

Abstract
Citrate is critical for acid-base homeostasis and to prevent calcium nephrolithiasis. Both metabolic acidosis and hypokalemia decrease citrate excretion and increase expression of Na+-dicarboxylate cotransporter 1 (NaDC1; SLC13A2), the primary protein involved in citrate reabsorption. However, the mechanisms transducing extracellular signals and mediating these responses are incompletely understood. The purpose of the present study was to determine the role of the Na+-coupled electrogenic bicarbonate cotransporter (NBCe1) A variant (NBCe1-A) in citrate metabolism under basal conditions and in response to acid loading and hypokalemia. NBCe1-A deletion increased citrate excretion and decreased NaDC1 expression in the proximal convoluted tubules (PCT) and proximal straight tubules (PST) in the medullary ray (PST-MR) but not in the PST in the outer medulla (PST-OM). Acid loading wild-type (WT) mice decreased citrate excretion. NaDC1 expression increased only in the PCT and PST-MR and not in the PST-MR. In NBCe1-A knockout (KO) mice, the acid loading change in citrate excretion was unaffected, changes in PCT NaDC1 expression were blocked, and there was an adaptive increase in PST-MR. Hypokalemia in WT mice decreased citrate excretion; NaDC1 expression increased only in the PCT and PST-MR. NBCe1-A KO blocked both the citrate and NaDC1 changes. We conclude that 1) adaptive changes in NaDC1 expression in response to metabolic acidosis and hypokalemia occur specifically in the PCT and PST-MR, i.e., in cortical proximal tubule segments; 2) NBCe1-A is necessary for normal basal, metabolic acidosis and hypokalemia-stimulated citrate metabolism and does so by regulating NaDC1 expression in cortical proximal tubule segments; and 3) adaptive increases in PST-OM NaDC1 expression occur in NBCe1-A KO mice in response to acid loading that do not occur in WT mice.
Keywords: citrate, Na+-dicarboxylate cotransporter 1, proximal tubule
INTRODUCTION
Citrate excretion by the kidneys has a critical role in both acid-base homeostasis and the prevention of calcium nephrolithiasis. The former occurs because citrate is a critical substrate for the tricarboxylic acid (TCA) cycle, where its metabolism results in bicarbonate generation at a 1:3 molar ratio (51). Accordingly, each citrate molecule excreted equates to excretion of three bicarbonate anions. Citrate excretion decreases in response to metabolic acidosis (12, 19, 48), whereby decreasing bicarbonate precursor excretion contributes to the kidney’s maintenance of acid-base homeostasis. Citrate also plays a key role in the prevention of calcium nephrolithiasis. This is because urinary citrate reversibly chelates Ca2+ (20, 45). By doing so, it decreases the ionized Ca2+ concentration in the luminal fluid and urine, which thereby decreases calcium stone development and growth (22, 29). In addition, urinary citrate adsorption on crystal surfaces may induce crystal dissolution directly (11).
The cellular mechanisms regulating citrate excretion are partially understood. Circulating citrate is essentially 100% filtered at the glomerulus and then undergoes regulated reabsorption in the proximal tubule. Distal to the proximal tubule, there is little to no citrate transport (10, 21, 23). In the proximal tubule, citrate reabsorption occurs primarily as result of Na+-coupled citrate transport (10, 21, 23, 60) via the apical integral membrane protein Na+-dicarboxylate cotransporter 1 (NaDC1; SLC13A2) (3, 49, 52, 57). In metabolic acidosis, there is both increased proximal tubule luminal citrate reabsorption (3, 9, 28) and increased cellular metabolism of the reabsorbed citrate, resulting in increased intracellular bicarbonate generation (62). Increased proximal tubule citrate reabsorption is associated with, and likely results, at least in part, from increased NaDC1 expression (3, 42).
Several important aspects of NaDC1’s role in citrate transport remain unclear. Importantly, the molecular mechanisms that transduce extracellular signals and regulate NaDC1 expression are unknown. One potential mechanism that could mediate the NaDC1 response to metabolic acidosis involves the proximal tubule basolateral Na+-coupled electrogenic bicarbonate cotransporter (NBCe1; SLC4A4). We have recently shown that NBCe1 is necessary for normal NaDC1 expression (50). However, deletion of all splice variants of NBCe1 causes 100% perinatal mortality (18, 25, 50), precluding assessment of NBCe1’s role to regulate NaDC1 expression either in the adult kidney or in response to acid loading or K+ restriction. NBCe1 has five known splice variants (8, 43, 55), and the A-variant, NBCe1-A, is the primary variant expressed in the renal proximal tubule. Deletion of only NBCe1-A, in contrast to deletion of all splice variants, does not cause early mortality (36). This enables examination of NBCe1-A’s role in the regulation of NaDC1 expression in adult mice under basal conditions and in response to experimental stimuli.
Thus, the purpose of the present study was to determine the role of NBCe1-A in the signal transduction pathway regulating basal, metabolic acidosis-stimulated and hypokalemia-stimulated citrate excretion. We found under basal conditions that NaDC1 expression is largely present in the brush border at similar intensity throughout the proximal tubules. We then determined whether acid loading and/or hypokalemia altered NaDC1 expression in those proximal tubule segments that express NBCe1-A, i.e., cortical proximal tubule segments, and whether there was a change in expression in proximal tubule segments that do not express NBCe1-A under basal conditions, i.e., the proximal straight tubule (PST) in the outer medulla (PST-OM). Our next experiments determined whether NBCe1-A deletion altered the proximal tubule NaDC1 response to acid loading and hypokalemia Finally, we determined whether changes in citrate excretion paralleled changes in NaDC1 expression. The results of this study show that NBCe1-A has a critical role in the signal transduction mechanisms regulating NaDC1 expression and citrate excretion. They also show that alternative/adaptive mechanisms responsive to acid loading are activated in the PST-OM in response to NBCe1-A deletion.
METHODS
Animals.
We obtained 4-mo-old wild-type (WT) male and female C57BL/6 mice from the Jackson Laboratory. Mice were allowed to acclimate for 3–4 days before study. To study the role of NBCe1-A in the regulation of citrate metabolism and NaDC1 expression, we used mice with selective deletion of NBCe1-A (36). Briefly, NBCe1-A knockout (KO) mice have an 11-bp deletion in the 5′-portion of the coding sequence specific to NBCe1-A (and NBCe1-D) that is not present in the coding sequence of NBCe1-B, NBCe1-C, or NBCe1-E. This causes a downstream frameshift mutation beginning at amino acid residue 30 and the generation of multiple early stop codons beginning at amino acid residue 50 that prevent NBCe1-A protein expression (36). Because NBCe1-D mRNA is not expressed in the mouse kidney (16), we refer to these mice as NBCe1-A KO mice. All breeding involved heterozygous sires with heterozygous dams, and homozygous deletion mice were compared with age-matched WT mice derived from the same breeding regimen. We genotyped all mice using tail-clip samples as previously described (36). The tissues used for the evaluation of the response to metabolic acidosis were randomly selected from a larger set of mice prepared for another project examining ammonia metabolism (36).
The Institutional Animal Care and Use Committees of the University of Florida and North Florida/South Georgia Veterans Health System approved all animal experiments. Trained personnel in the University of Florida College of Medicine Cancer and Genetics Transgenic Animal Core Facility oversaw all animal breeding.
Antibodies.
Antibodies to NaDC1 were obtained from ProteinTech Group (Rosemont, IL). We have previously shown that this antibody was specific to NaDC1 when used for immunohistochemistry studies by showing an absence of detectable immunolabel in mice with NaDC1 deletion (50). This antibody, however, was not specific when used for immunoblot analysis of mouse renal tissues, i.e., the same immunoblot findings were observed in proteins from WT and NaDC1 KO mouse kidneys (data not shown), thereby precluding immunoblot analysis in the present project.
Acid loading.
An acid diet was prepared as we previously described (6, 32, 37, 39, 58, 59). Briefly, we added 0.4 M HCl to powdered standard rodent chow at a ratio of 1 ml/g chow. The control diet was identical except that we substituted deionized water for HCl. Mice were housed in metabolic cages throughout the duration of the experiment, and daily 24-h urine collections were made.
Hypokalemia.
We induced hypokalemia as we have previously described (5, 38), with the exception that mice received the hypokalemia diet for only 4 days. Briefly, hypokalemic mice received K+-control diet (TD.88238, Envigo Teklad Diets, Envigo) for 2–3 days and then were changed to a nominally K+-free diet (TD.88239, Envigo Teklad Diets, Envigo). Control mice received only K+-control diet. Mice were housed in metabolic cages throughout the duration of the experiment, and daily 24-h urine collections were made.
Tissue preparation for immunohistochemistry.
Kidney tissues were fixed for immunohistochemistry using standard approaches (24, 33, 34, 37, 39). Briefly, mice were anesthetized with inhalant isoflurane, after which kidneys were preserved by in vivo cardiac perfusion with PBS (pH 7.4) containing 6,000 U/l of Na-heparin and 120 mg/l of lidocaine followed by periodate-lysine-2% paraformaldehyde (PLP), cut transversely into several 2- to 3-mm-thick slices, and immersed for 24–30 h at 4°C in the same fixative. Kidney samples from each animal were embedded in polyester wax made using polyethylene glycol 400 distearate (Polysciences, Warrington, PA) with 10% 1-hexadecanol, and 2-μm-thick sections were cut and mounted on gelatin-coated glass slides.
Immunohistochemistry.
Immunolocalization was accomplished using standard immunoperoxidase procedures we have previously described in detail (25, 34, 35, 39). Briefly, sections were dewaxed in ethanol, rehydrated, heated in Trilogy (Cell Marque, Rocklin, CA) to 88°C for 30 min and then to 96°C for 30 min, cooled for 30 min, and rinsed in PBS. Endogenous peroxidase activity was blocked by incubating sections in 3% H2O2 in distilled water for 45 min. Sections were blocked for 15 min with Serum-Free Protein Block (DakoCytomation) and then incubated at 4°C overnight with primary antibody. Sections were washed in PBS and incubated for 30 min with polymer-linked, peroxidase-conjugated goat anti-rabbit IgG (MACH2, Biocare Medical, Concord, CA), washed again with PBS, and then exposed to diaminobenzidine (DAB) for 5 min. Sections were washed in distilled water, dehydrated with xylene, mounted, and observed by light microscopy. All immunohistochemistry was performed simultaneously in the four sets of experimental mouse kidneys for each model used in this report, that is, for both genotypes fed control versus acid-loading diet or fed K+-control versus K+-free diet. Sections were examined on a Nikon E600 microscope equipped with DIC optics and photographed using a DXM1200F digital camera and ACT-1 software (Nikon).
Quantitative immunohistochemistry.
Quantitative immunohistochemistry was performed as previously described (30). Briefly, high-resolution, 36-megapixel, digital micrographs were taken of defined tubular segments. DIC and other contrast enhancement techniques were not used. We used freely available software (version 1.34s, ImageJ, National Institutes of Health) to quantify pixel intensity across a line drawn from the tubule lumen through the center of an individual cell. Background pixel intensity, calculated as mean pixel intensity in regions of the cell with no detectable immunolabel, was subtracted from absolute pixel intensity to yield net pixel intensity. Total cellular expression was determined by integrating net pixel intensity. All images were obtained and analyzed by an individual blinded to the treatment group of the slides being examined.
Images were obtained of proximal tubule epithelial cells in the following three defined regions to assess axial variations in NaDC1 expression, as we have previously described (34): 1) the proximal convoluted tubule (PCT) in the cortical labyrinth, 2) PST in the medullary ray (PST-MR), and 3) PST-OM. At least four profiles of each tubule segment were quantified in each kidney and were averaged to yield a single measurement. Results were then normalized such that mean expression in PCT segments in the cortical labyrinth in nonstimulated same-sex WT mice was 100.0. All quantitative immunohistochemistry micrographs for either the acid-loading protocol or hypokalemia protocol were obtained in a single microscopy session by a single investigator.
NMR spectroscopy for citrate quantification.
Proton 1D NMR spectra were collected using a 14.1-T NMR magnet equipped with a CP TXI CryoProbe and an Avance II Console (Bruker Biospin, Billerca, MA). Each NMR sample consists of 100 μl [50% (vol/vol), centrifuged and filtered] urine, 80 μl [40% (vol/vol)] deuterated 20× PBS, and 20 μl [10% (vol/vol)] of internal standard (5 mM DSS-D6 and 0.2% NaN3 in D2O, Chenom), making a total volume of 200 μl for a sample. The pH was controlled at 7.2. A 1D NOESY pulse sequence (tnnoesy.c) was used to acquire 1D proton spectra, with a 3-s relaxation delay, 100-ms mixing time, 64 scans, 12-ppm spectral width, and 4 s of acquisition time (54). All experiments were acquired at 25°C. Spectra were processed and analyzed using MestReNova 11.0.0 (Mestrelab Research, S.L., Santiago de Compostela, Spain). They were zero filled to 131,072 points with exponential line broadening of 0.5 Hz and Whittaker Smoother baseline correction. Chemical shifts were calibrated with respect to DSS singlet signal at 0 ppm. Citrate peak multiplets were fitted to a mixed Gaussian/Lorentzian line shape and compared with the DSS standard to determine the concentration.
Statistics.
Results are presented as mean ± SE; n refers to the number of animals studied. Statistical analysis was performed using IBM SPSS Statistics. Statistical comparison of groups with multiple comparisons used multivariate general linear model analysis ANOVA techniques. Specific comparisons used Bonferroni correction. Both male and female mice were studied; in no case did consideration of sex alter the statistical conclusions. P values of <0.05 were taken as statistically significant.
RESULTS
Axial NaDC1 expression.
Our first set of experiments determined NaDC1 immunolabel expression along the proximal tubule in the normal kidney. Immunohistochemistry using a validated anti-NaDC1 antibody showed apical NaDC1 immunolabel throughout the entire proximal tubule. High-power micrographs examining the PCT, PST-MR, and PST-OM showed similar NaDC1 immunolabel intensity in each of these portions of the proximal tubule (Fig. 1). Quantitative immunohistochemistry showed that there were no significant differences in NaDC1 immunolabel between the different regions of the proximal tubule. Thus, at least under baseline conditions, NaDC1 expression does not appear to differ in different regions of the proximal tubule.
Fig. 1.

Na+-dicarboxylate cotransporter 1 (NaDC1) immunolabel in the normal mouse kidney. Top left: low-power micrograph of NaDC1 immunolabel in the normal mouse kidney. Top right: high-power micrograph of the cortical labyrinth. Apical immunolabel was present only in proximal convoluted tubule (PCT) segments. Bottom left: high-power micrograph in the medullary ray in the cortex. Apical immunolabel was present only in proximal straight tubules in the medullary ray (PST-MR). Bottom right: high-power micrograph in the outer stripe of the outer medulla. Apical immunolabel was present only in proximal straight tubules in the outer medulla (PST-OM). In no region was significant NaDC1 immunolabel evident in nonproximal tubule cells. *Proximal tubule lumen. Results are representative of findings in 10 mice.
NaDC1 response to acid loading.
Acid loading decreases citrate excretion (12, 19, 48), and this is associated with increased NaDC1 expression (3, 42). The next set of experiments was designed to determine whether the effect of acid loading on NaDC1 expression was uniform throughout the mouse proximal tubule or whether there was axial heterogeneity in the NaDC1 response. To do so, we compared normal C57BL/6 mice that had been acid loaded for 7 days with control mice (used for the previous experiment examining NaDC1 expression under control conditions) that had been treated identically except that H2O was substituted for HCl.
In acid-loaded C57BL/6 mice, the general pattern of NaDC1 immunolabel was similar to that observed in control diet-fed mice. Apical immunolabel was present only in the proximal tubule, with no detectable expression in nonproximal tubule cells. However, compared control mice, NaDC1 immunolabel intensity appeared increased by acid loading in cortical proximal tubule segments, i.e., the PCT and PST-MR, but not in outer medullary proximal tubule segments, i.e., the PST-OM (Fig. 2).
Fig. 2.

Na+-dicarboxylate cotransporter 1 (NaDC1) immunolabel in acid-loaded normal mouse kidney. Top left: low-power micrograph of NaDC1 immunolabel. Increased immunolabel intensity in the cortex compared with the nonacid-loaded wild-type mouse was apparent. Top right: NaDC1 immunolabel involving the brush border in proximal convoluted tubule (PCT) segments in the cortical labyrinth. Moderate to more intense apical NaDC1 immunolabel was evident. Bottom left: apical NaDC1 immunolabel involving the brush border in proximal straight tubules in the medullary ray (PST-MR). Immunolabel intensity in both PCT and PST-MR appeared greater than in nonacid-loaded mouse kidneys (see Fig. 1). Bottom right: apical NaDC1 immunolabel involving the brush border in proximal straight tubules in the outer medulla (PST-OM). Immunolabel intensity appeared unchanged compared with nonacid-loaded mouse kidneys (see Fig. 1). Results are representative of findings in 12 mice. *Proximal tubule segments. In no region was significant NaDC1 immunolabel evident in nonproximal tubule cells.
Because of the novelty of this qualitative observation, that acidosis increases NaDC1 immunolabel in cortical but not in outer medullary proximal tubule segments, we used unbiased quantitative immunohistochemical approaches (30, 34, 64) to further assess NaDC1 immunolabel intensity in specific proximal tubule portions. Figure 3 shows these data. Acid loading increased NaDC1 immunolabel expression significantly in the PCT (P < 0.001) and PST-MR (P < 0.001) but did not alter PST-OM NaDC1 immunolabel significantly [P = not significant (NS)]. This axial heterogeneity indicates that differing signaling mechanisms are triggered by acid loading in cortical proximal tubule segments, i.e., the PCT and PST-MR, versus outer medullary PT segments, i.e., the PST-OM.
Fig. 3.
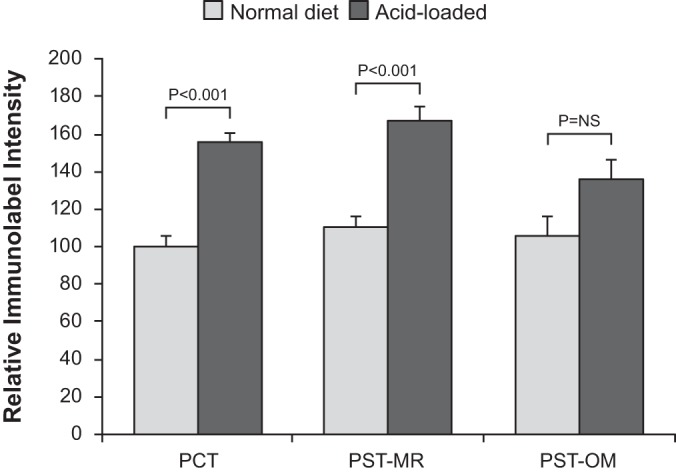
Quantitative assessment of Na+-dicarboxylate cotransporter 1 (NaDC1) expression in the proximal tubule of normal animals comparing basal conditions with an acid-loading diet. Cell-specific NaDC1 immunolabel intensity was determined in the proximal convoluted tubules (PCT), proximal straight tubules in the medullary ray (PST-MR), and proximal straight tubules in the outer medulla (PST-OM) of normal mice that received either control or acid-loading diet (representative immunohistochemistry shown in Figs. 1 and 2). Acid loading increased expression significantly in the PCT and PST-MR but not in the PST-OM. n = 10 normal diet-fed mice and 12 acid diet-fed mice. NS, not significant.
NBCe1-A regulates basal NaDC1 expression.
The sites of altered NaDC1 expression during acid loading, the cortical proximal tubule segments, PCT and PST-MR, correspond to the location of NBCe1-A (14, 36, 44, 56, 66). NBCe1-A is the primary NBCe1 splice variant present in the kidney (16, 31, 55), and it has a key role in determining the proximal tubule ammonia metabolism response to acid loading (36). Because ammonia and citrate metabolism are tightly correlated, we postulated that NBCe1-A might also mediate the NaDC1 response to acid loading.
To test this, we determined the effect of NBCe1-A deletion on NaDC1 expression. Low-power micrographs of the NBCe1-A KO kidney identified only faint apical NaDC1 immunolabel in the cortical proximal tubule segments, but intact expression in proximal tubule segments in the outer medulla; high-power micrographs confirmed these observations (Fig. 4). Compared with WT littermates, NBCe1-A deletion significantly decreased overall NaDC1 expression (P < 0.001 by ANOVA); however, this effect was present only in cortical proximal tubule segments, i.e., PCT (n = 8 WT mice and 8 KO mice, P < 0.001) and PST-MR (n = 8 WT mice and 8 KO mice, P < 0.001); NaDC1 immunolabel intensity in the PST-OM did not differ significantly between WT and KO mice (n = 8 WT mice and 8 KO mice, P = NS).
Fig. 4.

Na+-dicarboxylate cotransporter 1 (NaDC1) immunolabel expression in kidneys from Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) knockout (KO) mice on a normal diet. Top left: low-power micrograph of NaDC1 immunolabel in a NBCe1-A KO kidney. Relatively low cortical immunolabel, but intact outer medullary NaDC1 immunolabel, was evident. Top right: high-power micrograph of NaDC1 immunolabel in proximal convoluted tubules (PCT) illustrating low-intensity apical NaDC1 immunolabel. Bottom left: low-intensity apical NaDC1 immunolabel involving the brush border in proximal straight tubules in the medullary ray (PST-MR). Bottom right: modestly more intense apical NaDC1 immunolabel involving the brush border in proximal straight tubules in the outer medulla (PST-OM). Results are representative of findings in 11 separate mice. *Proximal tubule segments. In no region was significant NaDC1 immunolabel evident in nonproximal tubule cells.
NBCe1-A deletion causes a spontaneous and relatively severe metabolic acidosis (36). The present study used a randomly selected subset of mice from our previous report (36); the serum electrolytes from the subset of mice used for this project did not differ significantly from those previously reported (data not shown). Because experimentally induced metabolic acidosis increased NaDC1 expression in the PCT and PST-MR in normal C57BL/6 mice, the decreased NaDC1 expression in cortical proximal tubule segments of spontaneously acidotic NBCe1-A KO mice indicates substantial dysregulation of NaDC1 expression.
NaDC1 response to acid loading in NBCe1-A KO mouse.
We next determined whether NBCe1-A had a role in the NaDC1 response to exogenous acid loading. NaDC1 expression was examined in NBCe1-A KO mice that had been acid loaded for 7 days. Serum bicarbonate levels decreased with acid loading (see Ref. 36; similar findings were observed in the subset of animals used in this study). The mice were apparently healthy and exhibited normal grooming and motor activities. To ensure that any differences seen were specific to NBCe1-A deletion, a parallel set of experiments was performed in WT littermates. The response of WT littermates did not differ either qualitatively or quantitatively from the response observed in control C57BL/6 mice presented above (data not shown). In acid-loaded NBCe1-A KO mice, similar to findings in nonacid-loaded mice, NaDC1 immunolabel was present only in proximal tubule cells. High-power micrographs showed low-intensity immunolabel in the PCT and moderate-level intensity immunolabel in the PST-MR and PST-OM (Fig. 5). Compared with nonacid-loaded NBCe1-A KO mice, immunolabel intensity appeared more intense in the PST-MR and PST-OM but not in the PCT.
Fig. 5.

Na+-dicarboxylate cotransporter 1 (NaDC1) immunolabel in acid-loaded Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) knockout (KO) mouse kidneys. Top left: low-power micrograph of NaDC1 immunolabel in an acid-loaded NBCe1-A KO kidney. Low-intensity NaDC1 immunolabel was present in the cortical labyrinth, where intensity appeared more intense compared with that observed in nonacid-loaded KO mice (see Fig. 4) in proximal straight tubule segments in both the medullary ray (PST-MR) and outer medulla (PST-OM). Top right: higher magnification of low-intensity apical NaDC1 immunolabel in proximal convoluted tubules (PCT). Bottom left: substantially more intense apical NaDC1 immunolabel involving the brush border in the PST-MR compared with that observed in nonacid-loaded KO mice (see Fig. 4). Bottom right: intense apical NaDC1 immunolabel involving the brush border in the PST-OM. Results are representative of findings in 11 separate mice. *Proximal tubule segments. In no region was significant NaDC1 immunolabel evident in nonproximal tubule cells.
Quantitative analysis of the effect of NBCe1-A deletion on the NaDC1 response to acid loading.
Quantitative analysis showed that NBCe1-A KO deletion significantly altered basal and acid-loading NaDC1 expression. First, acid loading only minimally increased PCT NaDC1 expression in NBCe1-A KO mice (normal diet: 44 ± 4 and acid diet: 79 ± 5, n = 8 and 11, respectively, P < 0.001). This contrasts with the finding that acid loading substantially increased PCT NaDC1 immunolabel intensity in both normal C57BL/6 mice (vide supra) and WT littermates (normal diet: 100 ± 4 and acid diet: 207 ± 11, n = 8 and 11, respectively, P < 0.001). NBCe1-A deletion significantly blunted the magnitude of the PCT NaDC1 response to acid loading (P < 0.001 by ANOVA). Figure 6 shows these quantitative findings. Thus, NBCe1-A is necessary for the normal PCT NaDC1 response to acid loading.
Fig. 6.

Effect of Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) deletion on the Na+-dicarboxylate cotransporter 1 (NaDC1) response to acid loading in proximal convoluted tubules (PCT). Left: quantitative analysis of NaDC1 immunolabel in the PCT of wild-type (WT) and NBCe1-A knockout (KO) mice under control conditions and after acid loading for 7 days. NBCe1-A deletion decreased NaDC1 immunolabel intensity significantly both under control conditions and after acid loading. Acid loading increased NaDC1 expression significantly in both genotypes. Right: the increase in NaDC1 immunolabel intensity in response to acid loading was significantly less in NBCe1-A KO mice than in WT mice. n = 8 for WT mice on normal and acid-loading diets and n = 11 for KO mice on normal and acid-loading diets.
The effect of NBCe1-A deletion on the NaDC1 response to acid loading was different in the PST-MR. In this portion of the proximal tubule, acid loading increased NaDC1 expression significantly in both WT mice (normal diet: 153 ± 7 and acid diet: 228 ± 13, n = 7 and 11, respectively, P < 0.002) and NBCe1-A KO mice (normal diet: 61 ± 7 and acid diet: 182 ± 22, n = 7 and 10 in each group, P < 0.001). NBCe1-A deletion did not alter the response to acid loading significantly (P = NS by ANOVA; Fig. 7). Thus, in contrast to the PCT, in the PST-MR, NBCe1-A expression is not necessary for the NaDC1 response to acid loading.
Fig. 7.

Effect of Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) deletion on the Na+-dicarboxylate cotransporter 1 (NaDC1) response to acid loading in proximal straight tubules in the medullary ray (PST-MR). Left: quantitative analysis of NaDC1 expression in wild-type (WT) and NBCe1-A knockout (KO) mice under control conditions and after acid loading. NBCe1-A deletion decreased NaDC1 expression under control conditions and after acid loading. Acid loading increased NaDC1 expression in both WT and NBCe1-A KO mice. Right: the increase in response to acid loading did not differ significantly between WT and NBCe1-A KO mice. n = 7 for WT mice on normal and acid-loading diets and n = 11 for KO mice on normal and acid-loading diets. NS, not significant.
Finally, the response of the PST-OM differed from both the PCT and PST-MR. In this region of the proximal tubule, where NBCe1-A is not expressed, acid loading increased NaDC1 immunolabel significantly in NBCe1-A KO mice (normal diet: 180 ± 17 and acid diet: 266 ± 15, N = 8 and 12, respectively, P < 0.01; Fig. 8). This contrasts with findings in WT mice where acid loading did not alter expression significantly (normal diet: 185 ± 23 and acid diet: 202 ± 14, n = 8 and 11, respectively, P = NS). This effect of NBCe1-A deletion to induce an NaDC1 response to acid loading in the PST-OM was statistically significant (P < 0.05 by ANOVA).
Fig. 8.

Effect of Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) deletion on the Na+-dicarboxylate cotransporter 1 (NaDC1) response to acid loading in proximal straight tubules in the outer medulla (PST-OM). Left: quantitative analysis of NaDC1 expression in the PST-OM in WT and NBCe1-A KO mice under control conditions and after acid loading. NBCe1-A deletion did not alter NaDC1 expression under control conditions significantly. Acid loading increased NaDC1 expression significantly in NBCe1-A KO mice but not in WT mice. Right: the NaDC1 response in the PST-OM to acid loading was significantly greater in NBCe1-A KO mice than in WT mice. n = 8 for WT mice on normal and acid-loading diets, n = 11 for KO mice under control conditions, and n = 12 for NBCe1-A KO mice after acid loading. NS, not significant.
Effect of acid loading and NBCe1-A deletion on citrate excretion.
Finally, because NaDC1 is a primary determinant of citrate excretion, we determined the effect of acid loading and NBCe1-A deletion on urinary citrate excretion. Under basal conditions, citrate excretion in WT mice averaged 8.63 ± 1.59 µmol/day (n = 11). Citrate excretion by NBCe1-A KO mice under basal conditions was significantly greater, averaging 21.81 ± 4.74 µmol/day (n = 11 in each group, P < 0.02). Mice were then acid loaded for 7 days, and citrate excretion was measured. In WT mice, acid loading decreased citrate excretion significantly, to 0.12 ± 0.02 µmol/day (n = 11, P < 0.001 by paired t-test). In NBCe1-A KO mice, acid loading also decreased citrate excretion significantly, to 0.28 ± 0.15 µmol/day (n = 11, P < 0.001). After acid loading, urinary citrate did not differ significantly between WT and KO mice (P = NS). Also, the relative change in citrate excretion was not altered significantly by NBCe1-A deletion (WT mice: −97.9 ± 0.6% and KO mice: −98.2 ± 0.7%, n = 11 in each group, P = NS). Figure 9 shows these results. Thus, in WT mice, acid loading increases NaDC1 expression in cortical proximal tubule segments and decreases citrate excretion. In NBCe1-A KO mice, acid loading increases NaDC1 expression in a pattern different from WT mice, resulting in an intact ability to decrease citrate excretion in response to acid loading to identical levels as observed in WT mice.
Fig. 9.
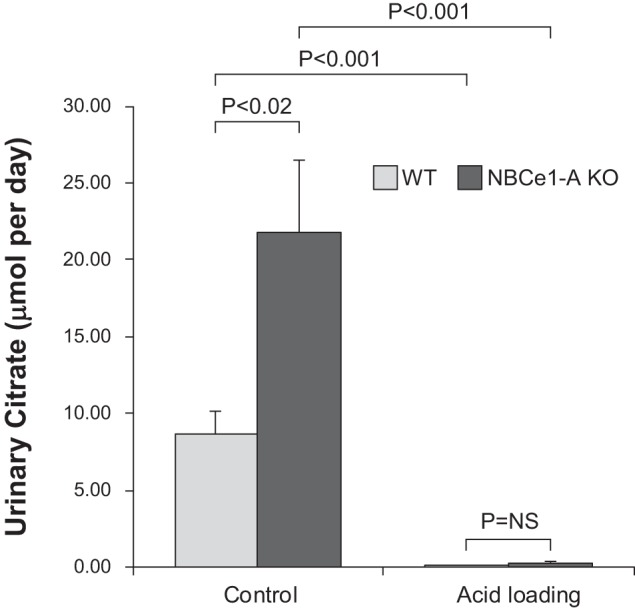
Effect of acid loading and Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) deletion on urinary citrate excretion. Urinary citrate excretion was determined by NMR spectroscopy of 24-h urine collections in wild-type (WT) and NBCe1-A knockout (KO) mice before and after acid loading for 7 days. Urinary citrate excretion was significantly greater in NBCe1-A KO mice than in WT mice fed a normal diet. Acid loading significantly suppressed citrate excretion in both WT and NBCe1-A KO mice. After acid loading, there was no significant difference in citrate excretion between WT and NBCe1-A KO mice. n = 11 mice for each genotype. NS, not significant.
Effect of hypokalemia on NaDC1 expression in WT mice.
Another common cause of altered citrate metabolism is hypokalemia (12, 15, 17). To determine whether the finding that acid loading increased NaDC1 expression only in cortical proximal tubule segments in WT mice was specific to acid loading or whether it might reflect a generalized finding regarding NaDC1 regulation, we determined the effect of hypokalemia on NaDC1 expression along the proximal tubule. Exposure of both WT and NBCe1-A KO mice to a K+-free diet for 4 days decreased plasma K+ significantly in both genotypes (data not shown). Immunohistochemistry of the hypokalemic WT kidney showed increased NaDC1 immunolabel intensity in the PCT and PST-MR but not in the PST-OM (Fig. 10). Quantitative immunohistochemistry confirmed these qualitative observations. Thus, hypokalemia, just as does metabolic acidosis, increases NaDC1 expression only in cortical proximal tubule segments in the mouse. Notably, this is the site of NBCe1-A expression.
Fig. 10.
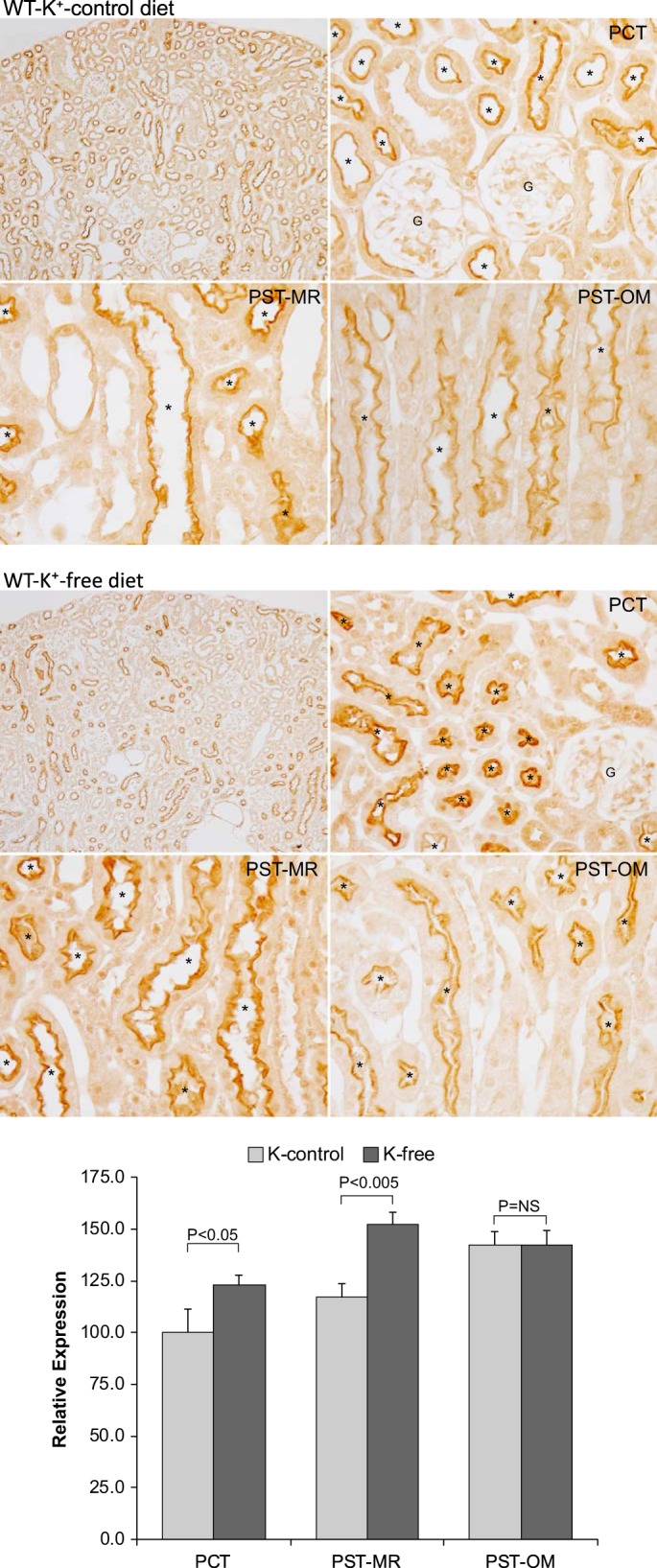
Effect of hypokalemia on Na+-dicarboxylate cotransporter 1 (NaDC1) expression in wild-type (WT) mice. Top and middle: NaDC1 immunohistochemistry in WT mice on the K+ control diet (top) and after the K+-free diet (middle) for 4 days. Bottom: results of quantitative immunohistochemistry for NaDC1 immunolabel. Results are normalized to mean proximal convoluted tubule (PCT) expression in WT mice on the K+ control diet equal to 100.0. K+-free diet increased NaDC1 expression significantly in PCT and proximal straight tubules in the medullary ray (PST-MR) but not in proximal straight tubules in the in the outer medulla (PST-OM). Results are from 5 mice on the K+ control diet and 6 mice on the K+-free diet. *Proximal tubule.
Effect of hypokalemia on NaDC1 expression in NBCe1-A KO mice.
The NaDC1 response to hypokalemia in NBCe1-A KO mice differed significantly from that observed in WT mice. NaDC1 immunolabel intensity did not appear to differ in any proximal tubule segment between that observed in mice exposed to K+-control diet and those exposed to K+-free diet (Fig. 11). Quantitative immunohistochemistry confirmed these qualitative findings; hypokalemia did not significantly alter NaDC1 expression in the PCT, PST-MR, or PST-OM (P = NS for each comparison). Comparison of WT and NBCe1-A KO mouse responses to hypokalemia showed that NBCe1-A deletion significantly altered the response to hypokalemia in the PCT and PST-MR (n = 5 K+-control diet and 6 K+-free diet; PCT: P < 0.05 and PST-MR: P < 0.05) but did not all alter the response in the PST-OM (n = 5 K+-control diet and 6 K+-free diet, P = NS).
Fig. 11.
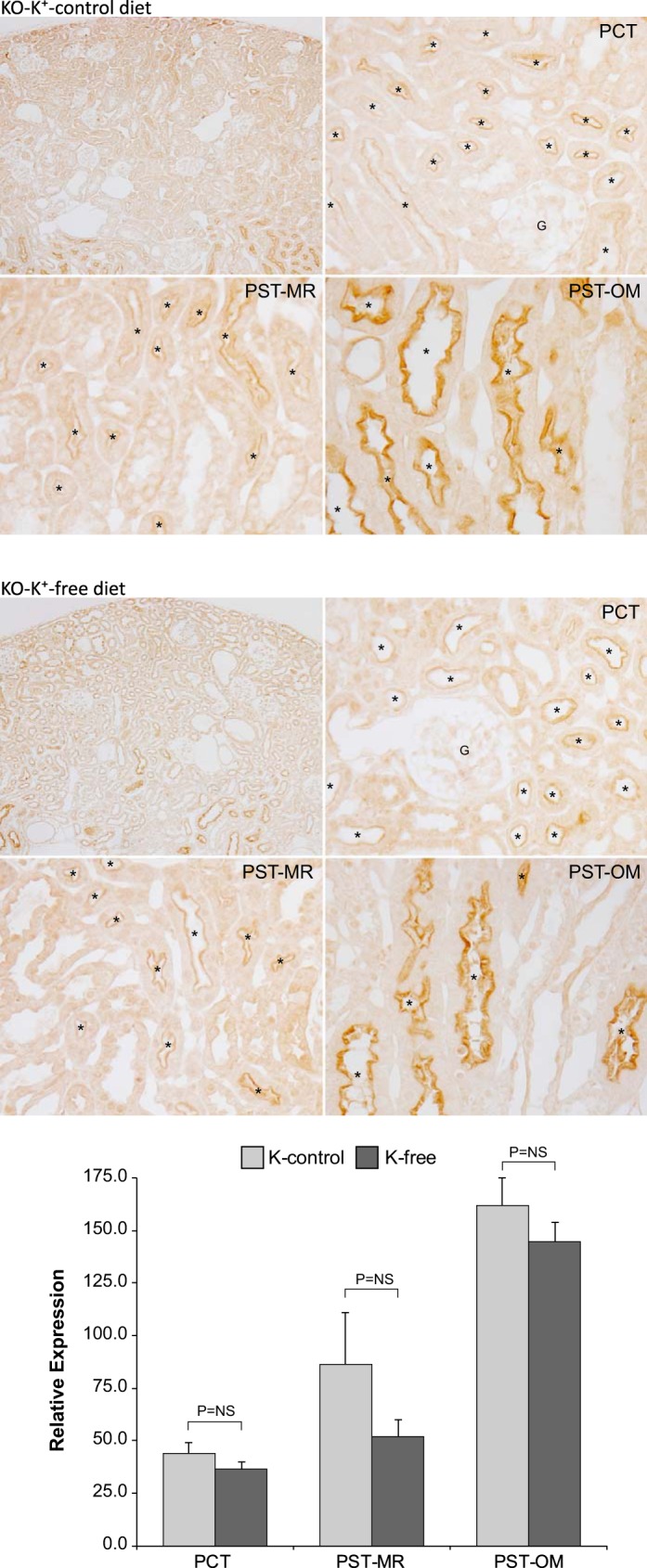
Na+-dicarboxylate cotransporter 1 (NaDC1) immunohistochemistry in response to hypokalemia in Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) knockout (KO) mice. Top and middle: NaDC1 immunohistochemistry in NBCe1-A KO mice on the K+control diet (top) and after the K+-free diet (middle) for 4 days. Bottom: results of quantitative immunohistochemistry for NaDC1 immunolabel. Results are normalized to mean proximal convoluted tubule (PCT) expression in wild-type (WT) mice on the K+ control diet equal to 100.0. The K+-free diet did not alter NaDC1 expression significantly in either the PCT, proximal straight tubules in the medullary ray (PST-MR), or proximal straight tubules in the in the outer medulla (PST-OM). Results are from 5 mice on the K+ control diet and 6 mice on the K+-free diet. *Proximal tubule.
Effect of NBCe1-A deletion on citrate response to hypokalemia.
The typical response to hypokalemia is decreased citrate excretion. We determined the effect of NBCe1-A deletion on this response by comparing citrate excretion in mice on the last day of the K+-control diet and then on the last day of the K+-free diet. WT mice exhibited the expected response, decreasing from 72 ± 10.6 to 3.3 ± 2.4 µmol/day (n = 6, P < 0.001; Fig. 12). In NBCe1-A mice, citrate excretion on the last day of the K+-control diet was significantly greater than in WT mice (KO mice: 96 ± 21.0 µmol/day, n = 6, P < 0.05 vs. WT mice). Hypokalemia decreased citrate excretion in NBCe1-A KO mice (16.6 ± 8.9 µmol/day, n = 6, P < 0.001 vs. K+-control diet), but citrate excretion in response to the K+-free diet was significantly greater than observed in WT mice (P < 0.01). The relative decrease in citrate excretion induced by the K+-free diet was significantly blunted by NBCe1-A KO (WT mice: 95 ± 3% and KO mice: 83 ± 6%, n = 6 in each group, P < 0.002).
Fig. 12.

Effect of Na+-coupled electrogenic bicarbonate cotransporter A variant (NBCe1-A) deletion on the urinary citrate response to a K+-free diet. Left: urine citrate excretion on the last day of the K+-control diet and then on day 4 of the K+-free diet in wild-type (WT) and NBCe1-A knockout (KO) mice. NBCe1-A deletion increased urine citrate excretion significantly in mice fed either the K+ control diet or K+-free diet. Right: the percent decrease in citrate excretion in response to the K+-free diet was significantly less in NBCe1-A KO mice than in WT mice. Results are from 6 WT mice and 6 KO mice.
DISCUSSION
This study identifies several important aspects of NaDC1 and citrate excretion as well as the role of NBCe1-A to regulate each of these processes. First, this study shows that expression of the primary protein involved in the regulation of urinary citrate excretion, NaDC1, in response to acid loading and hypokalemia exhibits significant axial heterogeneity along the proximal tubule. In WT mice, both experimental conditions increased expression in the PCT and PST-MR, i.e., in cortical proximal tubule segments, but not in the PST-OM. Second, this study shows that NBCe1-A mediates a critical role in the regulation of NaDC1 expression, namely, NBCe1-A is necessary for both the normal basal levels of NaDC1 in cortical proximal tubule segments, the PCT and PST-MR, and it is necessary for increased NaDC1 expression in the PCT in response to both acid loading and hypokalemia. Third, in the absence of NBCe1-A, alternative regulatory mechanisms, which are not active in the WT mouse, enable an increase in PST-OM NaDC1 expression in response to acid loading. Changes in urinary citrate excretion paralleled these NaDC1 findings under basal conditions, in response to acid loading and in response to hypokalemia. These findings significantly advance our understanding of renal citrate metabolism.
The first major finding in the present study is that there is significant axial heterogeneity in the proximal tubule NaDC1 response to metabolic acidosis and hypokalemia. In normal mice, both acid loading and hypokalemia increased NaDC1 immunolabel only in the PCT and PST-MR and not in the PST-OM. This parallels findings that metabolic acidosis has greater effects on another critical proximal tubule function, ammoniagenesis, in the earlier portions of the rat proximal tubule, the S1 and S2 segments, than in the S3 segment (46, 47, 67). This parallel regulation suggests similar mechanisms may be involved in the regulation of these two key proximal tubule functions in acid-base homeostasis.
At least one of the signaling pathways regulating cortical proximal tubule segment NaDC1 expression involves NBCe1-A. NBCe1-A deletion decreased NaDC1 expression in cortical proximal tubule segments, i.e., PCT and PST-MR, and its deletion significantly impaired the PCT NaDC1 response to acid loading. These effects occurred despite the concomitant metabolic acidosis that NBCe1-A deletion causes, whereas metabolic acidosis in mice with intact NBCe1-A expression increases PCT and PST-MR NaDC1 expression. These effects of NBCe1-A to regulate NaDC1 expression completely correspond with the cellular localization of NBCe1-A, which is expressed at detectable levels only in the PCT and PST-MR in the rat (14, 44, 56, 65), rabbit (1, 56), human (66), and mouse kidney (36).
The observation that NBCe1-A deletion decreases NaDC1 expression in cortical proximal tubule segments, the PCT and PST-MR, appears to be physiologically important. Urinary citrate excretion was almost threefold greater in NBCe1-A KO mice than in WT mice under basal conditions. Because reabsorbed citrate is metabolized through the TCA cycle, generating three bicarbonate ions per citrate, increased citrate excretion in NBCe1-A KO mice likely contributes to the metabolic acidosis observed in these mice under basal conditions (Ref. 36 and the present study). The citrate excreted by spontaneously acidotic NBCe1-A KO mice, because of this 1:3 conversion ratio, is equivalent to loss of ~65 µmol/day alkali, which decreases net acid excretion by ~20%. Moreover, since NBCe1-A deletion causes substantial metabolic acidosis (36), which normally essentially abolishes citrate excretion, the finding of increased citrate excretion in NBCe1-A KO mice indicates substantial dysregulation of citrate metabolism. Indeed, citrate excretion by NBCe1-A KO mice under basal conditions was ~200-fold greater than the citrate excretion by acid-loaded WT mice, even though nonacid-loaded NBCe1-A KO mice had more severe metabolic acidosis, i.e., their serum bicarbonate concentration was lower, than did acid-loaded WT mice.
This critical role of NBCe1-A in citrate metabolism appears directly relevant to human conditions. Mutations causing dysfunctional NBCe1 protein result in proximal renal tubular acidosis (RTA) in animal models (18, 36), and they are the only known genetic cause of human familial proximal RTA (2, 13, 26, 27, 31, 61). Studies in people with familial proximal RTA have shown that citrate excretion is inappropriately elevated when considered in view of their spontaneous metabolic acidosis (40). Although genetic identification of the cause of the proximal RTA was not evaluated in Ref. 40, the only known genetic cause of familial proximal RTA involves NBCe1 mutations. Thus, we suggest that NBCe1 mutations underlie the abnormal citrate excretion observed in the previous study (40).
In addition to regulating basal NaDC1 expression, NBCe1-A expression is also a key component of the signaling pathway through which metabolic acidosis increases NaDC1 expression. The present study shows that acid loading increases NaDC1 expression in the PCT, where NBCe1-A is expressed, and that NBCe1-A expression is necessary for this normal response. NBCe1-A also appears to have a key role regulating PST-MR NaDC1 expression. The present study shows that in the presence of NBCe1-A, acid loading increases PST-MR NaDC1 expression. In the absence of NBCe1-A, NaDC1 expression in the PST-MR under nonstimulated conditions was significantly less than in WT mice, either under basal conditions or after acid loading, and was even less than the increased induced by acid loading in WT mice. Because NBCe1-A KO mice under basal conditions had significantly greater metabolic acidosis than did NBCe1-A WT mice after acid loading, this suggests that NBCe1-A was necessary for both basal and acidosis-induced increases in PST-MR NaDC1 expression.
NBCe1-A also has a critical role in NaDC1 and citrate excretion responses to hypokalemia. In WT mice, hypokalemia increased NaDC1 expression in cortical proximal tubule segments, where NBCe1-A is expressed, but not in the PST-OM, where NBCe1-A is not found under normal conditions. NBCe1-A deletion completely blocked these responses in cortical proximal tubule segments. Hypokalemia decreased citrate excretion in WT mice, and NBCe1-A deletion, likely through its effects on NaDC1 expression, significantly blunted this response. Thus, NBCe1-A is necessary for normal NaDC1 and citrate responses to hypokalemia. There also appears to be an NBCe1-A-independent mechanism through which hypokalemia decreases citrate excretion in the absence of detectable changes in NaDC1 expression. Possible etiologies include differences in luminal pH, which can alter citrate reabsorption through mechanisms independent of changes in NaDC1 expression, posttranslational regulation of NaDC1, and the possibility of citrate reabsorption through proteins other than NaDC1.
Several other proteins regulate NaDC1 expression. One is the calcineurin inhibitor-targeted protein cyclophilin (4), which likely mediates the effects of calcineurin inhibitors to cause hypocitraturia (63). Other studies have shown that protein kinase C, Na+/H+ exchanger regulating factor 2, serum and glucocorticoid-inducible kinase, and protein kinase B can regulate NaDC1 (7, 53). Recent studies have also shown that acid stimulation of NaDC1 expression involves endothelin-1/endothelin type B receptor signaling (42) and the protein tyrosine kinase Pyk2 (68). However, NBCe1-A has a unique role in this regulation, as it is the only integral membrane protein presently identified that transduces extracellular signals and enables regulated NaDC1 expression and urinary citrate excretion.
There are several potential mechanisms through which NBCe1-A might regulate NaDC1 expression. First, because NBCe1-A typically functions in a bicarbonate exit mode in the renal proximal tubule, it is possible that NBCe1-A deletion decreases basolateral bicarbonate exit, resulting in intracellular alkalization, and that intracellular pH is the primary mechanism regulating NaDC1 expression. Supporting this possibility is that maneuvers expected to cause intracellular acidification, such as metabolic acidosis and hypokalemia, increase NaDC1 expression and/or activity (3, 41), and that the pH-sensitive cytosolic signaling protein Pyk2 regulates NaDC1 expression (68). It is also possible that decreased basolateral exit via NBCe1-A alters the cytoplasmic Na+ concentration, which may have direct and/or indirect mechanisms that alter NaDC1 expression. Another consideration is that because NBCe1-A is an electrogenic transporter, basolateral plasma membrane voltage may be altered by NBCe1-A deletion, resulting in altered cellular responsiveness. However, it is also possible that NBCe1-A deletion results in a coordinate decrease in apical H+ secretion by Na+/H+ exchanger isoform 3 and/or H+-ATPase and thus does not alter intracellular pH, that basolateral Na+-K+-ATPase prevents changes in intracellular Na+, and that its deletion does not alter basolateral plasma membrane voltage. Thus, it is possible that mechanisms other than those discussed above mediate the effect of NBCe1-A on NaDC1 expression. Finally, it is also possible that lack of NBCe1-A during development alters NaDC1 expression.
The observation that NBCe1-A regulates proximal tubule citrate handling through effects on NaDC1 expression add to the growing list of proximal tubule functions that NBCe1-A regulates. NBCe1-A deletion, in addition to being associated with proximal RTA, also causes impaired proximal tubule ammonia metabolism under basal conditions and, similar to the present study, in response to acid loading (36). Thus, NBCe1-A expression appears to be necessary for multiple components of the proximal tubule contribution to acid-base homeostasis, filtered bicarbonate reabsorption, ammonia metabolism, and the regulation of citrate reabsorption and excretion.
The present study also identifies that NBCe1-A-independent mechanisms can regulate NaDC1 expression. Acid loading did not alter PST-OM NaDC1 expression in the WT mouse, but it did in the NBCe1-A KO mouse. This observation indicates the involvement of a signaling pathway in the PST-OM that regulates NaDC1 expression in NBCe1-A KO mice that was not active in WT mice. At present, the specific signaling pathway in the PST-OM of NBCe1-A KO mice that is activated by acid loading is unclear. This signaling pathway could be induced either as a direct cellular response to NBCe1-A deletion or it could be indirectly induced by the significantly lower serum bicarbonate observed in NBCe1-A KO mice.
In summary, the present study identifies important new aspects related to renal citrate metabolism. In normal mice, acid loading and dietary K+ restriction increase NaDC1 only in cortical proximal tubule segments. NBCe1-A expression is necessary for normal basal NaDC1 expression in the same proximal tubule sites and is necessary for the PCT response to acid loading and to hypokalemia. Thus, NBCe1-A is a key component of the signaling pathways that regulate NaDC1 expression and citrate excretion. Finally, NBCe1-A deletion induces an alternative signaling pathway that is not active in WT mice that enables increased PST-OM NaDC1 expression in response to acid loading. These findings significantly advance our understanding of renal citrate metabolism.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R01-DK-045788 (to I. D. Weiner) and R01-DK-107798 (to I. D. Weiner and J. W. Verlander). A. N. Harris was supported by NIH Grants 5-T32-DK-104721 and 5-K08-DK-120873. Citrate measurements were performed in the McKnight Brain Institute at the National High Magnetic Field Laboratory’s AMRIS Facility, which is supported by National Science Foundation Cooperative Agreement DMR-1157490* and State of Florida Grants DMR-1157490 and DMR-1644779. This work was also supported in part by NIH Grant S10-RR-031637 for magnetic resonance instrumentation and NIH Grant U24-DK-097209 for NMR data analysis.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.O., K.L.W., A.N.H., J.W.V., and I.D.W. conceived and designed research; G.O., K.L.W., A.N.H., H.-W.L., C.C., L.F., R.B.K., and M.E.M. performed experiments; G.O., K.L.W., A.N.H., H.-W.L., C.C., R.B.K., M.E.M., J.W.V., and I.D.W. analyzed data; G.O., K.L.W., A.N.H., H.-W.L., J.W.V., and I.D.W. interpreted results of experiments; G.O., K.L.H., A.N.H., H.-W.L., and I.D.W. prepared figures; A.N.H., H.-W.L., L.F., J.W.V., and I.D.W. edited and revised manuscript; G.O., K.L.W., A.N.H., H.-W.L., M.F.R., R.B.K., M.E.M., J.W.V., and I.D.W. approved final version of manuscript.
ACKNOWLEDGMENTS
L. Fang passed away before the submission of this paper. Her contribution to this study is gratefully acknowledged, and her contribution to our lives will be missed.
REFERENCES
- 1.Abuladze N, Lee I, Newman D, Hwang J, Pushkin A, Kurtz I. Axial heterogeneity of sodium-bicarbonate cotransporter expression in the rabbit proximal tubule. Am J Physiol Renal Physiol 274: F628–F633, 1998. doi: 10.1152/ajprenal.1998.274.3.F628. [DOI] [PubMed] [Google Scholar]
- 2.Alper SL. Familial renal tubular acidosis. J Nephrol 23, Suppl 16: S57–S76, 2010. [PubMed] [Google Scholar]
- 3.Aruga S, Wehrli S, Kaissling B, Moe OW, Preisig PA, Pajor AM, Alpern RJ. Chronic metabolic acidosis increases NaDC-1 mRNA and protein abundance in rat kidney. Kidney Int 58: 206–215, 2000. doi: 10.1046/j.1523-1755.2000.00155.x. [DOI] [PubMed] [Google Scholar]
- 4.Bergeron MJ, Bürzle M, Kovacs G, Simonin A, Hediger MA. Synthesis, maturation, and trafficking of human Na+-dicarboxylate cotransporter NaDC1 requires the chaperone activity of cyclophilin B. J Biol Chem 286: 11242–11253, 2011. doi: 10.1074/jbc.M110.171728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop JM, Lee HW, Handlogten ME, Han KH, Verlander JW, Weiner ID. Intercalated cell-specific Rh B glycoprotein deletion diminishes renal ammonia excretion response to hypokalemia. Am J Physiol Renal Physiol 304: F422–F431, 2013. doi: 10.1152/ajprenal.00301.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop JM, Verlander JW, Lee HW, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the Rhesus glycoprotein, Rh B glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol 299: F1065–F1077, 2010. doi: 10.1152/ajprenal.00277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehmer C, Embark HM, Bauer A, Palmada M, Yun CH, Weinman EJ, Endou H, Cohen P, Lahme S, Bichler KH, Lang F. Stimulation of renal Na+ dicarboxylate cotransporter 1 by Na+/H+ exchanger regulating factor 2, serum and glucocorticoid inducible kinase isoforms, and protein kinase B. Biochem Biophys Res Commun 313: 998–1003, 2004. doi: 10.1016/j.bbrc.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Boron WF, Chen L, Parker MD. Modular structure of sodium-coupled bicarbonate transporters. J Exp Biol 212: 1697–1706, 2009. doi: 10.1242/jeb.028563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brennan S, Hering-Smith K, Hamm LL. Effect of pH on citrate reabsorption in the proximal convoluted tubule. Am J Physiol Renal Physiol 255: F301–F306, 1988. doi: 10.1152/ajprenal.1988.255.2.F301. [DOI] [PubMed] [Google Scholar]
- 10.Brennan TS, Klahr S, Hamm LL. Citrate transport in rabbit nephron. Am J Physiol Renal Physiol 251: F683–F689, 1986. [DOI] [PubMed] [Google Scholar]
- 11.Chung J, Granja I, Taylor MG, Mpourmpakis G, Asplin JR, Rimer JD. Molecular modifiers reveal a mechanism of pathological crystal growth inhibition. Nature 536: 446–450, 2016. doi: 10.1038/nature19062. [DOI] [PubMed] [Google Scholar]
- 12.Crawford MA, Milne MD, Scribner BH. The effects of changes in acid-base balance on urinary citrate in the rat. J Physiol 149: 413–423, 1959. doi: 10.1113/jphysiol.1959.sp006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinour D, Chang MH, Satoh J, Smith BL, Angle N, Knecht A, Serban I, Holtzman EJ, Romero MF. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem 279: 52238–52246, 2004. doi: 10.1074/jbc.M406591200. [DOI] [PubMed] [Google Scholar]
- 14.Endo Y, Yamazaki S, Moriyama N, Li Y, Ariizumi T, Kudo A, Kawakami H, Tanaka Y, Horita S, Yamada H, Seki G, Fujita T. Localization of NBC1 variants in rat kidney. Nephron Physiol 104: 87–94, 2006. doi: 10.1159/000094003. [DOI] [PubMed] [Google Scholar]
- 15.Evans BM, Macintyre I, MacPherson CR, Milne MD. Alkalosis in sodium and potassium depletion; with especial reference to organic acid excretion. Clin Sci 16: 53–65, 1957. [PubMed] [Google Scholar]
- 16.Fang L, Lee H-W, Chen C, Harris AN, Romero MF, Verlander JW, Weiner ID. Expression of the B splice variant of NBCe1 (SLC4A4) in the mouse kidney. Am J Physiol Renal Physiol 315: F417–F428, 2018. doi: 10.1152/ajprenal.00515.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fourman P, Robinson JR. Diminished urinary excretion of citrate during deficiencies of potassium in man. Lancet 265: 656–657, 1953. doi: 10.1016/S0140-6736(53)90375-4. [DOI] [PubMed] [Google Scholar]
- 18.Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/ cotransporter. J Biol Chem 282: 9042–9052, 2007. doi: 10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- 19.Grollman AP, Harrison HC, Harrison HE. The renal excretion of citrate. J Clin Invest 40: 1290–1296, 1961. doi: 10.1172/JCI104358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halperin ML, Cheema Dhadli S, Kamel KS. Physiology of acid-base balance: links with kidney stone prevention. Semin Nephrol 26: 441–446, 2006. doi: 10.1016/j.semnephrol.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Hamm LL. Renal handling of citrate. Kidney Int 38: 728–735, 1990. doi: 10.1038/ki.1990.265. [DOI] [PubMed] [Google Scholar]
- 22.Hamm LL, Hering-Smith KS. Pathophysiology of hypocitraturic nephrolithiasis. Endocrinol Metab Clin North Am 31: 885–93, viii, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol Renal Physiol 253: F595–F605, 1987. doi: 10.1152/ajprenal.1987.253.4.F595. [DOI] [PubMed] [Google Scholar]
- 24.Han KH, Mekala K, Babida V, Kim HY, Handlogten ME, Verlander JW, Weiner ID. Expression of the gas-transporting proteins, Rh B glycoprotein and Rh C glycoprotein, in the murine lung. Am J Physiol Lung Cell Mol Physiol 297: L153–L163, 2009. doi: 10.1152/ajplung.90524.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Handlogten ME, Osis G, Lee HW, Romero MF, Verlander JW, Weiner ID. NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal Physiol 309: F658–F666, 2015. doi: 10.1152/ajprenal.00219.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Igarashi T, Inatomi J, Sekine T, Cha SH, Kanai Y, Kunimi M, Tsukamoto K, Satoh H, Shimadzu M, Tozawa F, Mori T, Shiobara M, Seki G, Endou H. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet 23: 264–266, 1999. doi: 10.1038/15440. [DOI] [PubMed] [Google Scholar]
- 27.Inatomi J, Horita S, Braverman N, Sekine T, Yamada H, Suzuki Y, Kawahara K, Moriyama N, Kudo A, Kawakami H, Shimadzu M, Endou H, Fujita T, Seki G, Igarashi T. Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflugers Arch 448: 438–444, 2004. doi: 10.1007/s00424-004-1278-1. [DOI] [PubMed] [Google Scholar]
- 28.Jenkins AD, Dousa TP, Smith LH. Transport of citrate across renal brush border membrane: effects of dietary acid and alkali loading. Am J Physiol Renal Physiol 249: F590–F595, 1985. doi: 10.1152/ajprenal.1985.249.4.F590. [DOI] [PubMed] [Google Scholar]
- 29.Khan SR, Kok DJ. Modulators of urinary stone formation. Front Biosci 9: 1450–1482, 2004. doi: 10.2741/1347. [DOI] [PubMed] [Google Scholar]
- 30.Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293: F1238–F1247, 2007. doi: 10.1152/ajprenal.00151.2007. [DOI] [PubMed] [Google Scholar]
- 31.Kurtz I, Zhu Q. Structure, function, and regulation of the SLC4 NBCe1 transporter and its role in causing proximal renal tubular acidosis. Curr Opin Nephrol Hypertens 22: 572–583, 2013. doi: 10.1097/MNH.0b013e328363ff43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009. doi: 10.1152/ajprenal.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HW, Osis G, Handlogten ME, Guo H, Verlander JW, Weiner ID. Effect of dietary protein restriction on renal ammonia metabolism. Am J Physiol Renal Physiol 308: F1463–F1473, 2015. doi: 10.1152/ajprenal.00077.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee HW, Osis G, Handlogten ME, Lamers WH, Chaudhry FA, Verlander JW, Weiner ID. Proximal tubule-specific glutamine synthetase deletion alters basal and acidosis-stimulated ammonia metabolism. Am J Physiol Renal Physiol 310: F1229–F1242, 2016. doi: 10.1152/ajprenal.00547.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HW, Osis G, Handlogten ME, Verlander JW, Weiner ID. Proximal tubule glutamine synthetase expression is necessary for the normal response to dietary protein restriction. Am J Physiol Renal Physiol 313: F116–F125, 2017. doi: 10.1152/ajprenal.00048.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HW, Osis G, Harris AN, Fang L, Romero MF, Handlogten ME, Verlander JW, Weiner ID. NBCe1-A regulates proximal tubule ammonia metabolism under basal conditions and in response to metabolic acidosis. J Am Soc Nephrol 29: 1182–1197, 2018. doi: 10.1681/ASN.2017080935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HW, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID. Effect of intercalated cell-specific Rh C glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 299: F369–F379, 2010. doi: 10.1152/ajprenal.00120.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HW, Verlander JW, Bishop JM, Handlogten ME, Han KH, Weiner ID. Renal ammonia excretion in response to hypokalemia: effect of collecting duct-specific Rh C glycoprotein deletion. Am J Physiol Renal Physiol 304: F410–F421, 2013. doi: 10.1152/ajprenal.00300.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HW, Verlander JW, Handlogten ME, Han K-H, Weiner ID. Effect of collecting duct-specific deletion of both Rh B Glycoprotein (Rhbg) and Rh C Glycoprotein (Rhcg) on renal response to metabolic acidosis. Am J Physiol Renal Physiol 306: F389–F400, 2014. doi: 10.1152/ajprenal.00176.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int 58: 1267–1277, 2000. doi: 10.1046/j.1523-1755.2000.00282.x. [DOI] [PubMed] [Google Scholar]
- 41.Levi M, McDonald LA, Preisig PA, Alpern RJ. Chronic K depletion stimulates rat renal brush-border membrane Na-citrate cotransporter. Am J Physiol Renal Physiol 261: F767–F773, 1991. doi: 10.1152/ajprenal.1991.261.5.F767. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Zacchia M, Tian X, Wan L, Sakamoto A, Yanagisawa M, Alpern RJ, Preisig PA. Acid regulation of NaDC-1 requires a functional endothelin B receptor. Kidney Int 78: 895–904, 2010. doi: 10.1038/ki.2010.264. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Xu JY, Wang DK, Wang L, Chen LM. Cloning and identification of two novel NBCe1 splice variants from mouse reproductive tract tissues: a comparative study of NCBT genes. Genomics 98: 112–119, 2011. doi: 10.1016/j.ygeno.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Maunsbach AB, Vorum H, Kwon TH, Nielsen S, Simonsen B, Choi I, Schmitt BM, Boron WF, Aalkjaer C. Immunoelectron microscopic localization of the electrogenic Na/HCO(3) cotransporter in rat and ambystoma kidney. J Am Soc Nephrol 11: 2179–2189, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Moe OW, Preisig PA. Dual role of citrate in mammalian urine. Curr Opin Nephrol Hypertens 15: 419–424, 2006. doi: 10.1097/01.mnh.0000232882.35469.72. [DOI] [PubMed] [Google Scholar]
- 46.Nonoguchi H, Takehara Y, Endou H. Intra- and inter-nephron heterogeneity of ammoniagenesis in rats: effects of chronic metabolic acidosis and potassium depletion. Pflugers Arch 407: 245–251, 1986. doi: 10.1007/BF00585298. [DOI] [PubMed] [Google Scholar]
- 47.Nonoguchi H, Uchida S, Shiigai T, Endou H. Effect of chronic metabolic acidosis on ammonia production from l-glutamine in microdissected rat nephron segments. Pflugers Arch 403: 229–235, 1985. doi: 10.1007/BF00583592. [DOI] [PubMed] [Google Scholar]
- 48.Oestberg O. Studien uber die Zitronensaureausscheidung der Menschenniere in normalen und pathologischen Zustañnden. Acta Physiol (Oxf) 62: 81–222, 1931. [Google Scholar]
- 49.Ohana E, Shcheynikov N, Moe OW, Muallem S. SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J Am Soc Nephrol 24: 1617–1626, 2013. doi: 10.1681/ASN.2013010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osis G, Handlogten ME, Lee H-W, Hering-Smith KS, Huang W, Romero MF, Verlander JW, Weiner ID. Effect of NBCe1 deletion on renal citrate and 2-oxoglutarate handling. Physiol Rep 4: e12778, 2016. doi: 10.14814/phy2.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277: 30409–30412, 2002. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 52.Pajor AM. Citrate transport by the kidney and intestine. Semin Nephrol 19: 195–200, 1999. [PubMed] [Google Scholar]
- 53.Pajor AM, Sun N. Protein kinase C-mediated regulation of the renal Na(+)/dicarboxylate cotransporter, NaDC-1. Biochim Biophys Acta 1420: 223–230, 1999. doi: 10.1016/S0005-2736(99)00102-9. [DOI] [PubMed] [Google Scholar]
- 54.Ravanbakhsh S, Liu P, Bjorndahl TC, Mandal R, Grant JR, Wilson M, Eisner R, Sinelnikov I, Hu X, Luchinat C, Greiner R, Wishart DS. Accurate, fully-automated NMR spectral profiling for metabolomics. PLoS One 10: e0124219, 2015. doi: 10.1371/journal.pone.0124219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romero MF, Chen AP, Parker MD, Boron WF. The SLC4 family of bicarbonate () transporters. Mol Aspects Med 34: 159–182, 2013. doi: 10.1016/j.mam.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmitt BM, Biemesderfer D, Romero MF, Boulpaep EL, Boron WF. Immunolocalization of the electrogenic Na+- cotransporter in mammalian and amphibian kidney. Am J Physiol Renal Physiol 276: F27–F38, 1999. doi: 10.1152/ajprenal.1999.276.1.F27. [DOI] [PubMed] [Google Scholar]
- 57.Sekine T, Cha SH, Hosoyamada M, Kanai Y, Watanabe N, Furuta Y, Fukuda K, Igarashi T, Endou H. Cloning, functional characterization, and localization of a rat renal Na+-dicarboxylate transporter. Am J Physiol Renal Physiol 275: F298–F305, 1998. doi: 10.1152/ajprenal.1998.275.2.F298. [DOI] [PubMed] [Google Scholar]
- 58.Seshadri RM, Klein JD, Kozlowski S, Sands JM, Kim YH, Han KH, Handlogten ME, Verlander JW, Weiner ID. Renal expression of the ammonia transporters, Rhbg and Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F397–F408, 2006. doi: 10.1152/ajprenal.00162.2005. [DOI] [PubMed] [Google Scholar]
- 59.Seshadri RM, Klein JD, Smith T, Sands JM, Handlogten ME, Verlander JW, Weiner ID. Changes in subcellular distribution of the ammonia transporter, Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F1443–F1452, 2006. doi: 10.1152/ajprenal.00459.2005. [DOI] [PubMed] [Google Scholar]
- 60.Sheridan E, Rumrich G, Ullrich KJ. Reabsorption of dicarboxylic acids from the proximal convolution of rat kidney. Pflugers Arch 399: 18–28, 1983. doi: 10.1007/BF00652517. [DOI] [PubMed] [Google Scholar]
- 61.Shiohara M, Igarashi T, Mori T, Komiyama A. Genetic and long-term data on a patient with permanent isolated proximal renal tubular acidosis. Eur J Pediatr 159: 892–894, 2000. doi: 10.1007/PL00008363. [DOI] [PubMed] [Google Scholar]
- 62.Simpson DP. Pathways of glutamine and organic acid metabolism in renal cortex in chronic metabolic acidosis. J Clin Invest 51: 1969–1978, 1972. doi: 10.1172/JCI107003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stapenhorst L, Sassen R, Beck B, Laube N, Hesse A, Hoppe B. Hypocitraturia as a risk factor for nephrocalcinosis after kidney transplantation. Pediatr Nephrol 20: 652–656, 2005. doi: 10.1007/s00467-005-1831-y. [DOI] [PubMed] [Google Scholar]
- 64.Verlander JW, Chu D, Lee HW, Handlogten ME, Weiner ID. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal Physiol 305: F701–F713, 2013. doi: 10.1152/ajprenal.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang G, Li C, Kim SW, Ring T, Wen J, Djurhuus JC, Wang W, Nielsen S, Frøkiaer J. Ureter obstruction alters expression of renal acid-base transport proteins in rat kidney. Am J Physiol Renal Physiol 295: F497–F506, 2008. doi: 10.1152/ajprenal.00425.2007. [DOI] [PubMed] [Google Scholar]
- 66.Yamada H, Yamazaki S, Moriyama N, Hara C, Horita S, Enomoto Y, Kudo A, Kawakami H, Tanaka Y, Fujita T, Seki G. Localization of NBC-1 variants in human kidney and renal cell carcinoma. Biochem Biophys Res Commun 310: 1213–1218, 2003. doi: 10.1016/j.bbrc.2003.09.147. [DOI] [PubMed] [Google Scholar]
- 67.Yamada H, Nakada J, Aizawa C, Endou H. Intra- and inter-nephron heterogeneity of gluconeogenesis in the rat: effects of chronic metabolic acidosis and potassium depletion. Pflugers Arch 407: 1–7, 1986. doi: 10.1007/BF00580712. [DOI] [PubMed] [Google Scholar]
- 68.Zacchia M, Tian X, Zona E, Alpern RJ, Preisig PA. Acid stimulation of the citrate transporter NaDC-1 requires Pyk2 and ERK1/2 signaling pathways. J Am Soc Nephrol 29: 1720–1730, 2018. doi: 10.1681/ASN.2017121268. [DOI] [PMC free article] [PubMed] [Google Scholar]