

Abstract
Arginine-vasopressin (AVP)-mediated translocation of aquaporin-2 (AQP2) protein-forming water channels from storage vesicles to the membrane of renal collecting ducts is critical for the renal conservation of water. The type-1 PDZ-binding motif (PBM) in AQP2, “GTKA,” is a critical barcode for its translocation, but its precise role and that of its interacting protein partners in this process remain obscure. We determined that synapse-associated protein-97 (SAP97), a membrane-associated guanylate kinase protein involved in establishing epithelial cell polarity, was an avid binding partner to the PBM of AQP2. The role of PBM and SAP97 on AQP2 redistribution in response to AVP was assessed in LLC-PK1 renal collecting cells by confocal microscopy and cell surface biotinylation techniques. These experiments indicated that distribution of AQP2 and SAP97 overlapped in the kidneys and LLC-PK1 cells and that knockdown of SAP97 inhibited the translocation of AQP2 in response to AVP. Binding between AQP2 and SAP97 was mediated by specific interactions between the second PDZ of SAP97 and PBM of AQP2. Mechanistically, inactivation of the PBM of AQP2, global delocalization of PKA, or knockdown of SAP97 inhibited AQP2 translocation as well as AVP- and forskolin-mediated phosphorylation of Ser256 in AQP2, which serves as the major translocation barcode of AQP2. These results suggest that the targeting of PKA to the microdomain of AQP2 via SAP97-AQP2 interactions in association with cross-talk between two barcodes in AQP2, namely, the PBM and phospho-Ser256, plays an important role in the translocation of AQP2 in the kidney.
Keywords: aquaporin-2, confocal microscopy, phosphorylation, renal collecting ducts, synapse-associated protein-97, trafficking
INTRODUCTION
Aquaporin-2 (AQP2) water channels are primarily present in submembranous storage vesicles of renal collecting ducts (CDs). Stimulation of Gs-coupled vasopressin type-2 receptors (V2Rs) by arginine-vasopressin (AVP) increases cAMP and activates PKA. PKA-mediated phosphorylation of AQP2 at Ser256 induces the translocation of AQP2 in storage vesicles to and fusion with the apical membrane of CDs (25). PKA-mediated posttranslational modification of AQP2 at Ser256 generates the major barcode for the translocation of AQP2. Moreover, it appears that phospho-Ser256 is somehow involved in facilitating the phosphorylation of other serines in AQP2 and, through this cooperation, induces the translocation and insertion of AQP2 along with epithelial Na+ channels into the apical membrane of CDs (1, 5, 25, 54).
Congenital nephrogenic diabetes insipidus (NDI) is a recessive hereditary disorder characterized by the inability of the kidney to concentrate urine in response to AVP (1). Most patients with congenital forms of NDI have an X-linked pattern of V2R gene mutations that are responsible for NDI in ~90% of patients (28, 35). AQP2 gene mutations are also associated with varying degrees of NDI (13). These mutations are divided into autosomal dominant mutations in the carboxy-terminal tail of AQP2 (24, 29, 54b) or into autosomal recessive mutations that are associated with misfolding or misrouting of AQP2 (9, 36).
A major unanswered question is how the fidelity of PKA-mediated phosphorylation of AQP2 on the Ser256 barcode is attained. Fidelity of PKA signaling is enhanced by compartmentalization of PKA with its targets, such as AQP2 and others. How this compartmentalization might occur is unknown, but AQP2 contains an embedded type-1 PDZ-binding motif (PBM) barcode, “GTKA,” that is involved in regulating the translocation of AQP2 from intracellular vesicles to the membrane (39). Type-1 PDZ barcodes are involved in translocating several agonist-internalized G protein-coupled receptors (GPCRs) such as the β1-adrenergic receptor (AR), β2-AR, and other proteins from endosomes to the plasma membrane (15, 16, 19, 41–44). Based on our findings with human β1-ARs (42), we hypothesized that the PBM of AQP2 would mediate its effect on the AQP2 shuttle by compartmentalizing PKA and other signaling/trafficking proteins within the AQP2 microdomain (16). The concept of signaling via proteins clustered at the PBM is an emerging concept (43). This concept aims to expand the role of the PBM from a purely trafficking motif, which is involved in translocating the agonist-internalized GPCR or other proteins from endosomes to the plasma membrane into a motif that promotes fidelity of signaling by targeting PKA to its recipient (16, 43). A protein involved in compartmentalization of PKA at the PBM of β1-ARs is synapse-associated protein-97 (SAP97) (16, 41, 42). The PBM of the β1-AR (ESKV) and that of AQP2 were interchangeable in supporting the translocation of endosomal β1-AR-AQP2 chimera to the plasma membrane through their common binding to SAP97 (42). Therefore, we investigated whether targeting PKA to AQP2 via SAP97-PBM interactions is involved in the translocation of AQP2 in intracellular stores to the plasma membrane of kidney-derived LLC-PK1 cells.
METHODS
Cellular reagents, cDNAs, shRNAs, and antibodies.
LLC-PK1 cells, which are epithelial pig kidney cells, were cultured in medium 199 supplemented with 5% FBS. Transfection of enhanced green fluorescent protein (EGFP)-N1 into LLC-PK1 cells using Lipofectamine 3000 (Invitrogen, Eugene, OR) or TransIT-LT1 (Mirus Bio, Madison, WI) transfection reagents was carried out following the manufacturer’s protocol. c-Myc-tagged human AQP2 (NM_000486) cDNA was purchased from Genecopoiea (GC-OG05842). Mutations of Ser256 in AQP2 to either alanine (S256A) or aspartic acid (S256D) were generated by PCR and verified by sequencing. Similarly, mutating the type-1 PDZ “GTKA” sequence of AQP2 to “AAAA” (AQP2∆PDZ), as well as creating the double AQP2 mutant (S256D) AQP2∆PDZ, were generated by PCR and verified by sequencing. The sequence in pig SAP97 (XM_021070092) between 1805 and 1826 corresponding to 5′-GTCAGGGATTGAACTTCAAAT-3′ or its inactive control (5′-GTCTCCACGCGCAGTACATTT-3′) was cloned into RNA inhibition expression nonfluorescent vector pcDNA-6.2-GW-mir or the fluorescent RNA inhibition vector pcDNA-6.2-GW/EmGFP-mir (Invitrogen). LLC-PK1 cells stably expressing scrambled (Scr) or pig SAP97 shRNAs were grown in complete medium supplemented with 100 µg/ml blasticidin (42). Rat SAP97 (NM_012788) escapes degradation by this siRNA because the corresponding sequence at nt 1802, 5′-GTCAAGGGTTGAACTTCAAAT-3′, has two mismatches (underlined). Rat SAP97 was expressed in pIRES-EGFP II, which is a bicistronic vector that allows expression of the gene of interest to be monitored at the single cell level because of expression of hrGFP II (green) on the same transcript (31, 41–43).
Monoclonal anti-AQP2 (sc-515770) was from Santa Cruz Biotechnology (Santa Cruz, CA), and rabbit polyclonal anti-AQP2 antibody (orb10123) was from Biorbyt (Cambridge, UK). Anti-c-Myc (9E-10) IgG (no. 13-2500) and anti-c-Myc IgG-agarose (no. 20168) were from ThermoFisher (Schaumburg, IL). Monoclonal anti-SAP97 (VAM-PS005, Enzo Life Sciences, Farmingdale, NY), anti-sorting nexin (SNX)4 (ab198504), anti-SNX27(366–415) (ab178388), and anti-β-actin (ab8227) were from Abcam (Cambridge, MA). Cyanine-based fluorescent dye CF555 conjugated to 9E-10 anti-c-Myc IgG (BNC550596) or goat anti-mouse IgG (no. 20231) were from Biotium (Fremont, CA). Tetramethyl rhodamine goat anti-mouse antibody was from Invitrogen. Anti-phospho-Ser256 AQP2 rabbit IgG (orb155711) was from Biorbyt. H-89 and myristoylated PKA inhibitor 14–22 amide (mPKI) were from EMD-Millipore (Billerica, MA). st-Ht31 and st-Ht31-pro cell-permeable peptides were from Promega (Fitchburg, WI). All antibodies were validated for specificity by Western blot and immunohistochemical procedures (3).
Confocal microscopy and dual-fluorescence microscopy protocols.
For translocation experiments, cells on poly-l-lysine-coated glass coverslips were transfected with plasmids and then switched from complete medium to Opti-MEM with 1% serum for 6 h. Cells were then exposed to buffer or 40 nM AVP for 20 min at 37°C. Slides were fixed in PBS containing 4% paraformaldehyde and 4% sucrose (pH 7.4) for 10 min, permeabilized in ice-cold HBSS containing 0.2% Triton X-100 for 10 min at 4°C, and then blocked in 3% BSA in PBS for 30 min. Slides were incubated with 1:200 dilution (5 µl/ml) of CF555 conjugated to 9E-10 anti-c-Myc IgG in blocking buffer for 1 h at 37°C (31, 44). Confocal CF555 fluorescence microscopy was performed on coded slides (excitation wavelength: 555 nm and emission wavelength: 580 nm), and the optical section thickness of 1.0-µm images was acquired using an Olympus FV1000 confocal microscope equipped with 100-Å oil-immersion (numerical aperture: 1.30) and 60-Å oil-immersion (numerical aperture: 1.42) objective lenses (31, 42). Nomarski images were then taken under the same settings and magnification. Digital images were converted to TIFF files and then exported and analyzed for pixel intensity and distribution by Image J software ]National Institutes of Health (NIH), Bethesda, MD].
To estimate the distribution of AQP2 fluorescent pixels between the surface membrane and intracellular stores, each cell was partitioned by a line, every point of which was at a distance of 300 nm from the outer periphery, using Image J software (42). This line formed the outer limit of the area used to index pixel intensity of AQP2 in “storage vesicles,” whereas the area formed between the peripheral cell membrane and this line referenced the “membranous” population of AQP2. For each condition, we conducted no less than three experiments and analyzed the distribution of AQP2 in >10–15 images/experiment (n ≥ 30). These data are presented as means ± SD.
For colocalization between SAP97 and AQP2, LLC-PK1 cells were transfected with 1.5 µg of plasmids harboring SAP97-GFP (12) and c-Myc-AQP2 for 16 h, switched to Opti-MEM with 1% serum for 6 h, and then fixed. Cells were permeabilized, blocked, and incubated with CF555-anti-c-Myc and visualized using CF555/Cy3 (pseudo red for AQP2) and GFP (pseudo green for SAP97) laser settings.
Cell surface biotinylation and glutathione-S-transferase pulldown assays.
LLC-PK1 cells on 15-cm culture dishes were exposed to buffer or 40 nM AVP for 20 min at 37°C. The plates were quickly washed free of culture media and reagents with ice-cold HBSS and then incubated in a total volume of 5 ml HBSS supplemented with 3 mg/ml of the cell-impermeant biotin-amidohexanoic acid 3-sulfo-N-hydroxysuccinimide ester (B1022, Sigma-Aldrich, St. Louis, MO) for 60 min at 4°C (15). Cells were then washed free of unattached biotin by three washes with biotin quenching buffer (250 mM glycine in HBSS), with the last wash incubated for 20 min at 4°C. Cells were lysed in radioimmunoprecipitation buffer [150 mM NaCl, 50 mM Tris (pH 8.0), 5 mM EDTA, 1% (vol/vol) Triton X-100, 0.1% SDS, 10 mM NaF, 10 mM Na2-pyrophosphate, and protease inhibitors]. Equal amounts of protein from these cells were mixed with 25 µl of BSA-blocked ultralink-neutra avidin beads (Sigma-Aldrich) at 4°C overnight. The resin was collected by centrifugation, washed several times with lysis buffer, and then extracted with 2× Laemmli sample buffer with 200 mM dithiothreitol at 10 µl/100 µg of input protein for 40 min at 37°C. The supernatant was subjected to SDS-PAGE, transferred to nitrocellulose, probed with a 1:250 dilution of anti-AQP2 antibody, and visualized with horseradish peroxidase-conjugated donkey anti-rabbit IgG using Super Signal detection reagent (Sigma-Aldrich). Optical densities (ODs) of biotin-AQP2 from each condition were quantified using a Chemi DOC XRS densitometer (Bio-Rad) equipped with Quantity One software (16). ODs of biotin-AQP2 were corrected for minor differences in input proteins by calculating the OD of β-actin in 10 µg of input lysates that were run and quantified separately. The effect of AVP on AQP2 translocation was assessed by determining the fold increase in the ratio of the corrected OD of biotinylated AQP2 in AVP-treated cells over its OD in buffer-exposed cells. Ratios from n = 5 separate experiments were calculated and keyed into the statistical software program to estimate the average fold increase ± SD for each condition.
The creation of individual SAP97 PDZs fused to glutathione-S-transferase (GST) in pGEX-4T-2 vector was as previously described (41). GST (10 µg) or GST-SAP97 PDZ (12.5 µg) was incubated with 120 µg of cell lysates prepared from LLC-PK1 cells stably expressing c-Myc-AQP2 along with 10 µl of glutathione agarose beads at a total volume of 1 ml for 2 h at 4°C. The beads were washed three times in lysis buffer, and the proteins were eluted from the beads with 2× Laemmli sample buffer containing 200 mM dithiothreitol. Eluates were separated on SDS-polyacrylamide gels, electroblotted to nitrocellulose filters, and probed for AQP2 pulldown by immunoblot analysis.
AQP2 phosphorylation and coimmunoprecipitations.
LLC-PK1 cells expressing wild-type (WT) AQP2 or AQP2∆PDZ were treated overnight with 50 µM indomethacin and then switched to Opti-MEM with 1% serum and 50 µM indomethacin for 6 h. Cells were exposed to buffer (control) or stimulated with 40 nM AVP or 20 µM forskolin (FSK) for 10 min. Indomethacin was included in all phosphorylation conditions to reduce basal cAMP and phospho-Ser256 AQP2 levels so that the effect of AVP or FSK on this parameter could be observed (10). Cells were lysed in 500 μl of 20 mM HEPES, 150 mM NaCl, 2 mM EDTA, 10% glycerol, 0.5% Triton X-100, and complete protease and phosphatase inhibitor cocktail (Roche, Branchburg, NJ). Lysates were rotated at 4°C for 1 h followed by centrifugation at 14,000 g at 4°C for 20 min. Soluble extracts were precleared by 30-min incubation with 30 μl protein G-Sepharose. After protein concentrations were equalized across all samples, lysates were added to ~5 μl of anti-c-Myc 9E-10 IgG agarose beads for 4 h at 4°C. Immune complexes were washed five times, and proteins were eluted in 40 μl of elution buffer (200 μg/ml c-Myc peptide, 20 mM HEPES, 50 mM NaCl, 0.1% cholesterol, and 8 mM EDTA). Eluted immune complexes were separated by SDS-PAGE and subjected to Western blot analysis with anti-phospho-Ser256 AQP2 antibody as previously described (43).
Coimmunoprecipitations between c-Myc-AQP2 and SAP97 were performed as follows. LLC-PK1 cells expressing c-Myc-AQP2 were lysed, and insoluble cellular debris was removed by centrifugation. After protein concentrations were equalized across all samples, lysates were added to ~5 μl of anti-c-Myc agarose beads. Reverse experiments involved the addition of equal amounts of cell lysates to anti-SAP97 IgG bound to protein G-agarose beads (42). Control experiments were performed by incubating lysates with preimmune IgG conjugated to protein G-agarose or protein G-Sepharose beads at the same concentration for 4 h at 4°C. Immune complexes were washed five times in lysis buffer and eluted from the beads with 2× Laemmli sample buffer containing 40 mM dithiothreitol and subjected to Western blot analysis (43). To normalize for input protein levels, 5% of each cell lysate was subjected to Western blot analysis using anti-β-actin antibody. Luminescence was acquired using the Bio-Rad XRS chemiluminescence documentation system on short exposure blots, and densitometric changes were quantified as described above.
Animals and immunohistochemistry.
Animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committees of the University of Louisiana-Monroe and University of Tennessee Health Sciences Center. Experiments conformed with the NIH Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, Revised 1996). Eight-week-old male C57BL/6NTac mice (weighing between 24 and 28 g) were obtained from Taconic. Mice (n = 4) were euthanized by barbiturate overdose (pentobarbital, 150 mg/kg ip), and their kidneys were then quickly removed, fixed in 4% paraformaldehyde, and embedded in paraffin blocks. Tissues were sliced to 5- to 10-μm-thick sections and mounted on Superfrost plus glass slides. Sections were processed for AQP2 using rabbit anti-AQP2 antibody (1:500, orb10123) and SAP97 using monoclonal anti-SAP97 antibody (1:500, VAM-PS005). The secondary antibodies were tetramethyl rhodamine goat anti-mouse and AF-488-conjugated goat anti-rabbit antibodies (1:500, Invitrogen). Sections from all animals were processed simultaneously (52). Sections (at least 8 sections/experiment from 4 different animals) were observed under a Nikon Optiphot-2 microscope with epifluorescence attachment. The digital images were captured on a Mac computer by a Retiga 1300R digital camera attached to the microscope. Colocalization was calculated by Pearson’s coefficient between the SAP97 image channel and specific AQP2 channel using built-in iVision-Mac software (BioVision Tech). For each kidney, eight different optical areas of each section (from n = 4 mice) were evaluated.
Statistical analysis.
Data were derived from image analysis that determined specific total pixels and pixels outside versus pixels inside the 300-nm partition that was drawn around the inner circumference of LLC-PK1 cells. Ratios of pixels residing outside or inside the 300-nm partition to total pixels (in %) were calculated for each image. The average ± SD of percentile pixel ratios from 3 separate experiments derived from 10 images/experiment (n = 30 images) is presented. Statistical comparison between two groups was performed by unpaired t-tests and for multiple groups by ANOVA followed by a Bonferroni’s test with a single pooled variance test in which the family-wise error rate was set at 0.05 using GraphPad Prism 7 software (GraphPad Software, San Diego, CA).
RESULTS
Identification of a role for the type-1 PDZ of AQP2 in AVP-mediated translocation of AQP2.
We investigated the involvement of select amino acid sequences, termed “barcodes,” in AVP-mediated translocation of AQP2-bearing membranes from storage vesicles to the plasma membrane of LLC-PK1 cells (19). LLC-PK1 cells are a nontransformed epithelial cell line derived from porcine kidneys that express V2R and replicate many of the trafficking and biochemical processes involved in AQP2 translocation in response to AVP in renal CDs (2, 7, 21, 25, 33, 48). Transient expression of plasmids harboring c-Myc-tagged AQP2 in LLC-PK1 cells was carried out using the TransIT-LT1 reagent because it provided high transfection efficiencies (~80%) accompanied with >90% cell viability, as assessed by GFP fluorescence and trypan blue exclusion (Fig. 1A).
Fig. 1.

Role of the type-1 PDZ domain of aquaporin-2 (AQP2) in its translocation from storage vesicles to the plasma membrane of LLC-PK1 cells. A: effect of two transfection reagents on c-Myc-AQP2 expression in LLC-PK1 cells. B: distribution of wild-type (WT) c-Myc-AQP2 in LLC-PK1 cells that were exposed either to buffer (a) or 40 nM arginine-vasopressin (AVP; b) was determined by confocal microscopy. Nomarski images (inset) were acquired at the same magnification. Scale bars = 5 μm. C: averages ± SD of AQP2 pixels outside a 300-nm partition derived from 30 control (open bars) or AVP-exposed (closed bars) images from n = 3 experiments. Unpaired t-tests between the percentile of pixels outside the 300-nm partition in control versus AVP-treated cells are expressed as no significant difference (NS) or *P < 0.05, **P < 0.01, and **P < 0.001, respectively. D: total surface biotinylated WT AQP2 levels (arrows) in control versus AVP-treated LLC-PK1 cells. E: combined densitometry of biotinylated AQP2 in control versus AVP-treated cells that were corrected for β-actin levels in input lysates from five separate experiments (mean ± SD) were compared by Student’s t-test. F and H: distribution of AQP2∆PDZ in LLC-PK1 cells in response to buffer (c) or AVP (d) was determined either by confocal microscopy (F) or cell surface biotinylation (H), as described above. G and I: percentile of AQP2∆PDZ pixels from 30 images (n = 3) residing outside the 300-nm partition (G) and combined densitometry of the biotinylated samples (I; n = 5) are shown as means ± SD. Statistical comparisons were conducted as described in C and E. IB, immunoblot; IP, immunoprecipitation.
LLC-PK1 cells expressing c-Myc-AQP2 were grown on coverslips, exposed to buffer or 40 nM AVP for 20 min. They were then fixed, permeabilized, and stained with CF555-conjugated (9E-10) anti-c-Myc IgG to label AQP2. Slides were imaged, and their fluorescence is presented as pseudo red color along with Nomarski images (Fig. 1B, left inset). As described in methods, a 300-nm partition was drawn around the inner circumference of cells to index membrane versus storage vesicle distribution of AQP2 (Fig. 1C). In quiescent LLC-PK1 cells, 89 ± 10% of AQP2 pixel intensities (pseudo red, Fig. 1Ba) were distributed inside the 300-nm partition (P < 0.001), indicating that AQP2 was distributed in intracellular storage vesicles (n = 30 images from 3 separate experiments). Exposure of these cells to AVP induced varied degrees of pixel translocation from inside to outside the 300-nm partition that ranged from 45% to 80% with an average ± SD of 67 ± 19% (Fig. 1Bb, arrows).
These findings were supplemented by surface biotinylation experiments to detect membrane densities of total AQP2 in buffer- or AVP-treated cells (Fig. 1D). Biotinylation of LLC-PK1 cells stably expressing c-Myc-AQP2 detected AQP2 protein bands with Mr ranging from 31 to 28 KDa, indicating that some degradation of AQP2 has occurred during this procedure (47). The results show that the OD of total WT AQP2 biotin/β-actin bands in AVP-treated cells was significantly higher than in the corresponding bands of the buffer-treated group (P < 0.01; Fig. 1E), indicating that AVP induced the redistribution of AQP2 to the surface-exposed membrane of LLC-PK1 cells.
Repeating these experiments with an AQP2 construct with an inactivated “GTKA” PBM (AQP2∆PDZ) indicated that this mutation did not affect the intracellular distribution of AQP2 in quiescent LLC-PK1 cells but prevented the translocation of AQP2∆PDZ in response to AVP (Fig. 1, F, c and d, and G). These findings were replicated by cell surface biotinylation of LLC-PK1 cells expressing AQP2∆PDZ, wherein AVP failed to significantly increase the membrane density of AQP2∆PDZ-biotin/β-actin in these cells (Fig. 1, H and I).
Involvement of SAP97 in AVP-mediated translocation of AQP2 in LLC-PK1 cells.
The involvement of the PBM motif of AQP2 in regulating its translocation led us to investigate whether SAP97 was involved in this phenomenon because trafficking experiments indicated that the PBM of the β1-AR (ESKV), which supports the trafficking of the β1-AR in a SAP97-dependent manner, was interchangeable with the PBM of AQP2 (42). SAP97 is a major compartmentalizing protein with three PDZ domains and several protein-interacting domains (Fig. 2A) that is abundantly expressed in the kidneys (37, 55) and in LLC-PK1 cells (Fig. 2B, lane 1).
Fig. 2.

Role of synapse-associated protein-97 (SAP97) and its PDZ domains in binding to and translocating aquaporin-2 (AQP2) in response to arginine-vasopressin (AVP). A: schematic of SAP97. B: LLC-PK1 cells stably expressing AQP2 and either scrambled (Scr) or pig SAP97 RNA inhibitor were transfected with empty pIRES-EGFPII or rat-SAP97 in the pIRES-EGFPII vector and then probed by Western blot analysis for SAP97 expression. C: statistical comparisons of mean ± SD values of SAP97 levels in Scr RNA inhibitor (open bars) or SAP97 RNA inhibitor (closed bars) from n = 5 determinations were carried out and analyzed by Student’s t-test. D: effect of scrambled shRNA (a and b) or shRNA-mediated knockdown of SAP97 (c and d) and its subsequent rescue (e and f) with a shRNA-resistent SAP97 construct (pseudo green) on AQP2 distribution (pseudo red) in the cells described in Fig. 1B were determined by dual confocal microscopy. Scale bars = 5 μm. E: effect of endogenous SAP97 (lanes 1 and 2) or its knockdown (lanes 3 and 4) and SAP97 reexpression (lanes 7 and 8) on total surface biotinylated AQP2 levels (arrows) in control versus AVP-treated LLC-PK1 cells. F: combined densitometry of biotinylated AQP2 in control versus AVP-treated cells that were corrected for β-actin levels in input lysates from 5 separate experiments (mean ± SD) were compared by one-way ANOVA followed by Bonferroni’s test. NS, nonsignificant difference. *P < 0.05; **P < 0.01; ***P < 0.001.
To find out if SAP97 was involved in AVP-mediated translocation of AQP2, we developed a SAP97 knockdown and rescue strategy. This strategy involved the creation of an LLC-PK1 cell line that stably expressed AQP2 with either Scr or pig SAP97 RNA inhibitor to selectively knock down SAP97 in LLC-PK1 cells. Into these cells, we transiently expressed either the “empty” GFP-expressing pIRES-EGFP vector or pIRES-EGFP that expresses rat SAP97 cDNA, which differs by two nucleotides from the corresponding sequence in the pig SAP97 RNA inhibitor.
In LLC-PK1 cells stably expressing pig SAP97 shRNA along with the empty pIRES-EGFP vector, there was an 85 ± 13% reduction in endogenous SAP97 expression compared with SAP97 levels in Scr shRNA-expressing cells (P < 0.001 compared with cells expressing Scr shRNA; Fig. 2, B, lanes 1 and 2, and C). Expression of rat SAP97 in Scr shRNA-expressing cells increased SAP97 expression by 2.4 ± 0.7-fold above basal levels (n = 4, P < 0.05 compared with cells expressing empty pIRES-EGFP; Fig. 2B, lane 3). Expression of rat SAP97 in SAP97 knockdown LLC-PK1 cells increased SAP97 levels by 1.8 ± 0.5-fold over their basal level (Fig. 2B, lane 4), indicating that rat SAP97 (SAP97*) was resistant to degradation by the RNA inhibitor to pig SAP97.
The effect of knock down and rescue of SAP97 on AVP-mediated translocation of AQP2 was examined by confocal microscopy (Fig. 2D) and surface biotinylation (Fig. 2E). Transient expression of pIRES-EGFP into cells coexpressing AQP2 and Scr shRNA or in those expressing AQP2 and pig SAP97 shRNA did not alter the intracellular punctate distribution of AQP2 (pseudo red), which amounted to ~90 ± 13% of total fluorescence (Fig. 2D, a and c). Similarly, expression of rat SAP97 in pIRES-EGFP into these naïve cells did not alter the percentile of intracellular AQP2 fluorescence (Fig. 2De). In Scr shRNA-expressing cells, AVP induced the translocation of 63 ± 15% of AQP2 fluorescence to the peripheral membrane (Fig. 2Db). However, exposure of SAP97 knockdown cells to AVP failed to induce translocation of AQP2, which remained intracellular (92 ± 12%, P < 0.02 compared with Scr shRNA from n = 30 images pooled from 3 separate experiments; Fig. 2De). Expression of rat SAP97* in SAP97 knockdown cells restored translocation of 62 ± 18% of AQP2 fluorescence from inside to outside the 300-nm partition in response to AVP (representing n = 30 images from 3 separate determinations; Fig. 2Df).
These findings were supplemented by biotinylation experiments, which revealed that exposure of cells stably expressing AQP2 and Scr shRNA to AVP resulted in a 3.8 ± 1.9-fold increase in the total density of biotinylated AQP2/β-actin compared with control cells (Fig. 2E, lanes 1 and 2, and Fig. 3E). Exposure of cells stably expressing WT AQP2 and pig SAP97 shRNA to AVP increased the density of biotinylated AQP2/β-actin by 1.4 ± 0.86-fold, which was not significantly different than in buffer-treated cells (Fig. 2, E, lanes 3 and 4, and F). Transient expression of GFP in AQP2 and SAP97 shRNA-expressing cells followed by AVP did not significantly increase the density of biotinylated AQP2/β-actin compared with buffer-treated cells (Fig. 2, E, lanes 5 and 6, and F). However, in cells expressing SAP97*, we observed a 5.4 ± 2.1-fold increase in the OD of biotinylated AQP2/β-actin (P < 0.001) in response to AVP compared with buffer-treated cells (Fig. 2E, lanes 5 and 6, and F). These findings indicate that SAP97 played a major role in AVP-mediated translocation of AQP2 in LLC-PK1 cells.
Fig. 3.
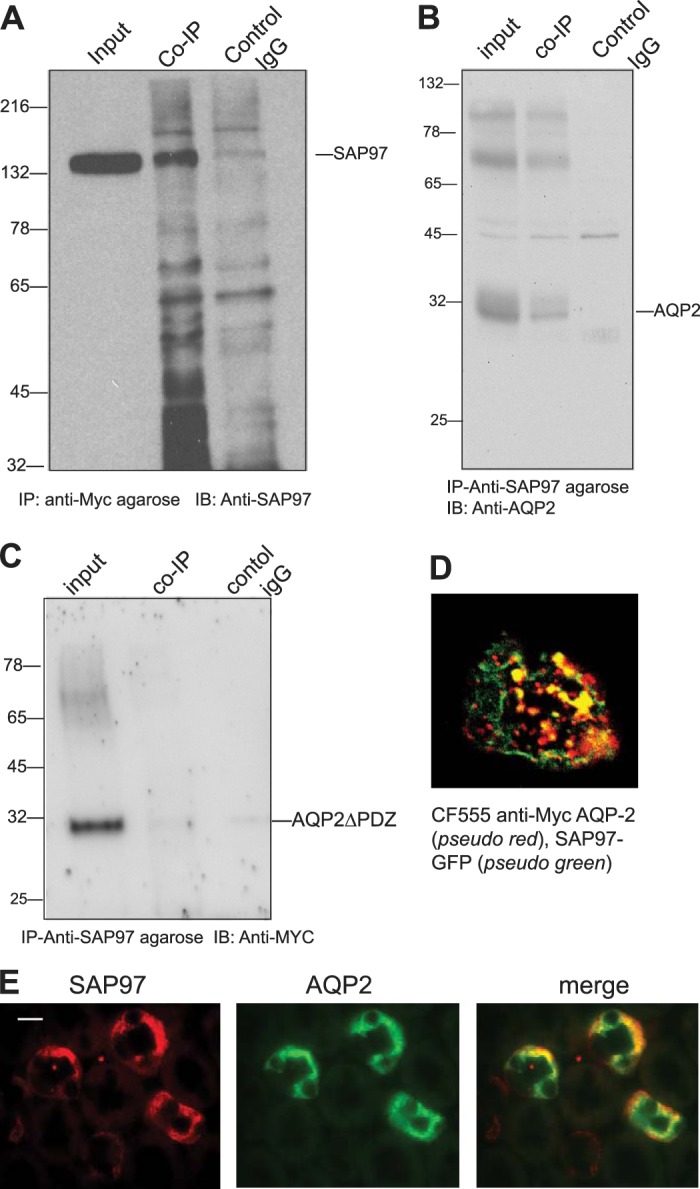
Characterization of synapse-associated protein-97 (SAP97) and aquaporin-2 (AQP2) interactions in LLC-PK1 cells and in mouse kidneys. A and B: coimmunoprecipitations (co-IPs) between c-Myc-AQP2 and SAP97. A: anti-c-Myc resin (lane 2) but not preimmune IgG (lane 3) coimmunoprecipitated SAP97 from c-Myc-AQP2-expressing cells. B: anti-SAP97 IgG resin coimmunoprecipitated AQP2 from c-Myc-AQP2-expressing cells. C: co-IPs between AQP2∆PDZ and SAP97. Inputs (lane 1) represent ~4% of total cell lysate from each condition. D: LLC-PK1 cells expressing c-Myc-WT AQP2 (pseudo red) and SAP97-green fluorescent protein (GFP) (pseudo green) were fixed, permeabilized, stained with CF555 anti-c-Myc IgG, and visualized by dual confocal microscopy. E: medullary slices from C57 black mouse kidneys were fixed, incubated with monoclonal anti-SAP97 and rabbit anti-AQP2 antibodies, and then stained with secondary tetramethyl rhodamine goat anti-mouse (1:500) and AF-488-conjugated goat anti-rabbit (1:500) antibodies, respectively. WT, wild-type.
Fig. 4.

Identification of the PDZ in synapse-associated protein-97 (SAP97) that is involved in the aquaporin-2 (AQP2) shuttle. A: glutathione-S-transferase (GST) pulldowns between AQP2 and the individual PDZ domains of SAP97 were conducted as described in methods and probed for AQP2 pulldown by Western blot [immunoblot (IB)]. B: LLC-PK1 cells stably expressing c-Myc-AQP2 (pseudo red) were transfected with the indicated PDZ of SAP97 in pIRES-EGFP (pseudo green) before the distribution of AQP2 in response to buffer (a, c, e, and g) or arginine-vasopressin (AVP; b, d, f, and h) was determined by dual fluorescence microscopy. Scale bars = 5 μm. C: average ± SD values of AQP2 pixels outside a 300-nm partition derived from 30 control (open bars) or AVP-exposed (closed bars) images from n = 3 experiments. Statistical comparisons were carried out by one-way ANOVA followed by a Bonferroni’s test. WT, wild-type.
Fig. 5.

Effect of inhibition of PKA and calcineurin pathways or mutagenesis of Ser256 in aquaporin-2 (AQP2) on the AQP2 shuttle. A: LLC-PK1 cells stably expressing c-Myc-AQP2 (pseudo red) were preexposed to buffer, 0.3 µM myristoylated PKA inhibitor (mPKI), 50 µM st-Ht31, 50 µM st-Ht31-pro, or 1 µM CN-585 for 30 min at 37°C before exposure to buffer (a, c, e, g, and i) or 40 nM arginine-vasopressin (AVP; b, d, f, h, and j) for 20 min. Confocal images from fixed permeabilized cells were acquired as described in Fig. 1B. Scale bars = 5 μm. B: average ± SD values of AQP2 pixels outside a 300-nm partition of n = 30 control or AVP-exposed cells from 3 separate experiments. C: LLC-PK1 cells transiently expressing wild-type (WT) c-Myc-AQP2 or the indicated point mutations in Ser256 or type-1 PDZ of c-Myc-AQP2 (pseudo red) were exposed to buffer (k, m, o, q, and s) or 40 nM AVP (l, n, p, r, and t) and processed as in A. D: average ± SD values of AQP2 pixels outside a 300-nm partition of n = 30 images/condition from n = 3 separate experiments. Statistical comparisons in B or D were carried out by one-way ANOVA followed by a Bonferroni’s test. NS indicates nonsignificant differences between the column pairs. ***P < 0.001.
Fig. 6.

Effect of A-kinase-anchoring protein-PKA and aquaporin 2 (AQP2)-synapse-associated protein-97 (SAP97) interactions on forskolin (FSK)- and arginine-vasopressin (AVP)-mediated phosphorylation of AQP2 on Ser256. A and B: LLC-PK1 cells expressing wild-type (WT) c-Myc-AQP2 or c-Myc-AQP2∆PDZ were incubated with 50 µM indomethacin (Indo) overnight and then exposed to 80 nM AVP (I/AVP) or 20 µM FSK (I/FSK) for 10 min at 37°C. C: cells stably expressing WT c-Myc-AQP2 exposed to Indo overnight were incubated with 50 µM st-Ht31 or st-Ht31-pro for 30 min before exposure to AVP or FSK for 10 min. D and E: cells stably expressing scrambled (Scr) or pig SAP97 shRNA were transiently transfected with WT c-Myc-AQP2. These cells were incubated with Indo overnight and then exposed to AVP or FSK and processed as described in A and B. A′–E′: graphical representation of combined densitometry results of phospho-Ser256 AQP2/β-actin in means ± SD from n = 5 experiments. Open bars indicate Indo (control), lightly shaded bars indicate I/AVP, and dark shaded bars indicate I/FSK. Statistical comparisons in A′−E′ between Indo, I/AVP, and I/FSK were carried out by one-way ANOVA followed by a Bonferroni’s test. NS, nonsignificant differences. *P < 0.05; **P < 0.01; ***P < 0.001.
Identification of SAP97 as a binding partner to AQP2.
To find out if SAP97 could bind AQP2, we performed immunoprecipitation experiments between endogenous SAP97 and AQP2 in LLC-PK1 cells stably expressing c-Myc-AQP2 (Fig. 3). The data shown in Fig. 3A demonstrate that input lysates contained significant amounts of endogenous SAP97 (input). c-Myc affinity-purified eluates coimmunoprecipitated endogenous SAP97 (n = 3), whereas preimmune or “control” IgG did not coimmunoprecipitate SAP97 from these cells (Fig. 3A, lanes 2 and 3, respectively). We repeated the opposite experiment, wherein anti-SAP97-purified eluates were probed for coimmunoprecipitation of WT AQP2 (Fig. 3B). The data indicated that endogenous SAP97 specifically coimmunoprecipitated AQP2 (Fig. 3B, lane 2), whereas preimmune IgG eluates were devoid of AQP2 (representing n = 3 determinations; Fig. 3B, lane 3). SAP97-mediated coimmunoprecipitation of AQP2 was dependent on its PBM because anti-c-Myc-purified lysates from c-Myc-AQP2∆PDZ-expressing cells did not coimmunoprecipitate SAP97 (Fig. 3C, lane 2). These findings were supplemented by dual confocal colocalization microscopy in LLC-PK1 cells expressing c-Myc-AQP2 (pseudo red) and SAP97-GFP (pseudo green), which revealed >70 ± 19% overlap between these proteins in unstimulated cells (n = 10; Fig. 3D). Immunohistochemical staining of mouse kidney sections (Fig. 3E) simultaneously with fluorescently labeled anti-SAP97 IgG (pseudo red) and anti-AQP2 IgG (pseudo green) indicated that ~50% of SAP97-positive cells colocalized with AQP2, whereas all of the AQP2-positive cells showed colocalization with SAP97 with a Pearson's coefficient and colocalization ratio values of 0.6 and 0.8, respectively (from 8 different optical areas per section from 3 different mice). These findings indicate that the distribution of renal SAP97 overlapped with AQP2 and that SAP97 interacted with the PBM of AQP2.
Characterization of the mechanism of SAP97 in regulating AQP2 translocation in LLC-PK1 cells.
The domains in SAP97 that bind to the PBM of AQP2 have not been identified but are thought to comprise one or more of its three PDZ domains (Fig. 2A) (37, 41, 56). To identify the AQP2-binding domain in SAP97, equal amounts of protein lysate from LLC-PK1 cells stably expressing full-length c-Myc-AQP2 were mixed with 10 µg GST or 12.5 µg GST fusions of PDZ1, PDZ2, or PDZ3 of SAP97 in a total volume of 1 ml (Fig. 4A). GST pulldowns revealed that PDZ2 of SAP97 selectively pulled down the ~30-kDa AQP2 protein (Fig. 4A, lane 3).
To examine the effect of each isolated PDZ of SAP97 on the translocation of AQP2 in response to AVP, we hypothesized that overexpression of the isolated AQP2-binding PDZ, i.e., PDZ2 of SAP97, would destabilize the AQP2-SAP97 scaffold and thus interfere with the translocation of AQP2 (Fig. 4B). Expression of GFP from the empty pIRES-EGFP vector alone or with the individual PDZs of SAP97 did not alter the distribution of AQP2 in storage vesicles (Fig. 4B, a, c, e, and g, and Table 1). Expression of GFP alone, GFP-PDZ1, or GFP-PDZ3 of SAP97 did not interfere with AVP-mediated translocation of AQP2 (Fig. 4, B and C). However, expression of GFP-PDZ2 of SAP97 abrogated the effect of AVP on AQP2 translocation, and the majority of AQP2-associated fluorescence remained inside the 300-nm partition (Fig. 4, B and C, and Table 1).
Table 1.
Effect of overexpression of isolated PDZ domains of synapse-associated protein-97 on aquaporin-2 trafficking in LLC-PK1 cells
| Buffer |
Arginine-Vasopressin |
|||
|---|---|---|---|---|
| Construct | Inside | Outside | Inside | Outside |
| GFP | 89 ± 18 | 10 ± 7 | 33 ± 9 | 67 ± 18 |
| PDZ1-GFP | 90 ± 13 | 12 ± 7 | 34 ± 10 | 69 ± 16 |
| PDZ2-GFP | 92 ± 11 | 12 ± 8 | 87 ± 16* | 19 ± 12* |
| PDZ3-GFP | 93 ± 14 | 11 ± 6 | 36 ± 11 | 65 ± 17 |
Data are means ± SD and were obtained from the analysis of n = 20 images/conditions from 2 separate experiments. Inside and outside refer to pixel distribution on either side of the 300-nm partition drawn around the inner circumference of the cell. GFP, green fluorescent protein.
P < 0.05 compared with GFP.
SAP97-mediated AKAP-PKA interactions with the type-1 PDZ of AQP2 are involved in regulating the translocation of AQP2 in response to AVP.
Tethering SAP97 to the PBM of a given protein creates multimeric complexes because several domains in SAP97 bind to other proteins (38, 56). For example, bipartite binding of SAP97 to the β1-AR (via PDZ2) and to the A-kinase anchoring protein (AKAP)5/PKA complex (via the SH3/HOOK domain) targeted PKA to the β1-AR microdomain (38, 41, 56). To find out if PKA targeting to AQP2 was required for its translocation in response to AVP, we first determined whether PKA or its association to an AKAP were involved in AQP2 trafficking (Fig. 5A). These experiments revealed that pretreatment of LLC-PK1 cells stably expressing c-Myc-AQP2 with the PKA inhibitor mPKI or with st-Ht31 (8), which globally destabilizes PKA-AKAP interactions, prevented the translocation of AQP2. Only 13 ± 9% and 15 ± 11% of CF555-AQP2 pixels were detected outside the 300-nm partition (P < 0.05 compared with control or st-Ht31-pro preexposed cells; Fig. 5, A and B). However, in cells exposed to diluent or to st-Ht-31-pro, which is the inactive counterpart of st-Ht31, AVP induced the redistribution of 65 ± 24% and 55 ± 30% of AQP2 pixels to outside of the 300-nm partition, respectively (Fig. 5, A, compare a with b and e with f, and B).
We also determined that inactivation of Ser256 [(S256A) AQP2] or the PBM (AQP2∆PBM) inhibited translocation of AQP2 in response to AVP to regions outside the 300-nm border (Fig. 5, C, n and p, and D) (14, 32). Constitutive activation of Ser256 in the context of the active PBM [(S256D) AQP2] or inactivated PBM [(S256A) AQP2∆PDZ] resulted in 76 ± 20% and 65 ± 22% distribution of these constructs in the region outside the 300-nm partition of naïve cells, respectively (Fig. 5C, q and s; data analysis in Fig. 5D). AVP had little effect on the distribution of the (S256D) mutated AQP2 constructs [73 ± 23% and 60 ± 22%, respectively, toward the outside of the 300-nm boundary (P > 0.05 compared with control unstimulated cells; Fig. 5D)]. Although these findings are in agreement with the notion that phosphorylation of AQP2 on Ser256 was essential for the redistribution of AQP2 from storage vesicles to the membrane (14, 32, 34), they are the first to show that SAP97-AQP2 PBM interactions were involved in this phenomenon.
Characterization of AQP2-PBM/SAP97 interactions on AVP-mediated phosphorylation of AQP2 at Ser256.
Phosphorylation of AQP2 on Ser256 appears to be the primary barcode for AQP2 translocation in response to AVP because this modification was not dependent on the state of phosphorylation of other serines or threonines in AQP2 (6, 34). To determine if AQP2-SAP97 interactions were involved in AVP-mediated phosphorylation of AQP2 at Ser256, we assessed the status of Ser256 in LLC-PK1 cells stably expressing equivalent amounts of WT c-Myc-AQP2 or c-Myc-AQP2∆PDZ. Cells were treated with indomethacin overnight to lower endogenous cAMP levels (54a) and then exposed for 10 min to buffer, 80 nM AVP, or 20 µM FSK, which maximally increases cAMP levels by activating all isoforms of adenylyl cyclase. AVP or FSK increased phospho-Ser256 WT AQP2 by 2.3 ± 0.7-fold and 3.1 ± 1.1-fold, respectively (Fig. 6A). Pooled data from five experiments that were normalized to β-actin indicated that AVP significantly increased phosphorylation of AQP2 on Ser256 (P < 0.05 compared with the indomethacin control; Fig. 6A′). However, phosphorylation of Ser256 in response to AVP or FSK in AQP2∆PDZ-expressing cells was not statistically different from that in indomethacin control cells (P > 0.05; Fig. 6, B and B′).
These experiments were followed by experiments that examined the role of PKA-AKAP interactions on the phosphorylation of WT c-Myc-AQP2 at Ser256 (Fig. 6C). On the one hand, pretreatment of cells with the inactive PKA-AKAP disruptor st-Ht31-pro and then with AVP or FSK was associated with increased AQP2 phosphorylation on Ser256 by 2.2 ± 0.7-fold and 3.3 ± 1.3-fold, respectively (P < 0.05 compared with the indomethacin control; Fig. 6C′). On the other hand, phosphorylation of WT AQP2 on Ser256 in response to AVP or FSK in st-Ht31-pretreated cells was not statistically different from that in cells pretreated with indomethacin (P > 0.05; Fig. 6, C′ and C′′). Therefore, trafficking and phosphorylation of AQP2 on Ser256 both required the PBM of AQP2 and anchorage of PKA to an AKAP.
To determine the role of SAP97 in this phenomenon, LLC-PK1 cells stably expressing either Scr shRNA or SAP97 shRNA were transfected with WT c-Myc-AQP2 (Fig. 6, D and E). In cells expressing Scr shRNA, phosphorylation of AQP2 on Ser256 in response to AVP or FSK increased by 2.6 ± 1-fold and 4.0 ± 1.2-fold, respectively (P < 0.05 compared with indomethacin). Downregulation of SAP97 abrogated AVP-mediated effects on the phosphorylation of AQP2 to 1.1 ± 0.5-fold and reduced the effect of FSK to 1.6 ± 0.6-fold, which were not statistically significant (n = 5, P > 0.05 compared with the indomethacin control; Fig. 6, E and E′). These findings suggest that SAP97 was also involved in imprinting the phospho-Ser256 translocation barcode of AQP2.
The salient features of AVP-mediated effects on the redistribution of AQP2 were remarkably similar to the effects of β-agonists on β1-AR trafficking, which required SNX27 and calcineurin (22, 42, 43). As shown in Fig. 5, A, i and j, and B, the selective calcineurin inhibitor CN-585 (11) inhibited the AQP2 shuttle, as previously reported (22). SNX27 is an endosomal protein that possesses a multivalent PDZ, which is essential for the trafficking of PBM-containing cargoes from early endosomes to the plasma membrane (30, 46). To find out if SNX27 was involved in the AQP2 shuttle, we probed cell extracts prepared from LLC-PK1 cells and human embryonic kidney (HEK)-293 cells for SNX27 (Fig. 7A). Western blots indicated that SNX27 was expressed in HEK-293 cells but was not expressed in LLC-PK1 cells (Fig. 7A). However, SNX4, which is another SNX involved in GPCR trafficking, was expressed at relatively high levels in both cell lines (Fig. 7A).
Fig. 7.
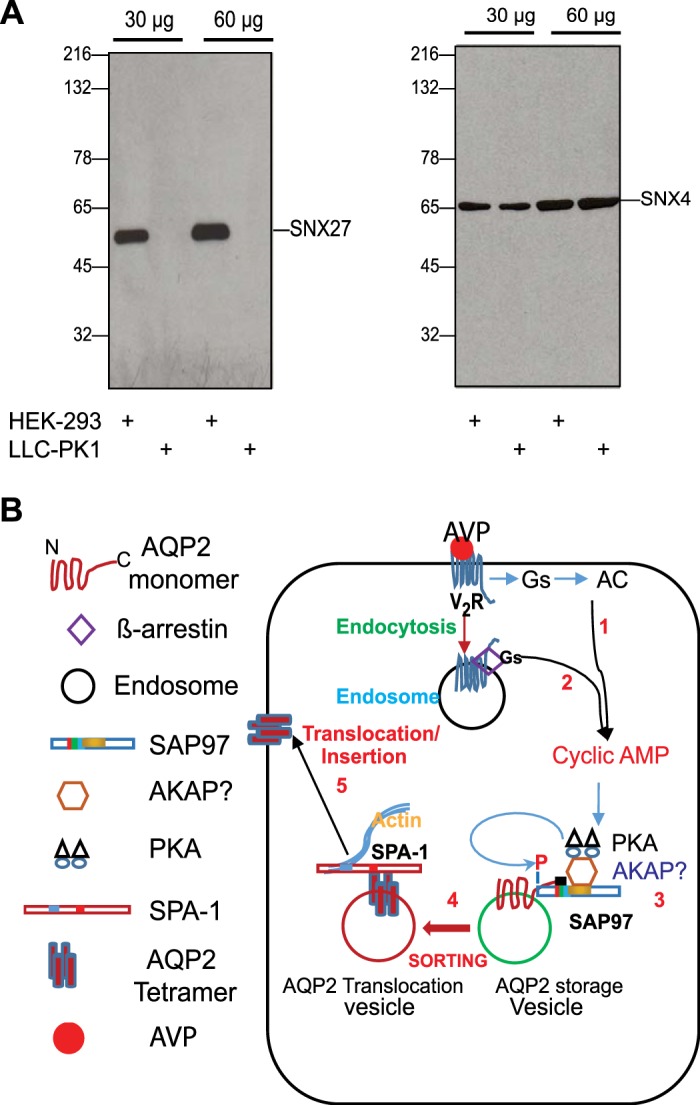
Schematic illustration of aquaporin-2 (AQP2) translocation paradigms in response to arginine-vasopressin (AVP). A: Western blot analysis of 30 or 60 µg cell lysates prepared from human embryonic kidney (HEK)-293 or LLC-PK1 renal cells with anti-sorting nexin (SNX)27 or anti-SNX4 antibodies. B: schematic illustrating the putative role of synapse-associated protein-97 (SAP97) in the AQP2 shuttle. Activation of membranous vasopressin type 2 receptors (V2Rs) with AVP increases the generation of cAMP (1). In addition, agonist-activated V2Rs bind tightly to β-arrestin/Gs and are endocytosed together into endosomes to signal from there to cAMP (2). Consequently, membranous and endosomal-generated cAMP sustains the activation of PKA. We hypothesize that the pool of PKA involved in phosphorylating AQP2 in storage vesicles is A-kinase-anchoring protein (AKAP) bound and that the AKAP/PKA complex is tethered to the PDZ-binding motif (GTKA) of AQP2 through SAP97 (3). This organization enhances the fidelity of PKA-mediated phosphorylation of AQP2 on Ser256 in storage vesicles and induces its translocation to the cell membrane via a type-1 PDZ-binding motif (PBM)-relay sorting mechanism (4). In this regard, signal-induced proliferation-associated gene (SPA)-1 and/or other PDZ proteins bind to the PBM of phosphorylated AQP2 and catalyze its translocation along actin filaments for eventual insertion of AQP2 tetramers into the membrane of collecting ducts (5).
DISCUSSION
It appears that four major stipulations are mechanistically involved in AVP-mediated translocation of AQP2 in renal cells. First is its reliance on AVP-mediated persistent stimulation of the cAMP-PKA axis to maintain the phosphorylation of AQP2 on Ser256 (6, 14, 32). Second is the involvement of the PBM of AQP2 and its interaction with signal-induced proliferation-associated gene (SPA)-1 (39). Third is the involvement of a pool of PKA that is compartmentalized with a hitherto uncharacterized AKAP (22, 50). Fourth is the involvement of calcineurin in this process (18, 22).
Progress in the first area indicates that sustained signaling of AVP-stimulated V2Rs to cAMP was mediated by an endosomal complex between the V2R with β-arrestin and Gs (12, 53). Recently, Gilbert et al. (17) showed that genetic expression of dominant negative PKA in the kidneys elicited diabetes insipidus in mice by inhibiting AQP2 translocation in response to AVP. This study and others provide conclusive proof that the cAMP-PKA axis is a prime system involved in the translocation of AQP2 in renal CDs (20, 22, 26, 27, 50).
Concerning the mechanism of type-1 PDZ in the translocation of AQP2, this report shows that the type-1 PDZ is involved in PKA-mediated phosphorylation of AQP2 on Ser256. Furthermore, we identified a novel role for SAP97 in this phenomenon by building on our serendipitous discovery concerning the interchangeability between type-1 PDZs of the β1-AR and AQP2 in supporting the trafficking of the β1-AR through their shared binding to SAP97 (42). These two findings add a mechanistic angle to the role of the AQP2 PBM by indicating that SAP97 binding to the AQP2 PBM was involved in PKA-mediated phosphorylation of AQP2 on Ser256 and in its shuttling between storage vesicles and the plasma membrane.
The involvement of PKA-AKAP interactions in phosphorylating AQP2 on Ser256 and in shuttling AQP2 from storage vesicles to the membrane have been well documented (20, 22, 26, 27, 50), but the relationship between the PBM and the pool of AKAP-compartmentalized PKA in phosphorylating AQP2 on Ser256 is still obscure (51). This study suggests a role for SAP97 in the AQP2 shuttle in LLC-PK1 cells through its bipartite binding to the type-1 PDZ of AQP2 via its PDZ2 domain and possibly to AKAP-PKA complexes via one of its many protein-protein-interacting modules (16, 41, 42). Whether the effect of SAP97 on the AQP2 shuttle was mediated through this mechanism is unknown because the PKA anchoring AKAP is still obscure. The involvement of AKAP18δ (an isoform of AKAP7) in this process has been reported (20), but transgenic mice with deletion of all the isoforms of AKAP7 did not manifest polyuria or urinary hypoosmolality associated with AQP2 trafficking defects (23). In light of the obscurity of the AKAP involved in the AQP2 shuttle, we attempted to add credence to the idea that SAP97 was involved in associating an AKAP-PKA complex with the AQP2 PBM by showing that SAP97-AQP2 interactions were dependent on the type-1 PDZ of AQP2 and were mediated by PDZ2 of SAP97 (Figs. 2–4). Moreover, we showed, for the first time, that SAP97 is intimately involved in PKA-mediated phosphorylation of AQP2 on Ser256 and that the distribution of SAP97 overlaps with the distribution of AQP2 in quiescent LLC-PK1 cells and in medullary regions of the mouse kidney (Figs. 3 and 6). Therefore, based on the β1-AR model, we propose that targeting of PKA to the PBM of AQP2 through a SAP97-AKAP/PKA complex appears to be involved in AVP-mediated phosphorylation of AQP2 on Ser256, which is a primary signal for the translocation of AQP2 from storage vesicles to the surface membrane of CDs, as shown in Fig. 7B.
In addition to SAP97, the PDZ protein SPA-1 was involved in the AQP2 shuttle (39). The effect of SPA-1 was mediated through the binding of its type-1 PDZ to phosphorylated AQP2 and by its Rap1 GAP region, which is involved in F-actin assembly (39, 40). This would suggest that SAP97-mediated phosphorylation of AQP2 preceded SPA-1 interactions with AQP2 because the binding of SPA-1 to phosphorylated AQP2 would occur downstream of the effects of SAP97 on AQP2 phosphorylation. Therefore, we speculated that SAP97 and SPA-1 might play complementary roles, whereby SAP97 would be involved in compartmentalizing PKA to faithfully phosphorylate AQP2 on Ser256, whereas SPA-1 binding to phosphorylated AQP2 would be involved in maintaining F-actin assembly around AQP2-containing vesicles as they translocate toward the cell membrane, as shown in Fig. 7B (40).
Concerning the role of calcineurin in AQP2 trafficking, recent findings indicate that Wnt5a induced phosphorylation of AQP2 and its translocation in LLC-PK1 cells by activating the calcineurin signaling pathway without activating the AVP-V2R-cAMP-PKA pathway (1a, 22). Thus, the Wnt5a-calcineurin pathway might provide a PKA-independent avenue for the translocation of AQP2, and its inhibition by CN-585 circumvented part, but not all, of the AQP2 shuttle.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-085848 and by Bridge Funding support by the University of Tennessee Health Sciences Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.W.B. conceived and designed research; M.M.N. and A.K. performed experiments; M.M.N., A.K., and S.W.B. analyzed data; ; A.K. and S.W.B. interpreted results of experiments M.M.N. and S.W.B. prepared figures; S.W.B. drafted manuscript; M.M.N. and S.W.B. edited and revised manuscript; M.M.N., A.K., and S.W.B. approved final version of manuscript.
REFERENCES
- 1.Alpern RJ, Moe OW, Caplan M (editors). Polyuria and diabetes insipidus. In: Seldin and Giebisch’s The Kidney (5th ed.). New York: Elsevier, 2013, p. 1571–1600. [Google Scholar]
- 1a.Ando F, Sohara E, Morimoto T, Yui N, Nomura N, Kikuchi E, Takahashi D, Mori T, Vandewalle A, Rai T, Sasaki S, Kondo Y, Uchida S. Wnt5a induces renal AQP2 expression by activating calcineurin signalling pathway. Nat Commun 7: 13636, 2016. doi: 10.1038/ncomms13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arthur J, Huang J, Nomura N, Jin WW, Li W, Cheng X, Brown D, Lu HJ. Characterization of the putative phosphorylation sites of the AQP2 C terminus and their role in AQP2 trafficking in LLC-PK1 cells. Am J Physiol Renal Physiol 309: F673–F679, 2015. doi: 10.1152/ajprenal.00152.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahouth SW. Western blot detection of adrenergic receptors. Methods Mol Biol 126: 301–314, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol 19: 1845–1854, 2008. doi: 10.1681/ASN.2008020225. [DOI] [PubMed] [Google Scholar]
- 6.Bradford D, Raghuram V, Wilson JL, Chou CL, Hoffert JD, Knepper MA, Pisitkun T. Use of LC-MS/MS and Bayes’ theorem to identify protein kinases that phosphorylate aquaporin-2 at Ser256. Am J Physiol Cell Physiol 307: C123–C139, 2014. doi: 10.1152/ajpcell.00377.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown D. The ins and outs of aquaporin-2 trafficking. Am J Physiol Renal Physiol 284: F893–F901, 2003. doi: 10.1152/ajprenal.00387.2002. [DOI] [PubMed] [Google Scholar]
- 8.Carr DW, Stofko-Hahn RE, Fraser IDC, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem 266: 14188–14192, 1991. [PubMed] [Google Scholar]
- 9.Deen PM, Croes H, van Aubel RA, Ginsel LA, van Os CH. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest 95: 2291–2296, 1995. doi: 10.1172/JCI117920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deen PM, Rijss JP, Mulders SM, Errington RJ, van Baal J, van Os CH. Aquaporin-2 transfection of Madin-Darby canine kidney cells reconstitutes vasopressin-regulated transcellular osmotic water transport. J Am Soc Nephrol 8: 1493–1501, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Erdmann F, Weiwad M, Kilka S, Karanik M, Pätzel M, Baumgrass R, Liebscher J, Fischer G. The novel calcineurin inhibitor CN585 has potent immunosuppressive properties in stimulated human T cells. J Biol Chem 285: 1888–1898, 2010. doi: 10.1074/jbc.M109.024844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinstein TN, Yui N, Webber MJ, Wehbi VL, Stevenson HP, King JD Jr, Hallows KR, Brown D, Bouley R, Vilardaga JP. Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem 288: 27849–27860, 2013. doi: 10.1074/jbc.M112.445098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujiwara TM, Bichet DG. Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol 16: 2836–2846, 2005. doi: 10.1681/ASN.2005040371. [DOI] [PubMed] [Google Scholar]
- 14.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 272: 14800–14804, 1997. doi: 10.1074/jbc.272.23.14800. [DOI] [PubMed] [Google Scholar]
- 15.Gardner LA, Naren AP, Bahouth SW. Assembly of an SAP97-AKAP79-cAMP-dependent protein kinase scaffold at the type 1 PSD-95/DLG/ZO1 motif of the human β1-adrenergic receptor generates a receptosome involved in receptor recycling and networking. J Biol Chem 282: 5085–5099, 2007. doi: 10.1074/jbc.M608871200. [DOI] [PubMed] [Google Scholar]
- 16.Gardner LA, Naren AP, Bahouth SW. Assembly of an SAP97-AKAP79-cAMP-dependent protein kinase scaffold at the type 1 PSD-95/DLG/ZO1 motif of the human β1-adrenergic receptor generates a receptosome involved in receptor recycling and networking. J Biol Chem 282: 5085–5099, 2007. doi: 10.1074/jbc.M608871200. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert ML, Yang L, Su T, McKnight GS. Expression of a dominant negative PKA mutation in the kidney elicits a diabetes insipidus phenotype. Am J Physiol Renal Physiol 308: F627–F638, 2015. doi: 10.1152/ajprenal.00222.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gooch JL, Guler RL, Barnes JL, Toro JJ. Loss of calcineurin Aalpha results in altered trafficking of AQP2 and in nephrogenic diabetes insipidus. J Cell Sci 119: 2468–2476, 2006. doi: 10.1242/jcs.02971. [DOI] [PubMed] [Google Scholar]
- 19.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol 48: 537–568, 2008. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 20.Henn V, Edemir B, Stefan E, Wiesner B, Lorenz D, Theilig F, Schmitt R, Vossebein L, Tamma G, Beyermann M, Krause E, Herberg FW, Valenti G, Bachmann S, Rosenthal W, Klussmann E. Identification of a novel A-kinase anchoring protein 18 isoform and evidence for its role in the vasopressin-induced aquaporin-2 shuttle in renal principal cells. J Biol Chem 279: 26654–26665, 2004. doi: 10.1074/jbc.M312835200. [DOI] [PubMed] [Google Scholar]
- 21.Hull RN, Cherry WR, Weaver GW. The origin and characteristics of a pig kidney cell strain, LLC-PK. In Vitro 12: 670–677, 1976. doi: 10.1007/BF02797469. [DOI] [PubMed] [Google Scholar]
- 22.Jo I, Ward DT, Baum MA, Scott JD, Coghlan VM, Hammond TG, Harris HW. AQP2 is a substrate for endogenous PP2B activity within an inner medullary AKAP-signaling complex. Am J Physiol Renal Physiol 281: F958–F965, 2001. doi: 10.1152/ajprenal.2001.281.5.F958. [DOI] [PubMed] [Google Scholar]
- 23.Jones BW, Brunet S, Gilbert ML, Nichols CB, Su T, Westenbroek RE, Scott JD, Catterall WA, McKnight GS. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc Natl Acad Sci USA 109: 17099–17104, 2012. doi: 10.1073/pnas.1215219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamsteeg EJ, Wormhoudt TA, Rijss JP, van Os CH, Deen PM. An impaired routing of wild-type aquaporin-2 after tetramerization with an aquaporin-2 mutant explains dominant nephrogenic diabetes insipidus. EMBO J 18: 2394–2400, 1999. doi: 10.1093/emboj/18.9.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katsura T, Gustafson CE, Ausiello DA, Brown D. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am J Physiol 272: F816–F822, 1997. doi: 10.1152/ajprenal.1997.272.6.F816. [DOI] [PubMed] [Google Scholar]
- 26.Klussmann E, Maric K, Wiesner B, Beyermann M, Rosenthal W. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J Biol Chem 274: 4934–4938, 1999. doi: 10.1074/jbc.274.8.4934. [DOI] [PubMed] [Google Scholar]
- 27.Klussmann E, Rosenthal W. Role and identification of protein kinase A anchoring proteins in vasopressin-mediated aquaporin-2 translocation. Kidney Int 60: 446–449, 2001. doi: 10.1046/j.1523-1755.2001.060002446.x. [DOI] [PubMed] [Google Scholar]
- 28.Knoers NV, van Os CH. Molecular and cellular defects in nephrogenic diabetes insipidus. Curr Opin Nephrol Hypertens 5: 353–358, 1996. doi: 10.1097/00041552-199607000-00011. [DOI] [PubMed] [Google Scholar]
- 29.Kuwahara M, Iwai K, Ooeda T, Igarashi T, Ogawa E, Katsushima Y, Shinbo I, Uchida S, Terada Y, Arthus MF, Lonergan M, Fujiwara TM, Bichet DG, Marumo F, Sasaki S. Three families with autosomal dominant nephrogenic diabetes insipidus caused by aquaporin-2 mutations in the C-terminus. Am J Hum Genet 69: 738–748, 2001. doi: 10.1086/323643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauffer BE, Melero C, Temkin P, Lei C, Hong W, Kortemme T, von Zastrow M. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J Cell Biol 190: 565–574, 2010. doi: 10.1083/jcb.201004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Nooh MM, Bahouth SW. Role of AKAP79/150 protein in β1-adrenergic receptor trafficking and signaling in mammalian cells. J Biol Chem 288: 33797–33812, 2013. doi: 10.1074/jbc.M113.470559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu HJ, Matsuzaki T, Bouley R, Hasler U, Qin QH, Brown D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am J Physiol Renal Physiol 295: F290–F294, 2008. doi: 10.1152/ajprenal.00072.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamuya FA, Cano-Peñalver JL, Li W, Rodriguez Puyol D, Rodriguez Puyol M, Brown D, de Frutos S, Lu HA. ILK and cytoskeletal architecture: an important determinant of AQP2 recycling and subsequent entry into the exocytotic pathway. Am J Physiol Renal Physiol 311: F1346–F1357, 2016. doi: 10.1152/ajprenal.00336.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moeller HB, MacAulay N, Knepper MA, Fenton RA. Role of multiple phosphorylation sites in the COOH-terminal tail of aquaporin-2 for water transport: evidence against channel gating. Am J Physiol Renal Physiol 296: F649–F657, 2009. doi: 10.1152/ajprenal.90682.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol 63: 607–630, 2001. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- 36.Mulders SM, Bichet DG, Rijss JP, Kamsteeg EJ, Arthus MF, Lonergan M, Fujiwara M, Morgan K, Leijendekker R, van der Sluijs P, van Os CH, Deen PM. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J Clin Invest 102: 57–66, 1998. doi: 10.1172/JCI2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller BM, Kistner U, Veh RW, Cases-Langhoff C, Becker B, Gundelfinger ED, Garner CC. Molecular characterization and spatial distribution of SAP97, a novel presynaptic protein homologous to SAP90 and the Drosophila discs-large tumor suppressor protein. J Neurosci 15: 2354–2366, 1995. doi: 10.1523/JNEUROSCI.15-03-02354.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nikandrova YA, Jiao Y, Baucum AJ, Tavalin SJ, Colbran RJ. Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J Biol Chem 285: 923–934, 2010. doi: 10.1074/jbc.M109.033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noda Y, Horikawa S, Furukawa T, Hirai K, Katayama Y, Asai T, Kuwahara M, Katagiri K, Kinashi T, Hattori M, Minato N, Sasaki S. Aquaporin-2 trafficking is regulated by PDZ-domain containing protein SPA-1. FEBS Lett 568: 139–145, 2004. doi: 10.1016/j.febslet.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 40.Noda Y, Sasaki S. Trafficking mechanism of water channel aquaporin-2. Biol Cell 97: 885–892, 2005. doi: 10.1042/BC20040120. [DOI] [PubMed] [Google Scholar]
- 41.Nooh MM, Naren AP, Kim SJ, Xiang YK, Bahouth SW. SAP97 controls the trafficking and resensitization of the β-1-adrenergic receptor through its PDZ2 and I3 domains. PLoS One 8: e63379, 2013. doi: 10.1371/journal.pone.0063379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nooh MM, Chumpia MM, Hamilton TB, Bahouth SW. Sorting of β1-adrenergic receptors is mediated by pathways that are either dependent on or independent of type I PDZ, protein kinase A (PKA), and SAP97. J Biol Chem 289: 2277–2294, 2014. doi: 10.1074/jbc.M113.513481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nooh MM, Bahouth SW. Two barcodes encoded by the type-1 PDZ and by phospho-Ser312 regulate retromer/WASH-mediated sorting of the ß1-adrenergic receptor from endosomes to the plasma membrane. Cell Signal 29: 192–208, 2017. doi: 10.1016/j.cellsig.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 44.Nooh MM, Bahouth SW. Visualization and quantification of GPCR trafficking in mammalian cells by confocal microscopy. Methods Cell Biol 142: 67–78, 2017. doi: 10.1016/bs.mcb.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 46.Pavlos NJ, Friedman PA. GPCR signaling and trafficking: the long and short of it. Trends Endocrinol Metab 28: 213–226, 2017. doi: 10.1016/j.tem.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren H, Yang B, Ruiz JA, Efe O, Ilori TO, Sands JM, Klein JD. Phosphatase inhibition increases AQP2 accumulation in the rat IMCD apical plasma membrane. Am J Physiol Renal Physiol 311: F1189–F1197, 2016. doi: 10.1152/ajprenal.00150.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rice WL, Zhang Y, Chen Y, Matsuzaki T, Brown D, Lu HA. Differential phosphorylation dependent trafficking of AQP2 in LLCPK1 cells. PLoS One 7: e32843, 2012. doi: 10.1371/journal.pone.0032843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stefan E, Wiesner B, Baillie GS, Mollajew R, Henn V, Lorenz D, Furkert J, Santamaria K, Nedvetsky P, Hundsrucker C, Beyermann M, Krause E, Pohl P, Gall I, MacIntyre AN, Bachmann S, Houslay MD, Rosenthal W, Klussmann E. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol 18: 199–212, 2007. doi: 10.1681/ASN.2006020132. [DOI] [PubMed] [Google Scholar]
- 51.Tamma G, Robben JH, Trimpert C, Boone M, Deen PM. Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am J Physiol Cell Physiol 300: C636–C646, 2011. doi: 10.1152/ajpcell.00433.2009. [DOI] [PubMed] [Google Scholar]
- 52.Thakkar A, Bijnsdorp IV, Geldof AA, Shah GV. Profiling of the calcitonin-calcitonin receptor axis in primary prostate cancer: clinical implications and molecular correlates. Oncol Rep 30: 1265–1274, 2013. doi: 10.3892/or.2013.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ. GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 166: 907–919, 2016. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology 146: 5063–5070, 2005. doi: 10.1210/en.2005-0868. [DOI] [PubMed] [Google Scholar]
- 54a.van Balkom BW, Savelkoul PJ, Markovich D, Hofman E, Nielsen S, van der Sluijs P, Deen PM. The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J Biol Chem 277: 41473–41479, 2002. doi: 10.1074/jbc.M207525200. [DOI] [PubMed] [Google Scholar]
- 54b.van Os CH, Deen PM. Aquaporin-2 water channel mutations causing nephrogenic diabetes insipidus. Proc Assoc Am Physicians 110: 395–400, 1998. [PubMed] [Google Scholar]
- 55.Wu H, Reuver SM, Kuhlendahl S, Chung WJ, Garner CC. Subcellular targeting and cytoskeletal attachment of SAP97 to the epithelial lateral membrane. J Cell Sci 111: 2365–2376, 1998. [DOI] [PubMed] [Google Scholar]
- 56.Zhou W, Zhang L, Guoxiang X, Mojsilovic-Petrovic J, Takamaya K, Sattler R, Huganir R, Kalb R. GluR1 controls dendrite growth through its binding partner, SAP97. J Neurosci 28: 10220–10233, 2008. doi: 10.1523/JNEUROSCI.3434-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]