = 0.62). ****P < 0.0001 vs. respective control (Con) and ####P < 0.0001 vs. corresponding WT, analyzed by 2-way ANOVA and individual groups compared using Tukey’s multiple-comparisons test.
Table 3.
Mean BW (g), RVDP (mmHg), HR in beats/min, RV CI (1/s), and HCT (%red blood cells) in WT, ASIC2−/−, and ASIC3−/− mice exposed to Con or CH conditions
| WT |
ASIC2−/− |
ASIC3−/− |
||||
|---|---|---|---|---|---|---|
| Con (8/8) | CH (10/10) | Con (5/4) | CH (6/4) | Con (3/3) | CH (4/4) | |
| BW, g | 33 ± 1 | 28 ± 1 | 32 ± 3 | 30 ± 2 | 35 ± 4 | 27 ± 1 |
| RVDP, mmHg | −0.05 ± 0.36 | −0.33 ± 0.55 | 0.73 ± 0.43 | 0.15 ± 0.69 | −0.28 ± 0.42 | 0.21 ± 0.64 |
| HR, beats/min | 520 ± 7 | 486 ± 11 | 503 ± 10 | 505 ± 10 | 508 ± 20 | 491 ± 6 |
| RV CI, 1/s | 108 ± 7 | 87 ± 2* | 85 ± 6# | 87 ± 5 | 103 ± 10 | 88 ± 2* |
| HCT, % | 43.8 ± 0.5 | 58.2 ± 0.9* | 41.8 ± 0.9 | 60.6 ± 1.2* | 38.9 ± 1.3# | 52.4 ± 1.8*# |
Values are means ± SE; n indicated in parentheses next to group (males/females) and are the same animals as in Fig. 4. ASIC, acid-sensing ion channel; BW, body weight; CH, chronic hypoxia; Con, control; HCT, hematocrit; HR, heart rate; RV CI, right ventricular contractility index; RVDP, right ventricular diastolic pressure; WT, wild type.
P < 0.01 vs. control group;
P < 0.01 vs. corresponding WT mice.
RV hypertrophy was evident in all genotypes following exposure to CH, as indicated by a greater RV-to-(LV + S) ratio (Fig. 4B). Exposure to CH resulted in decreased RV contractility index in WT and ASIC3−/− CH mice, indicating RV dysfunction following CH consistent with development of cor pulmonale associated with pulmonary hypertension (Table 3). Interestingly, ASIC2−/− control mice exhibited a decreased RV contractility index compared with WT control mice. The contractility index in ASIC2−/− control mice was similar to values obtained from CH mice. Furthermore, exposure of ASIC2−/− mice to CH did not cause a further reduction in RV contractility index.
All mice exhibited polycythemia after CH, as indicated by a significantly greater hematocrit compared with control animals. However, the degree of polycythemia was significantly less in ASIC3−/− CH mice compared with WT CH mice (Table 3), similar to our previous observations in ASIC1−/− CH mice (36). Although still within normal range, hematocrit was also significantly lower in ASIC3−/− control mice compared with WT control-mice, suggesting ASIC3 may alter red blood cell homeostasis. In summary, these data suggest that loss of ASIC2, but not ASIC3, enhances CH-induced pulmonary hypertension.
Increased Pulmonary Arterial Muscularization in ASIC2−/− Mice
The degree of muscularization was greater in small pulmonary arteries (<100 µm outer diameter) from CH versus control mice in all three groups (Fig. 5). ASIC2−/− CH mice had significantly greater muscularization compared with WT or ASIC3−/− CH mice. The medial layer was also greater in small pulmonary arteries from ASIC2−/− control mice compared with WT controls, suggesting that alterations in pulmonary vascular function may exist, even though we did not observe differences in RVSP between WT and ASIC2−/− control mice (P = 0.9726; Fig. 4).
Fig. 5.
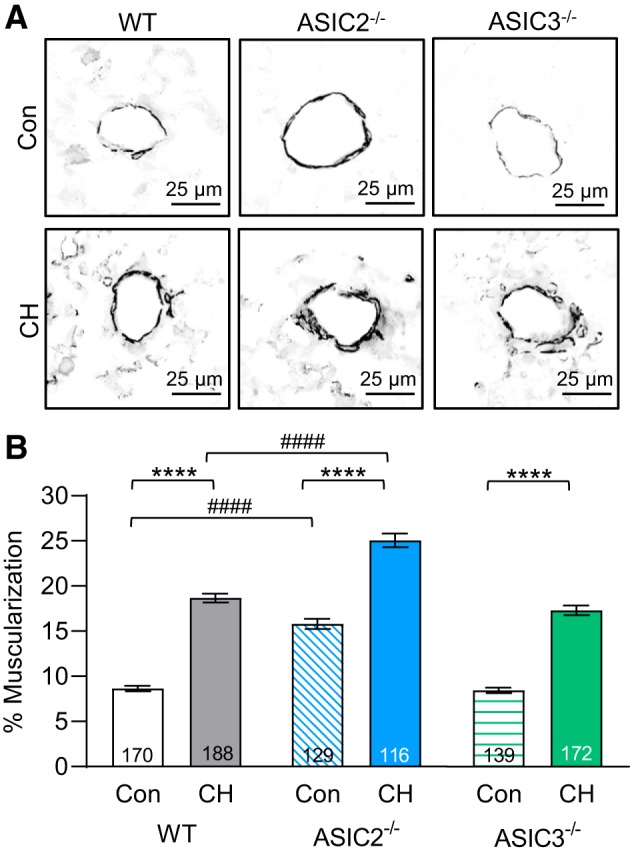
Small pulmonary arterial muscularization is greater in acid-sensing ion channel (ASIC)2−/− compared with wild-type (WT) mice. A: representative smooth muscle α-actin immunofluorescence images of lung sections from control (Con) and chronic hypoxia (CH) WT, ASIC2−/−, and ASIC3−/− mice. Fluorescence images were digitally inverted to provide better contrast and visibility of immunofluorescence. B: %muscularization calculated as %thresholded smooth muscle α-actin area divided by total arterial wall area. All vessels were <100 µm. Values are means ± SE; n = no. of vessels (indicated in bar) analyzed from 4 animals/group. ****P ≤ 0.0001 vs. the control group; ####P < 0.0001 vs. corresponding WT mice.
CH Augments Minute Ventilation in ASIC2−/− Mice
The involvement of ASICs in the peripheral chemoreceptor reflex may indirectly influence the development of hypoxic pulmonary hypertension. Our previous data demonstrate that baseline ventilatory responses in control ASIC2−/− and ASIC3−/− mice are similar to WT mice. However, ASIC2−/− control-mice have a blunted ventilatory response to isocapnic hypoxia (14). We used whole body plethysmography to determine whether loss of ASIC2 or ASIC3 alters ventilatory responses in CH-mice. Under normoxia (21% O2, 0% CO2), minute ventilation was greater in ASIC2−/− CH mice compared with WT CH mice (Fig. 6C), which results from an increase in tidal volume (Fig. 6B) but not respiratory rate (Fig. 6A). However, this increase in minute ventilation in ASIC2−/− mice did not correlate with altered arterial Po2, Pco2, or pH compared with WT (Fig. 6, D–F). Responses to hypocapnic hypoxia, isocapnic hypoxia, and hypercapnia were similar between WT and ASIC2−/− CH mice (Table 4), although there was a tendency for ASIC2−/− CH-mice to have a blunted ventilatory response to isocapnic hypoxia (P = 0.11), as previously observed in ASIC2−/− control-mice (14). Baseline normoxic respiratory rate was elevated in ASIC3−/− CH-mice (Fig. 6A), but this did not lead to greater minute ventilation (Fig. 6C). The increased respiratory rate in ASIC3−/− CH-mice was associated with a significant increase in pH (Fig. 6F) and tendency for lower arterial pCO2 (P = 0.24; Fig. 6E). This increase in pH persisted under hypocapnic and isocapnic hypoxic conditions (Table 4).
Fig. 6.

Effect of acid-sensing ion channel (ASIC) on baseline ventilation and blood gases following chronic hypoxia (CH). Whole body plethysmography was used to determine respiratory frequency (breaths/min; A), tidal volume (μl·breath−1·g body wt−1; B), and minute ventilation (ml·min−1·g body wt−1; C) in conscious wild-type (WT), ASIC2−/−, and ASIC3−/− CH mice breathing room air. Baseline arterial Po2 (mmHg; D), Pco2 (mmHg; E), and pH (F) in conscious WT, ASIC2−/−, and ASIC3−/− CH mice breathing room air. Values are means ± SE; n/group are indicated at the bottom of the bars as no. of males/females and as color-coded dots: black square, CH male; red square, CH female. There is no significant difference between males and females (P = 0.979). *P < 0.05 and **P < 0.01 vs. respective WT, analyzed by 1-way ANOVA and individual groups compared using Tukey’s multiple-comparisons test.
Table 4.
Blood gas measurements and ventilatory responses in response to hypoxia, isocapnic hypoxia, or hypercapnia in conscious, chronically catheterized WT, ASIC2−/−, and ASIC3−/− mice following 4 wk of CH
| Po2, mm Hg | Pco2, mm Hg | pH | Respiratory Rate, breaths/min | Tidal Volume, µl·breath−1·g−1 | Minute Ventilation, ml·min−1·g−1 | |
|---|---|---|---|---|---|---|
| Hypoxia (7% O2, 0% CO2) | ||||||
| CH WT | 30 ± 1# | 14 ± 1# | 7.40 ± 0.02# | 310 ± 6# | 11 ± 1# | 3.4 ± 0.2# |
| CH ASIC2−/− | 30 ± 2# | 14 ± 1# | 7.39 ± 0.01# | 302 ± 10# | 13 ± 1# | 3.9 ± 0.2# |
| CH ASIC3−/− | 31 ± 1# | 13 ± 1# | 7.47 ± 0.02#* | 321 ± 7# | 12 ± 1# | 3.7 ± 0.3# |
| Isocapnic Hypoxia (7% O2, 3.2% CO2) | ||||||
| CH WT | 37 ± 1# | 27 ± 1 | 7.26 ± 0.02 | 374 ± 6# | 20 ± 1# | 7.3 ± 0.4# |
| CH ASIC2−/− | 38 ± 1# | 30 ± 1 | 7.26 ± 0.02 | 349 ± 9# | 18 ± 1# | 6.4 ± 0.5# |
| CH ASIC3−/− | 38 ± 1# | 25 ± 1 | 7.32 ± 0.02* | 372 ± 6# | 19 ± 1# | 7.1 ± 0.5# |
| Hypercapnia (21% O2, 6% CO2) | ||||||
| CH WT | 103 ± 1# | 48 ± 1# | 7.13 ± 0.02# | 332 ± 7# | 18 ± 1# | 6.0 ± 0.5# |
| CH ASIC2−/− | 103 ± 2# | 52 ± 1#* | 7.14 ± 0.02# | 329 ± 13# | 18 ± 1# | 5.9 ± 0.5# |
| CH ASIC3−/− | 100 ± 3# | 46 ± 1# | 7.17 ± 0.02# | 333 ± 8# | 17 ± 1# | 5.7 ± 0.3# |
Enhanced Pulmonary Vasoconstrictor Reactivity in ASIC2−/− and ASIC3−/− Mice
Baseline pulmonary vascular resistance (PVR) during normoxic ventilation was higher in isolated saline-perfused lungs from ASIC2−/− control-mice compared with lungs from either WT or ASIC3−/− control-mice (Fig. 7A). Hypoxic ventilation increased PVR in lungs from all mice; however, the increase in PVR was significantly greater in lungs from ASIC2−/− and ASIC3−/− control mice (Fig. 7B). Increases in PVR to the depolarizing stimulus KCl were augmented in isolated lungs from ASIC2−/− control mice (Fig. 7C), and Emax responses to U-46619 were augmented in isolated lungs from both ASIC2−/− and ASIC3−/− control mice compared with WT (Fig. 7D). These data indicate that ASIC2 and ASIC3 act to attenuate pulmonary vasoconstrictor reactivity.
Fig. 7.

Enhanced pulmonary vasoconstrictor reactivity in control acid-sensing ion channel (ASIC)2−/− and ASIC3−/− mice. Baseline pulmonary vascular resistance (PVR; in mmHg·ml−1·min·kg−1; A) and changes (Δ) in PVR in response to hypoxic ventilation (6% CO2 and balance N2; B), KCl (30 mM; C), and U-46619 (10−9 – 10−6 M; D) in isolated lungs from control wild-type (WT), ASIC2−/−, and ASIC3−/− mice. All experiments were conducted in the presence of NG-nitro-l-arginine (300 μM) and meclofenamate (30 μM). Values are means ± SE; n/group indicated as color-coded dots: black circle, control male; red circle, control female. **P < 0.01 and ***P < 0.001 vs. corresponding WT, analyzed by 1-way ANOVA and individual groups compared using Tukey’s multiple comparisons test. NS, not significant.
PASMC from ASIC2−/− Mice Have Increased Basal [Ca2+]i and SOCE
To determine the mechanism by which ASIC2 and ASIC3 modulate vasoconstrictor reactivity, we assessed [Ca2+]i levels in PASMC. Basal [Ca2+]i levels were significantly higher in PASMC from ASIC2−/− than WT or ASIC3−/− control mice (Fig. 8A). Superfusion with Ca2+-free, HEPES-based PSS decreased the fura-2 ratio in all groups and normalized responses between genotypes, suggesting that the increase in [Ca2+]i levels is mediated by Ca2+ entry from the extracellular space. Treatment with the LTCC inhibitor diltiazem did not significantly alter basal [Ca2+]i levels in PASMC, suggesting that the increase in basal [Ca2+]i levels is not mediated by LTCC (Fig. 8B). Since we have shown previously ASIC1 contributes to Ca2+ influx in PASMC, we additionally examined the effect of the specific ASIC1a inhibitor PcTX1 on basal [Ca2+]i levels and SOCE. PcTX1 significantly reduced basal [Ca2+]i levels in PASMC from ASIC2−/− mice, normalizing responses to those of PASMC from WT mice (Fig. 8B). SOCE was augmented largely in PASMC from ASIC2−/− mice (Fig. 8C). Consistent with our previous data, the ASIC1 inhibitor PcTX1 reduced SOCE in PASMC from all groups and normalized responses between groups (23, 27, 28, 36, 37). These data suggest that loss of ASIC2 augments ASIC1-dependent increases in basal [Ca2+]i levels as well as SOCE in PASMC.
Fig. 8.
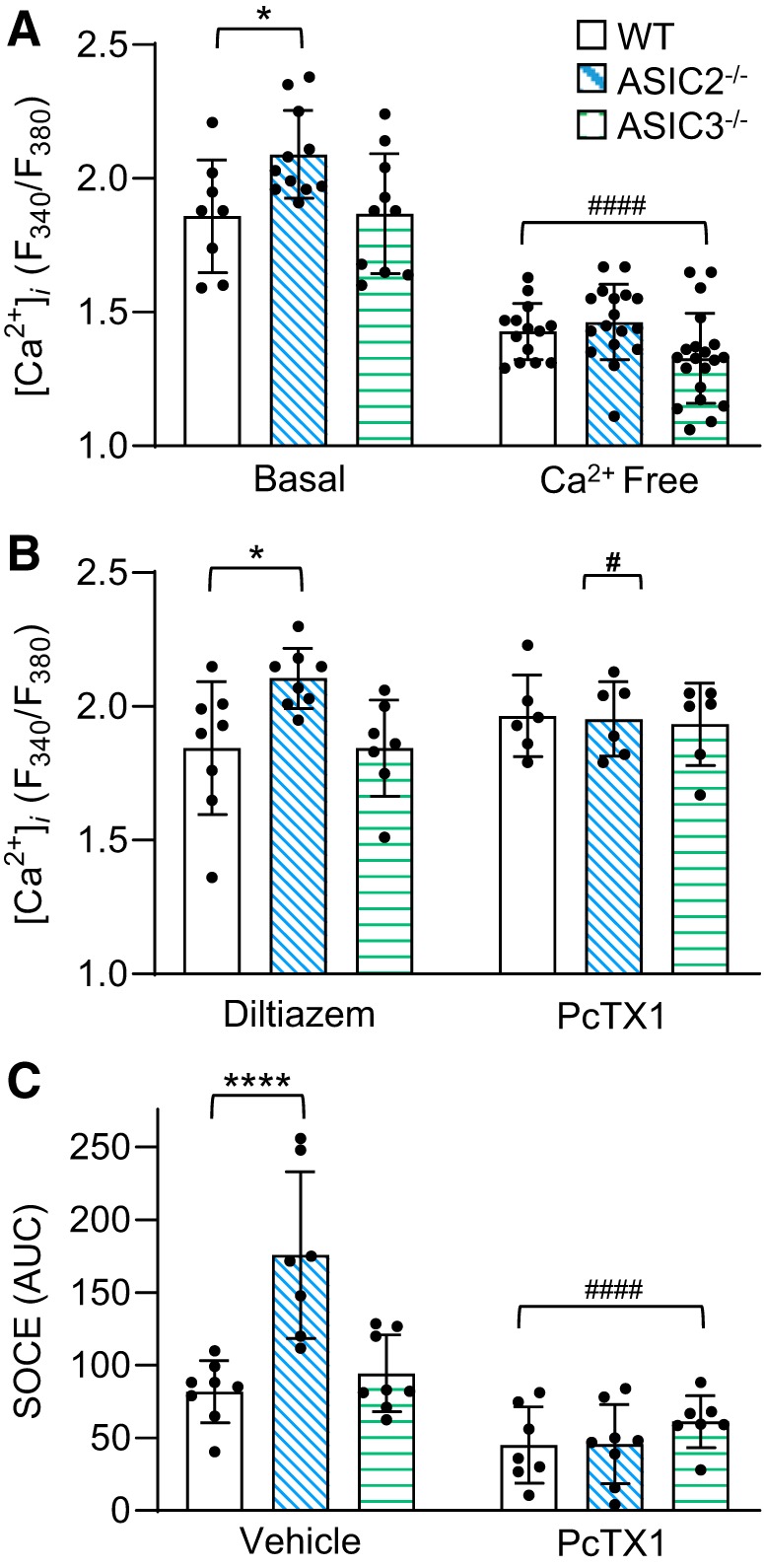
Augmented control acid-sensing ion channel (ASIC)1-dependent basal Ca2+ and store-operated calcium entry (SOCE) in pulmonary arterial smooth muscle cells (PASMC) from ASIC2−/− mice. A: summary data showing basal and Ca2+ free [Ca2+]i levels (fura2 340/380 ratio) in PASMC from control wild-type (WT), ASIC2−/−, or ASIC3−/− male mice. B: [Ca2+]i levels (fura2 340/380 ratio) following treatment with diltiazem (50 μM) or PcTX1 (20 nM). C: SOCE responses performed in the presence of cyclopiazonic acid (CPA; 10 μM), diltiazem (50 μM), and ± PcTX1. Values are means ± SE; ●, n of 6–10/group. *P < 0.05 and ****P < 0.0001 vs. WT; #P < 0.05 and ####P < 0.0001 vs. corresponding baseline/vehicle, analyzed by 2-way ANOVA and individual groups compared using Tukey’s multiple comparisons test.
Enhanced Vasoconstriction in Small Pulmonary Arteries from ASIC2−/− Mice is ASIC1 Dependent
We utilized isolated, pressurized, small pulmonary arteries to further determine the role of ASIC1 in the augmented vasoreactivity and [Ca2+]i levels we observed in the ASIC2−/− mice. Both basal tone (Fig. 9A) and [Ca2+]i levels (Fig. 9B) were significantly greater in small pulmonary arteries from ASIC2−/− compared with WT control mice. There was no significant difference between groups when comparing passive (Ca2+ free) inner diameters (WT: 157 ± 8 µm; ASIC2−/−: 154 ± 5 µm; P = 0.78) or [Ca2+]i levels during superfusion with Ca2+ free HEPES-based PSS (fura-2 ratios for WT: 0.36 ± 0.04; ASIC2−/−: 0.37 ± 0.05; P = 0.78). These data suggest that influx of Ca2+ via ASIC1 leads to greater basal tone and [Ca2+]i levels in small pulmonary arteries. Although vasoconstriction in response to KCl was not significantly different in small pulmonary arteries from ASIC2−/− compared with WT control mice (Fig. 9C), the KCl-induced change in vessel wall [Ca2+]i levels were significantly greater (Fig. 9D). Furthermore, PcTX1 significantly decreased both KCl-induced vasoconstriction and changes in vessel wall [Ca2+]i only in small pulmonary arteries from ASIC2−/− mice (Fig. 9C–D). The lack of effect of PcTX1 in small pulmonary arteries from WT mice is consistent with our previous findings in rats and WT/ASIC1−/− mice, where inhibition of ASIC1 (either pharmacologically with PcTX1 or genetic knockout) had no effect on KCl-induced vasoreactivity (27, 36). The present finding suggests a novel role for ASIC1 in depolarization-induced pulmonary vasoconstriction when there is a loss of ASIC2.
Fig. 9.
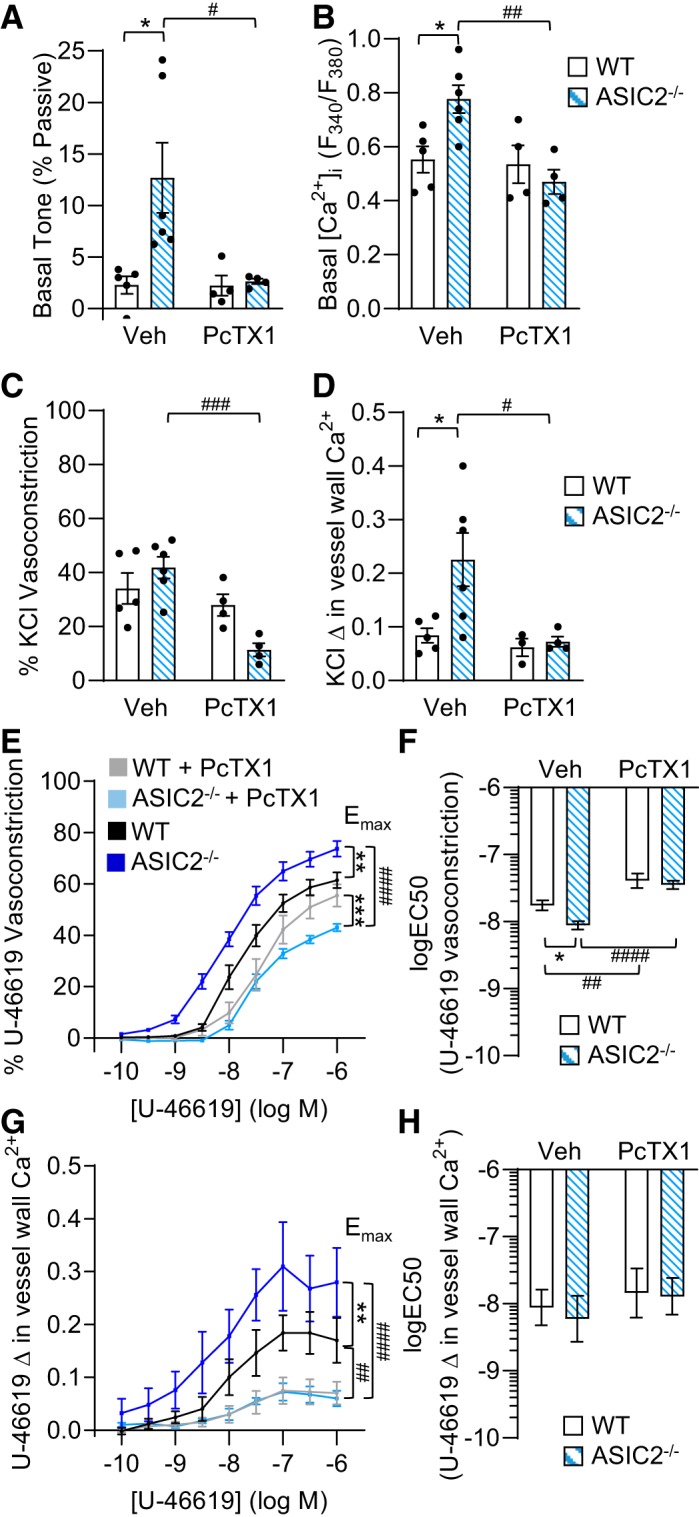
Enhanced pulmonary vasoconstrictor reactivity in control acid-sensing ion channel (ASIC)2−/− mice is mediated by ASIC1. A and B: basal arterial tone (%Ca2+ free diameter; A) and basal intracellular Ca2+ concentration ([Ca2+]i; B) expressed as vessel wall fura-2 ratio (F340/F380) in isolated, pressurized, small pulmonary arteries from control wild-type (WT) and ASIC2−/− mice in the presence or absence of psalmotoxin 1 (PcTX1; 20 nM). C and D: %vasoconstriction (C) and changes (Δ) in vessel wall Ca2+ (Δ in fura-2 ratio) (D) in response to KCl (30 mM). E–H: %vasoconstriction (E) and change in vessel wall Ca2+ (G) to U-46619 (10−10 to 10−6 M). U-46619 dose response curves were analyzed by evaluating maximum effect (Emax; E and G) and logEC50 (F and H). Values are means ± SE; ●, n/group. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. corresponding WT; #P < 0.05, ##P < 0.01, and ###P < 0.001, and ####P < 0.0001 vs. corresponding vehicle (Veh) treated, analyzed by 2-way ANOVA and individual groups compared using Tukey’s multiple comparisons test.
Similar to our findings in isolated lungs (Fig. 7), we found that vasoconstriction (Emax) and changes in vessel wall [Ca2+]i to U-46619 in small pulmonary arteries from ASIC2−/− control mice were significantly greater compared with those in WT mice. Consistently, the logEC50 for vasoconstriction to U-46619 was significantly shifted to the left. Similar to our previous data with other agonists (27, 36), we found that PcTX1 significantly attenuated vasoconstriction and changes in vessel wall [Ca2+]i levels in small pulmonary arteries from both WT and ASIC2−/− control mice. In small pulmonary arteries from WT mice, PcTX1 did not affect vasoconstrictor Emax but significantly blunted logEC50. PcTX1 had a greater effect in small pulmonary arteries from ASIC2−/− than from WT mice to decrease both vasoconstrictor Emax and logEC50. This was specified by a significant interaction (2-way ANOVA analysis), indicating that the effect of PcTX1 was dependent on genotype. PcTX1 largely blunted Emax but not logEC50 values for changes in vessel wall [Ca2+]i levels in small pulmonary arteries from both groups, thereby normalizing responses between groups.
Knockdown of ASIC2 and ASIC3 Does Not Significantly Alter ASIC1 Expression in PASMC
Because of the enhanced ASIC1-dependent Ca2+ responses in PASMC from ASIC2−/− and ASIC3−/−, we investigated whether knockout of ASIC2 or ASIC3 altered ASIC1 expression (Fig. 10). We did not detect significant differences in mRNA expression in PASMC between WT and ASIC2−/− (P = 0.29) or ASIC3−/− mice (P = 0.10; Fig. 10A) or protein expression in PASMC between WT and ASIC2−/− (P = 0.62) and ASIC3−/− mice (P = 0.64; Fig. 10, B–C).
Fig. 10.
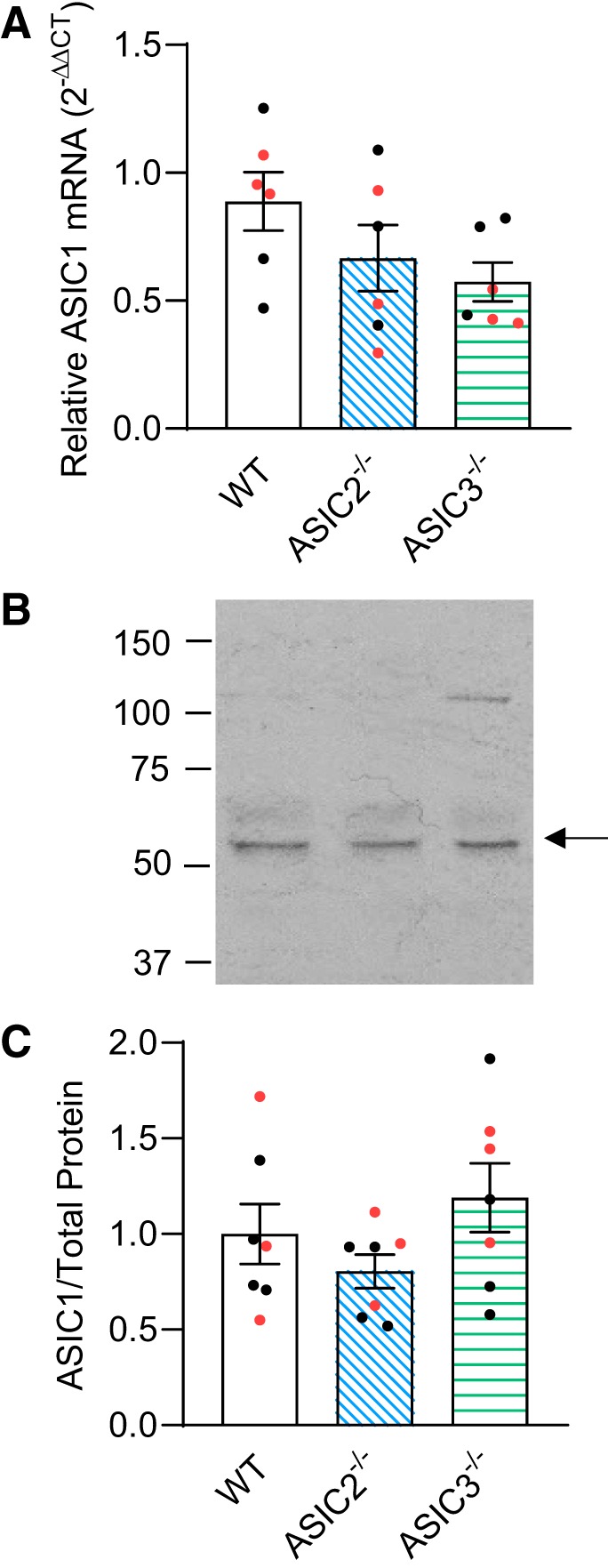
Effect of acid-sensing ion channel (ASIC)2−/− and ASIC3−/− on ASIC1 expression. A: summary data showing mRNA analysis by real-time quantitative PCR for ASIC1 in primary pulmonary arterial smooth muscle cells (PASMC) from control wild-type (WT), ASIC2−/−, and ASIC3−/− mice. ASIC1 expression was normalized to 18S rRNA as an internal control (). B and C: representative Western blots for ASIC1 (indicated by arrow; B) and summary data for protein analysis of ASIC1 (normalized to entire lane on Coomassie-stained blot; C) in primary PASMC from WT, ASIC2−/−, and ASIC3−/− mice. Values are means ± SE. Nos. of animals/group (6, 7) are indicated as color-coded dots: black circle, control male; red circle, control female, analyzed by 1-way ANOVA.
DISCUSSION
Our laboratory has previously shown that ASIC1 mediates an important component of acute hypoxic pulmonary vasoconstriction as well as the enhanced agonist-induced vasoconstrictor responsiveness associated with pulmonary hypertension (36). ASIC2 and ASIC3 are also expressed in PASMC, and the goal of this study was to determine the contribution of ASIC2 and ASIC3 to pulmonary vascular reactivity and development CH-induced pulmonary hypertension. Contrary to our hypothesis, the major findings from this study are that ASIC2−/− mice exhibit enhanced CH-induced pulmonary hypertension compared with WT. This response was not associated with differences in neuronal regulation of cardiovascular function but was associated with direct changes in PVR. Isolated saline-perfused lungs from control ASIC2−/− mice exhibited greater baseline PVR and enhanced vasoconstrictor reactivity to acute hypoxia, KCl, and U-46619. This increase in reactivity was associated with enhanced pulmonary arterial basal tone, elevated basal PASMC [Ca2+]i, and SOCE in ASIC2−/− mice. Interestingly, this increase in basal [Ca2+]i was not dependent on LTCC. Rather, inhibition of ASIC1 eliminated the elevation of basal [Ca2+]i and enhancement of SOCE in PASMC from ASIC2−/− mice. Although these data suggest greater activity of ASIC1, it was not contingent upon an increase in ASIC1 mRNA or protein expression in PASMC from ASIC2−/− mice. Together, the results from this study demonstrate an important role for ASIC2 to regulate pulmonary vascular reactivity and modulate the development of CH-induced pulmonary hypertension. These data further suggest that loss of ASIC2 enhances the contribution of ASIC1 to overall pulmonary vascular reactivity.
The identification of ASIC1, ASIC2, and ASIC3 in PASMC, along with our recent findings characterizing ASIC1 as a regulator of PASMC Ca2+ influx, has provided novel insight into the potential contribution of the ASIC channel family to normal vascular processes and to the pathophysiology of CH-induced pulmonary hypertension. Our initial studies demonstrate that ASIC1-mediated Ca2+ influx is activated through a store-operated mechanism and contributes to pulmonary vasoconstriction elicited by various agonists and alveolar hypoxia (27, 28, 36). Further studies in ASIC1-null mice indicate that global loss of ASIC1 results in largely diminished changes in RVSP and prevents RV hypertrophy and pulmonary arterial remodeling (36). Together, these data provide strong evidence that ASIC1 contributes to the active vasoconstriction, arterial muscularization, and right ventricular hypertrophy associated with the development of CH-induced pulmonary hypertension.
Whereas ASIC1 conducts both Na+ and Ca2+, ASIC2 and ASIC3 are characterized as more Na+-selective channels, and their activation would be expected to induce membrane depolarization and subsequent activation of LTCC in PASMC. In acutely cultured primary PASMC, we found that siRNA knockdown of ASIC1, but not ASIC2 or ASIC3, significantly diminished the activation of whole cell Ca2+ current in response to store depletion (28). We additionally examined the role of ASICs in whole cell Na+ current in response to store depletion. Targeted siRNA knockdown of ASIC1, and to a lesser degree ASIC3, inhibited Na+ current induced by store depletion. Whereas siRNA knockdown of ASIC2 tended to increase Na+ current, statistically significant differences were not detected (P = 0.09 (28). Taking into account that it is unlikely that we were achieving complete knockdown of ASIC protein with siRNA, these data suggest ASIC2 and ASIC3 may contribute to Na+ influx in PASMC. Because there are no specific pharmacological inhibitors of ASIC2 and ASIC3, we used ASIC2−/− and ASIC3−/− mice to determine the role of these channels in pulmonary vasoreactivity and development of pulmonary hypertension.
RVSP was similar in control mice of all genotypes. However, in contrast to our hypothesis and findings from ASIC1−/− mice (36), ASIC2−/− mice exhibited greater RVSP compared with both WT and ASIC3−/− following exposure to CH. Because these are global knockout animals, we first considered the possibility that ASIC2 regulates pulmonary vascular reactivity through other integrated nonvascular mechanisms. For example, ASIC2 has been shown to mediate the mechanosensitivity of aortic baroreceptors and ASIC3 the pH sensitivity of carotid body chemoreceptors (1).
ASIC2−/− mice have been reported to have elevated systemic MABP and heart rate that is associated with impaired baroreceptor reflex, leading to exaggerated sympathetic and depressed parasympathetic control of the circulatory system (33). Although the innervation of pulmonary vessels is relatively sparse compared with systemic vessels, increases in heart rate and cardiac output could potentially lead to greater RVSP. However, the reported effect of ASIC2−/− on systemic MABP regulation has been variable, as Gannon et al. (18) demonstrated no difference in MABP or heart rate between WT and ASIC2−/− mice. In both studies, measurements were made in conscious animals by radiotelemetry. In agreement with the latter reports, we did not detect significant cardiovascular changes under normal conditions in either ASIC2−/− or ASIC3−/− conscious mice. Furthermore, the similar heart rate and RV contractility between CH groups suggest that greater RSVP in ASIC2−/− CH mice is not likely a function of higher cardiac output.
Previously, we examined ventilatory responses in conscious, unrestrained control WT, ASIC1−/−, ASIC2−/−, and ASIC3−/− mice using whole body plethysmography (14). Respiratory frequency, tidal volume, and minute ventilation were similar in all groups under baseline conditions and in response to hypercapnia. However, minute ventilation in ASIC2−/− mice during isocapnic hypoxia was significantly lower compared with WT, suggesting that ASIC2−/− mice may have a blunted hypoxic ventilatory response (14). If ASIC2−/− mice had a similar decreased ventilatory response to CH, this could lead to greater hypoxemia and a more severe pulmonary hypertensive response. However, we observed that ASIC2−/− CH mice have a slightly enhanced minute ventilation, which appeared to be largely a function of greater tidal volume. These data indicate that the effect of ASIC2−/− to increase the severity of pulmonary hypertension does not appear to be explained by more severe hypoxemia. Furthermore, there were no differences in , , or pH. Together, these findings lead us to investigate local regulatory mechanisms within the lung.
The isolated lung preparation allows the study of pulmonary hemodynamic parameters in a controlled ventilatory setting and in the absence of other integrative mechanisms (both neural and cardiac). In this setting, baseline PVR was elevated in lungs from ASIC2−/− mice. In addition, the change in PVR in response to various stimuli, including hypoxia and U-46619, was significantly increased in lungs from both ASIC2−/− and ASIC3−/− mice compared with WT, suggesting that ASIC2 and ASIC3 normally act to inhibit pulmonary vasoconstrictor reactivity. In the pulmonary vasculature, the increased PVR could be attributed to loss of endothelial function, PASMC depolarization leading to LTCC activation, Ca2+ influx through other mechanisms (i.e., SOCE), or Ca2+ sensitization of the myofilament contractile apparatus, all of which are mechanisms known to contribute to pulmonary hypertension (4, 8, 10, 29, 31, 34, 35, 44–46, 48, 53, 57).
Further investigation into Ca2+ handling in PASMC revealed that basal [Ca2+]i levels in PASMC from control ASIC2−/− mice, but not ASIC3−/− mice, are elevated compared with WT mice. The lack of effect of the LTCC inhibitor (diltiazem) to reduce [Ca2+]i levels indicates that this increase in basal [Ca2+]i is not mediated by LTCC, even though the increase in [Ca2+]i was via an extracellular source. Remarkably, the coupling of SOCE to smooth muscle contraction appears to be of particular importance in pulmonary arteries over other vascular beds (25, 47). Indeed, similar to basal [Ca2+]i levels, we also found that SOCE was largely augmented in PASMC from ASIC2−/− mice. Consistent with our previous studies (22, 23, 25, 27, 28), this enhanced SOCE response is mediated largely by ASIC1. In addition, pretreatment with PcTX1 abolished the elevation in basal [Ca2+]i levels in PASMC from ASIC2−/− mice. Together, these data suggest ASIC2 negatively regulates ASIC1-mediated Ca2+ homeostasis in PASMC.
To determine whether greater PASMC [Ca2+]i in ASIC2−/− mice correlates with a functional increase in ASIC1-dependent vasoreactivity, we conducted additional studies using isolated, pressurized, small pulmonary arteries. In previous studies, we found that basal vessel wall [Ca2+]i is not altered in small pulmonary arteries from ASIC1−/− mice (36) or by treatment with PcTX1 in rat small pulmonary arteries (27). In the present study, we similarly found that PcTX1 did not alter basal tone or vessel wall [Ca2+]i levels in WT mice; however, in small pulmonary arteries from ASIC2−/− mice, the increases in basal vascular tone and vessel wall [Ca2+]i were abolished by PcTX1 and normalized to WT levels. These data suggest that loss of ASIC2 permits basal activity of ASIC1. Although we have found that inhibition of ASIC1 (either genetically or pharmacologically) diminishes vasoreactivity to a variety of agonists (27, 36), the effect of ASIC1 inhibition is always greater following CH-induced pulmonary hypertension than in control animals. In this study, we also observed a significantly greater effect of PcTX1 to inhibit U-46619-induced responses in arteries from ASIC2−/− than WT mice. Also similar to previous studies (27, 36), KCl-induced constriction and changes in vessel wall [Ca2+]i were unaffected by PcTX1 in arteries from WT mice. However, because ASICs are considered to be voltage insensitive (30), they would not be expected to respond to depolarizing stimuli (i.e., KCl). Interestingly, in arteries from ASIC2−/− mice, KCl-induced vasoconstriction and changes in vessel wall [Ca2+]i were sensitive to ASIC1 inhibition. Although the mechanism for this is unclear, it suggests that ASIC1 may contribute to depolarization-induced constriction following loss of ASIC2.
Although these data indicate greater basal and stimulated ASIC1 activity, they did not correspond with a compensatory increase in ASIC1 mRNA or protein expression in PASMC from ASIC2−/− mice. Similarly, we have shown that ASIC1 activity, but not expression, is increased following CH (23, 36). This increase in ASIC1 activity in pulmonary hypertensive animals corresponds with greater plasma membrane localization of ASIC1 in PASMC (23). Although we could speculate that ASIC2 levels are decreased following CH, leading to greater ASIC1 activity, we did not observe a significant change in ASIC2 expression following CH in isolated intrapulmonary arteries (Fig. 3). However, there is a difference between the enhanced ASIC1 activity associated with CH and that in PASMC from ASIC2−/− mice. The elevated basal [Ca2+]i in PASMC from pulmonary hypertensive animals is independent of ASIC1 (27, 36), suggesting that under these conditions ASIC1 is not constitutively active. In contrast, the increase in basal [Ca2+]i in PASMC from ASIC2−/− mice is ASIC1 dependent and implies that ASIC2 modulates ASIC1 function, which has been shown by a number of other investigators.
ASIC2a and ASIC2b interact with ASIC1a and can shift the pH sensitivity, desensitization kinetics, and ion selectivity of acid-evoked currents. ASIC1a/ASIC2a heterotrimers are less proton sensitive, desensitize faster, and are less Ca2+ permeable than ASIC1a homotrimers (2, 5–7, 24). Therefore, loss of ASIC2 expression can affect ASIC currents in PASMC by regulating the relative ratio of ASIC1a/ASIC2a heterotrimers to ASIC1a homotrimers. This may explain why there is a greater ASIC1-mediated Ca2+ component in ASIC2−/− PASMC. In aggressive brain tumors, the high-grade astrocytoma cells express only ASIC1, whereas normal cells express both ASIC1 and ASIC2, suggesting that ASIC2 may function as a suppressor of ASIC1 current (7). In vascular smooth muscle cells where ASIC mediates migration, specific knockdown of ASIC2 using siRNA conversely leads to increased chemotactic migration (20), suggesting that ASIC2 modulates the role of other ASIC subunits.
In the pulmonary vasculature, loss of ASIC2 enables some degree of basal ASIC1 activity that is not normally present. Under control conditions, the increase in PASMC and pulmonary arterial vessel wall [Ca2+]i levels correlates with increased small pulmonary arterial basal tone (Fig. 9), pulmonary vascular resistance (Fig. 7), and medial thickening of small pulmonary arteries (Fig. 5). Although this was also reflected by an increase in RVSP in ASIC2−/− CH mice, RVSP was not different between WT and ASIC2−/− control mice. These data imply that although loss of ASIC2 has a significant effect on pulmonary vascular function, in a more integrated setting, pulmonary pressure is influenced by cardiac function. Although heart rate was not different between groups, RV contractility in control ASIC2−/− mice was decreased compared with WT, which could potentially offset the effect of increased pulmonary vascular resistance observed in ASIC2−/− mice on RVSP. Although loss of ASIC2 modulates ASIC1-mediated responses in the pulmonary vasculature under control conditions, the effect is not sufficient to induce pulmonary hypertension by itself. Further investigation of aged animals could potentially help address this question.
In summary, we found that loss of ASIC2 enhanced baseline pulmonary vascular resistance and increased vasoconstrictor activity to a variety of stimuli. Furthermore, ASIC2−/− mice developed a greater degree of pulmonary hypertension. This response correlated with increased ASIC1 activity in the pulmonary vasculature and suggests that the function of ASIC2 in PASMC is to fine-tune the native ASIC current. Therefore, loss of ASIC2 may shift the properties of ASICs so that more homotrimeric ASIC1 is formed with greater Ca2+ permeability, leading to a greater role of ASIC1 in normal pulmonary vascular reactivity and development of pulmonary hypertension. However, future studies are needed to address this possibility.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-111084 and American Heart Association Grant 18TPA34110281 to N. L. Jernigan. Trainee support of this work was supported by National Institutes of Health Grants T32-HL-007736 (to T. C. Resta), K12-GM-088021 (to A. Wandinger-Ness), F31-HL-145836 (to S.M. Garcia), and R25-GM-060201 (to M. Werner-Washburne).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.D.D., T.C.R., B.R.W., and N.L.J. conceived and designed research; N.D.D., L.M.H., S.M.G., S.Y., K.G.V., J.R.S., and N.L.J. performed experiments; N.D.D., L.M.H., S.M.G., S.Y., K.G.V., J.R.S., and N.L.J. analyzed data; N.D.D., L.M.H., S.M.G., T.C.R., B.R.W., and N.L.J. interpreted results of experiments; N.D.D., L.M.H., S.M.G., S.Y., and N.L.J. prepared figures; N.L.J. drafted manuscript; N.D.D., L.M.H., S.M.G., S.Y., T.C.R., B.R.W., and N.L.J. edited and revised manuscript; N.D.D., L.M.H., S.M.G., S.Y., K.G.V., J.R.S., T.C.R., B.R.W., and N.L.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Anthony Gravagne (Department of Physics and Astronomy, University of New Mexico, Albuquerque, NM) for assistance with the design and fabrication of the plethysmography chamber and Dr. Tamara Howard (University of New Mexico Health Sciences Center, Albuquerque, NM) for assistance with the preparation of lung sections for immunofluorescence.
REFERENCES
- 1.Abboud FM, Benson CJ. ASICs and cardiovascular homeostasis. Neuropharmacology 94: 87–98, 2015. doi: 10.1016/j.neuropharm.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem 279: 18296–18305, 2004. doi: 10.1074/jbc.M312145200. [DOI] [PubMed] [Google Scholar]
- 3.Babinski K, Catarsi S, Biagini G, Séguéla P. Mammalian ASIC2a and ASIC3 subunits co-assemble into heteromeric proton-gated channels sensitive to Gd3+. J Biol Chem 275: 28519–28525, 2000. doi: 10.1074/jbc.M004114200. [DOI] [PubMed] [Google Scholar]
- 4.Barman SA. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muscle involves Rho-kinase and protein kinase C. Am J Physiol Lung Cell Mol Physiol 293: L472–L479, 2007. doi: 10.1152/ajplung.00101.2006. [DOI] [PubMed] [Google Scholar]
- 5.Bartoi T, Augustinowski K, Polleichtner G, Gründer S, Ulbrich MH. Acid-sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci USA 111: 8281–8286, 2014. doi: 10.1073/pnas.1324060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, Snyder PM. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc Natl Acad Sci USA 99: 2338–2343, 2002. [Erratum in: Proc Natl Acad Sci USA 99: 4752, 2002.] 10.1073/pnas.032678399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berdiev BK, Xia J, McLean LA, Markert JM, Gillespie GY, Mapstone TB, Naren AP, Jovov B, Bubien JK, Ji HL, Fuller CM, Kirk KL, Benos DJ. Acid-sensing ion channels in malignant gliomas. J Biol Chem 278: 15023–15034, 2003. doi: 10.1074/jbc.M300991200. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet S, Belus A, Hyvelin J-M, Roux E, Marthan R, Savineau J-P. Effect of chronic hypoxia on agonist-induced tone and calcium signaling in rat pulmonary artery. Am J Physiol Lung Cell Mol Physiol 281: L193–L201, 2001. doi: 10.1152/ajplung.2001.281.1.L193. [DOI] [PubMed] [Google Scholar]
- 9.Boscardin E, Alijevic O, Hummler E, Frateschi S, Kellenberger S. The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR Review 19. Br J Pharmacol 173: 2671–2701, 2016. doi: 10.1111/bph.13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broughton BRS, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 11.Chen C-C, England S, Akopian AN, Wood JN. A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci USA 95: 10240–10245, 1998. doi: 10.1073/pnas.95.17.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung W-S, Farley JM, Swenson A, Barnard JM, Hamilton G, Chiposi R, Drummond HA. Extracellular acidosis activates ASIC-like channels in freshly isolated cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 298: C1198–C1208, 2010. doi: 10.1152/ajpcell.00511.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Weille JR, Bassilana F, Lazdunski M, Waldmann R. Identification, functional expression and chromosomal localisation of a sustained human proton-gated cation channel. FEBS Lett 433: 257–260, 1998. doi: 10.1016/S0014-5793(98)00916-8. [DOI] [PubMed] [Google Scholar]
- 14.Detweiler ND, Vigil KG, Resta TC, Walker BR, Jernigan NL. Role of acid-sensing ion channels in hypoxia- and hypercapnia-induced ventilatory responses. PLoS One 13: e0192724, 2018. doi: 10.1371/journal.pone.0192724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deval E, Baron A, Lingueglia E, Mazarguil H, Zajac JM, Lazdunski M. Effects of neuropeptide SF and related peptides on acid sensing ion channel 3 and sensory neuron excitability. Neuropharmacology 44: 662–671, 2003. doi: 10.1016/S0028-3908(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 16.Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics 16: 81–87, 1955. [PubMed] [Google Scholar]
- 17.Drummond HA, Xiang L, Chade AR, Hester R. Enhanced maximal exercise capacity, vasodilation to electrical muscle contraction, and hind limb vascular density in ASIC1a null mice. Physiol Rep 5: e13368, 2017. doi: 10.14814/phy2.13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gannon KP, McKey SE, Stec DE, Drummond HA. Altered myogenic vasoconstriction and regulation of whole kidney blood flow in the ASIC2 knockout mouse. Am J Physiol Renal Physiol 308: F339–F348, 2015. doi: 10.1152/ajprenal.00572.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez Bosc LV, Plomaritas DR, Herbert LM, Giermakowska W, Browning C, Jernigan NL. ASIC1-mediated calcium entry stimulates NFATc3 nuclear translocation via PICK1 coupling in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 311: L48–L58, 2016. doi: 10.1152/ajplung.00040.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grifoni SC, Jernigan NL, Hamilton G, Drummond HA. ASIC proteins regulate smooth muscle cell migration. Microvasc Res 75: 202–210, 2008. doi: 10.1016/j.mvr.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gründer S, Geissler HS, Bässler EL, Ruppersberg JP. A new member of acid-sensing ion channels from pituitary gland. Neuroreport 11: 1607–1611, 2000. doi: 10.1097/00001756-200006050-00003. [DOI] [PubMed] [Google Scholar]
- 22.Herbert LM, Nitta CH, Yellowhair TR, Browning C, Gonzalez Bosc LV, Resta TC, Jernigan NL. PICK1/calcineurin suppress ASIC1-mediated Ca2+ entry in rat pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 310: C390–C400, 2016. doi: 10.1152/ajpcell.00091.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbert LM, Resta TC, Jernigan NL. RhoA increases ASIC1a plasma membrane localization and calcium influx in pulmonary arterial smooth muscle cells following chronic hypoxia. Am J Physiol Cell Physiol 314: C166–C176, 2018. doi: 10.1152/ajpcell.00159.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hesselager M, Timmermann DB, Ahring PK. pH Dependency and desensitization kinetics of heterologously expressed combinations of acid-sensing ion channel subunits. J Biol Chem 279: 11006–11015, 2004. doi: 10.1074/jbc.M313507200. [DOI] [PubMed] [Google Scholar]
- 25.Jernigan NL. Smooth muscle acid-sensing ion channel 1: pathophysiological implication in hypoxic pulmonary hypertension. Exp Physiol 100: 111–120, 2015. doi: 10.1113/expphysiol.2014.081612. [DOI] [PubMed] [Google Scholar]
- 26.Jernigan NL, Broughton BR, Walker BR, Resta TC. Impaired NO-dependent inhibition of store- and receptor-operated calcium entry in pulmonary vascular smooth muscle after chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 290: L517–L525, 2006. doi: 10.1152/ajplung.00308.2004. [DOI] [PubMed] [Google Scholar]
- 27.Jernigan NL, Herbert LM, Walker BR, Resta TC. Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol 302: C931–C940, 2012. doi: 10.1152/ajpcell.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jernigan NL, Paffett ML, Walker BR, Resta TC. ASIC1 contributes to pulmonary vascular smooth muscle store-operated Ca(2+) entry. Am J Physiol Lung Cell Mol Physiol 297: L271–L285, 2009. doi: 10.1152/ajplung.00020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82: 735–767, 2002. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 31.Lin M-J, Leung GPH, Zhang W-M, Yang X-R, Yip K-P, Tse C-M, Sham JSK. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 32.Lingueglia E, de Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, Lazdunski M. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem 272: 29778–29783, 1997. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Ma X, Sabharwal R, Snitsarev V, Morgan D, Rahmouni K, Drummond HA, Whiteis CA, Costa V, Price M, Benson C, Welsh MJ, Chapleau MW, Abboud FM. The ion channel ASIC2 is required for baroreceptor and autonomic control of the circulation. Neuron 64: 885–897, 2009. doi: 10.1016/j.neuron.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 35.Naik JS, Earley S, Resta TC, Walker BR. Pressure-induced smooth muscle cell depolarization in pulmonary arteries from control and chronically hypoxic rats does not cause myogenic vasoconstriction. J Appl Physiol (1985) 98: 1119–1124, 2005. doi: 10.1152/japplphysiol.00819.2004. [DOI] [PubMed] [Google Scholar]
- 36.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014. doi: 10.1152/ajpheart.00269.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plomaritas DR, Herbert LM, Yellowhair TR, Resta TC, Gonzalez Bosc LV, Walker BR, Jernigan NL. Chronic hypoxia limits H2O2-induced inhibition of ASIC1-dependent store-operated calcium entry in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 307: L419–L430, 2014. doi: 10.1152/ajplung.00095.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poirot O, Berta T, Decosterd I, Kellenberger S. Distinct ASIC currents are expressed in rat putative nociceptors and are modulated by nerve injury. J Physiol 576: 215–234, 2006. doi: 10.1113/jphysiol.2006.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price MP, Lewin GR, McIlwrath SL, Cheng C, Xie J, Heppenstall PA, Stucky CL, Mannsfeldt AG, Brennan TJ, Drummond HA, Qiao J, Benson CJ, Tarr DE, Hrstka RF, Yang B, Williamson RA, Welsh MJ. The mammalian sodium channel BNC1 is required for normal touch sensation. Nature 407: 1007–1011, 2000. doi: 10.1038/35039512. [DOI] [PubMed] [Google Scholar]
- 40.Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32: 1071–1083, 2001. doi: 10.1016/S0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 41.Price MP, Snyder PM, Welsh MJ. Cloning and expression of a novel human brain Na+ channel. J Biol Chem 271: 7879–7882, 1996. doi: 10.1074/jbc.271.14.7879. [DOI] [PubMed] [Google Scholar]
- 42.Schaefer L, Sakai H, Mattei M, Lazdunski M, Lingueglia E. Molecular cloning, functional expression and chromosomal localization of an amiloride-sensitive Na(+) channel from human small intestine. FEBS Lett 471: 205–210, 2000. doi: 10.1016/S0014-5793(00)01403-4. [DOI] [PubMed] [Google Scholar]
- 43.Sherwood TW, Lee KG, Gormley MG, Askwith CC. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J Neurosci 31: 9723–9734, 2011. doi: 10.1523/JNEUROSCI.1665-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 279: L884–L894, 2000. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- 45.Shimoda LA, Sylvester JT, Sham JS. Chronic hypoxia alters effects of endothelin and angiotensin on K+ currents in pulmonary arterial myocytes. Am J Physiol 277: L431–L439, 1999. doi: 10.1152/ajplung.1999.277.3.L431. [DOI] [PubMed] [Google Scholar]
- 46.Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol 266: H365–H370, 1994. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- 47.Snetkov VA, Aaronson PI, Ward JP, Knock GA, Robertson TP. Capacitative calcium entry as a pulmonary specific vasoconstrictor mechanism in small muscular arteries of the rat. Br J Pharmacol 140: 97–106, 2003. doi: 10.1038/sj.bjp.0705408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki H, Twarog BM. Membrane properties of smooth muscle cells in pulmonary arteries of the rat. Am J Physiol 242: H900–H906, 1982. doi: 10.1152/ajpheart.1982.242.5.H900. [DOI] [PubMed] [Google Scholar]
- 49.Vukicevic M, Kellenberger S. Modulatory effects of acid-sensing ion channels on action potential generation in hippocampal neurons. Am J Physiol Cell Physiol 287: C682–C690, 2004. doi: 10.1152/ajpcell.00127.2004. [DOI] [PubMed] [Google Scholar]
- 50.Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem 272: 20975–20978, 1997. doi: 10.1074/jbc.272.34.20975. [DOI] [PubMed] [Google Scholar]
- 51.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature 386: 173–177, 1997. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 52.Walker BR, Adams EM, Voelkel NF. Ventilatory responses of hamsters and rats to hypoxia and hypercapnia. J Appl Physiol (1985) 59: 1955–1960, 1985. doi: 10.1152/jappl.1985.59.6.1955. [DOI] [PubMed] [Google Scholar]
- 53.Weigand L, Sylvester JT, Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 290: L284–L290, 2006. doi: 10.1152/ajplung.00449.2004. [DOI] [PubMed] [Google Scholar]
- 54.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118: 687–698, 2004. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 55.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA 101: 6752–6757, 2004. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, Liu H, Jiang H, Xu TL. A nonproton ligand sensor in the acid-sensing ion channel. Neuron 68: 61–72, 2010. doi: 10.1016/j.neuron.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Yuan JX-J, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998. doi: 10.1161/01.CIR.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 58.Zha XM. Acid-sensing ion channels: trafficking and synaptic function. Mol Brain 6: 1, 2013. doi: 10.1186/1756-6606-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]