

Abstract
Acute kidney injury (AKI) significantly increases the risk of development of chronic kidney disease (CKD), which is closely associated with the severity of AKI. However, the underlying mechanisms for the AKI to CKD transition remain unclear. Several animal models with AKI to CKD transition have been generated and widely used in research; however, none of them exhibit the typical changes in glomerular filtration rate or plasma creatinine, the hallmarks of CKD. In the present study, we developed a novel model with a typical phenotype of AKI to CKD transition in C57BL/6 mice. In this model, life-threatening ischemia-reperfusion injury was performed in one kidney, whereas the contralateral kidney was kept intact to maintain animal survival; then, after 2 wk of recovery, when the renal function of the injured kidney restored above the survival threshold, the contralateral intact kidney was removed. Animals of this two-stage unilateral ischemia-reperfusion injury model with pedicle clamping of 21 and 24 min exhibited an incomplete recovery from AKI and subsequent progression of CKD with characteristics of a progressive decline in glomerular filtration rate, increase in plasma creatinine, worsening of proteinuria, and deleterious histopathological changes, including interstitial fibrosis and glomerulosclerosis. In conclusion, a new model of the AKI to CKD transition was generated in C57BL/6 mice.
Keywords: acute kidney injury, animal model, chronic kidney disease, ischemia-reperfusion injury
INTRODUCTION
Acute kidney injury (AKI), characterized by an abrupt loss of kidney function with a reduction in glomerular filtration rate (GFR) and retention of nitrogenous waste products, is a common clinical syndrome with substantial morbidity, mortality, and cost (1, 35, 45). It is estimated to occur in ~20% of all hospital admissions (6, 21, 36) and in >50% of patients in the intensive care unit (12, 37). AKI was previously considered as a short-term complication without long-term sequelae (9, 24, 32). However, several recent epidemiological studies have provided compelling evidence that AKI significantly increases the risk of development of chronic kidney disease (CKD) (8, 15, 31), and the progression to CKD is significantly associated with the severity of AKI (20, 44, 46). Despite extensive experimental and clinical research, the underlying mechanisms for the AKI to CKD transition have not been clarified. In addition, there is no cure for CKD and no effective treatment available to prevent the AKI to CKD transition. Therefore, further understanding the pathophysiological mechanisms for the AKI to CKD transition is essential.
Renal ischemia-reperfusion injury (IRI) is one of the leading causes of AKI, which temporarily interrupts the supply of oxygen and nutrients to the kidney, initiating a cascade of deleterious cellular and molecular responses primarily in tubular epithelial cells (2, 5, 29, 41). After IRI, the acutely damaged kidney experiences a self-healing process to restore structural integrity and clearance function, but if the injury is too severe, maladaptive repair may result in incomplete recovery and a progression to CKD (5, 19, 47, 48). There are several AKI to CKD transition animal models that have been generated and widely used in research, which have provided significant information on the post-AKI pathophysiological changes and molecular mechanisms (25, 43, 56, 57). However, none of them exhibit the typical changes in GFR or plasma creatinine (PCr), the hallmarks of CKD. In the present study, we developed a new model in C57BL/6 mice with a novel strategy that severe or fatal IRI was induced in one kidney and the contralateral kidney was kept intact; then, after the function of the injured kidney recovered to maintain survival of the animal, the intact kidney was removed. These animals exhibited the typical phenotype of an AKI to CKD transition, including a progressive decline in GFR, increase in PCr, persistent worsening of proteinuria, and continuous deleterious histopathological changes.
METHODS
Animal Models and Protocols
C57BL/6 mice (8–10 wk old, Jackson Laboratory, Bar Harbor, ME) were provided free access of food and water in the animal facility at the University of South Florida. All procedures in this study were approved by the Institutional Animal Care and Use Committee of the University of South Florida.
Protocols
Protocol I: the unilateral IRI model.
Unilateral clamping of the left renal pedicle plus contralateral uninephrectomy was performed as we have previously described (Fig. 1A) (14, 34). Briefly, mice were anesthetized with pentobarbital (50 mg/ml ip), and the body temperature was controlled at 36.8–37.2°C during surgery with a temperature-controlled operating table (no. 03-02, Vestavia Scientific, AL). Both kidneys were exposed through a single abdominal incision. The right kidney was removed, and the left renal pedicle was carefully dissected and clamped with a silver clip (FE690K, AESCULAP) for 15, 18, or 21 min (uIRI-15, uIRI-18, and uIRI-21 min groups), respectively. The wound was sutured after release of the clip. Animals that underwent only right kidney nephrectomy but not left renal pedicle clamping served as controls (uIRI-0 min group).
Fig. 1.

Protocols of the unilateral ischemia-reperfusion injury (IRI), bilateral IRI, and two-stage unilateral IRI models. Unilateral IRI (A) or bilateral IRI (B) was performed at day 0, and kidney function was monitored until day 84. In the two-stage unilateral IRI model (C), the left kidney was subjected to IRI at the beginning of stage I (day 0), and the right kidney was removed at the beginning of stage II (day 14). Biomarkers of chronic kidney disease were assessed until day 98.
Protocol II: the bilateral IRI model.
Bilateral clamping of renal pedicles was performed as we have previously described (Fig. 1B) (52, 53). Animal anesthesia and body temperature control were the same as for protocol I. Bilateral renal pedicles were clamped with silver clips for 15, 18, or 21 min (bIRI-15, bIRI-18, and bIRI-21 min groups), respectively. The wound was sutured after the release of the clip. Sham-operated animals without clamping served as controls (bIRI-0 min group).
Protocol III: the two-stage unilateral IRI model.
The strategy of the two-stage unilateral IRI model is shown in Fig. 1C. At the beginning of stage I (day 1), IRI was induced by unilateral clamping of the left renal pedicle with a silver clip for 15, 18, 21, 24, 27, or 30 min (two-stage uIRI-15, uIRI-18, uIRI-21, uIRI-24, uIRI-27, and uIRI-30 min groups), respectively, under the same conditions of animal anesthesia and body temperature control as in protocols I and II. The right kidney was kept intact. The wound was sutured after the release of the clip, and animals were allowed to recover for 14 days. At the beginning of stage II (day 14), the right kidneys were removed. Mice that were subjected to only right kidney nephrectomy at day 14 but not left renal pedicle clamping at day 1 served as controls (two-stage uIRI-0 min group).
Plasma and Urine Biochemistry
Blood samples (50 µl/each) were collected through the retroorbital venous sinus and centrifuged at 8,000 rpm for 5 min at 4°C to separate plasma (25 µl/each). PCr was measured by HPLC at the O’Brien Center Core of the University of Alabama at Birmingham. Urine samples were collected in metabolic cages for 24 h. The urinary albumin concentration was measured with an ELISA kit (catalog no. ab-108792, Abcam, Cambridge, MA), and the urinary creatinine concentration was also measured by HPLC at the University of Alabama at Birmingham. The urine albumin-to-creatinine ratio (ACR) was calculated by the urinary albumin concentration over urinary creatinine concentration, as we have previously described (52).
GFR Measurement
GFR was measured in conscious mice by the clearance of plasma FITC-sinistrin with a single bolus injection, as we have previously described (33, 50, 52). Briefly, FITC-sinistrin solution (5.6 mg/100 g body wt) was injected through the retroorbital venous sinus, and blood samples (10 µl/each) were then collected with heparinized capillary tubes from the tail vein at 3, 7, 10, 15, 35, 55, and 75 min after the FITC-sinistrin injection. Blood samples were centrifuged at 8,000 rpm for 5 min at 4°C to separate plasma (5 µl/each). The plasma FITC-sinistrin concentration was measured using a plate reader (Cytation3, BioTek) with excitation at 485 nm and emission at 538 nm. GFR was calculated with GraphPad Prism 7 software (GraphPad Software, San Diego, CA).
Blood Pressure Measurement
Mean arterial pressure was measured at the end of experiment as previously described (51, 58). Mice were anesthetized with pentobarbital (50 mg/ml ip), and body temperature was maintained at 37°C with temperature-controlled operating table. After the cannulation of the trachea was performed, the carotid artery was catheterized for blood pressure measurement via a PowerLab system (ADInstruments, Boulder, CO).
Morphological Evaluation With Light Microscopy and Transmission Electron Microscopy
The kidneys were harvested at the end of experiments, fixed in 4% paraformaldehyde solution, and then embedded in paraffin, as previously described (23, 52). Kidney slices (2 µm) were cut and treated with Masson’s trichrome stain or periodic acid-Schiff (PAS) stain. In Masson’s trichrome-stained slices, 10 randomly chosen fields were captured under ×400 magnification, and tubular atrophy, interstitial fibrosis, and inflammatory cell invasion were scored using the following criteria: 0, affecting 0–5% of the renal area; 1, 6–25%; 2, 26–50%; and 3, >50% (23, 30). In PAS-stained slices, 50 randomly chosen glomeruli/group were captured under ×1,000 magnification, and glomerulosclerosis, collapse of glomerular tufts, and dilatation of Bowman’s capsules were graded as present or absent (17, 23).
Ultrastructural changes of renal interstitium and glomerulus were assessed by transmission electron microscopy (TEM), as previously descripted (38, 52). Briefly, fresh kidney pieces (~1 mm3) were fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer overnight, postfixed with 1% osmium tetroxide for 2 h, dehydrated with a series of graded ethanol, and then embedded in resin. Ultrathin sections (100 nm) were cut and examined with TEM (JEM-1400Plus). All tissue processing and electron microscopy imaging were performed at the University of South Florida Health Lisa Muma Weitz Imaging Core Laboratory. Tubulointerstitial fibrosis was evaluated by the width of peritubular interstitium and accumulation of collagen (38). Glomerular injury was assessed by the thickness of the glomerular basement membrane, width of the podocyte foot process, and presence of cellular debris in Bowman's space (52). All morphometric analysis were performed in a blinded manner.
Statistics
Statistical analysis was performed using GraphPad Prism 7 software. The effects of interest were tested using t-test or two-factor ANOVA followed by a multiple-comparisons post hoc test. Data are expressed as means ± SE. The significance value for consideration was P < 0.05.
RESULTS
Protocol I: Kidney Function and Survival Rate in the Unilateral IRI Model
To determine whether the unilateral IRI model induces a transition from AKI to CKD, kidney function was evaluated by measuring PCr and GFR for 12 wk after surgery. PCr increased to 1.73 ± 0.08 mg/dl in the uIRI-15 min group, 2.28 ± 0.13 mg/dl in the uIRI-18 min group, and 2.72 ± 0.07 mg/dl in the uIRI-21 min group at 24 h after unilateral IRI plus contralateral uninephrectomy. PCr of the uIRI-15 min group returned to baseline levels in 3 wk and remained steady until the end of the experiment (Fig. 2A). All animals in the uIRI-18 min group and uIRI-21 min group died within 3 days after surgery (Fig. 2C). In the uIRI-0 min group, GFR decreased by 23.2 ± 4.2% after uninephrectomy without altering PCr. In the uIRI-15 min group, GFR decreased by 49.3 ± 5.4% at 7 days after unilateral IRI plus contralateral uninephrectomy and recovered to a level similar to that of the control group within 3 wk and maintained at a constant level until the end of the experiment (Fig. 2B).
Fig. 2.

Renal function and survival rate of the unilateral ischemia-reperfusion injury (uIRI) and bilateral ischemia-reperfusion injury (bIRI) models. Kidney function of the animals in the uIRI groups was evaluated by plasma creatinine (PCr; A) and glomerular filtration rate (GFR; B). The survival rate of the animals in each group was scored and presented as a percentage (C). n = 6–10/group. *P < 0.01 vs. the uIRI-0 min group. Kidney function of the animals in the bIRI groups was evaluated by PCr (D) and GFR (E). The survival of the mice in each group was scored and presented as a percentage (F). n = 8–10/group. *P < 0.01 vs. the bIRI-0 min group.
Protocol II: Kidney Function and Survival Rate in the Bilateral IRI Model
To determine whether the bilateral IRI model induces a transition from AKI to CKD, kidney function was evaluated by measuring PCr and GFR for 12 wk. PCr increased to 1.23 ± 0.08 mg/dl in the bIRI-15 min group, 1.84 ± 0.12 mg/dl in the bIRI-18 min group, and 2.52 ± 0.17 mg/dl in the bIRI-21 min group at 24 h after bilateral IRI. PCr of the bIRI-15 min and bIRI-18 min groups returned to baseline level in 3 wk and remained at a constant level until the end of the experiment (Fig. 2D). All animals in the bIRI-21 min group died within 3 days after surgery (Fig. 2F). GFR decreased by 30.8 ± 4.7% in the bIRI-15 min group and 37.5 ± 3.6% in the bIRI-18 min group at 7 days after bilateral IRI and then recovered within 4 wk and maintained until the end of experiment (Fig. 2E).
Protocol III: the Two-Stage Unilateral IRI Model
PCr, GFR, and survival rate.
Kidney function of the two-stage unilateral IRI model was continuously monitored by measuring PCr and GFR throughout the entire experiment. Because the right kidneys were kept intact during stage I, PCr was not significantly changed in any groups after unilateral IRI in the left kidneys. After the original insult of IRI-induced AKI, PCr gradually increased from 0.25 ± 0.08 mg/dl at 4 wk to 0.43 ± 0.05 mg/dl at 12 wk in the two-stage uIRI-21 min group and from 0.36 ± 0.09 mg/dl at 4 wk to 0.63 ± 0.09 mg/dl at 12 wk in the two-stage uIRI-24 min group. PCr in the two-stage uIRI-15 min and two-stage uIRI-18 min groups decreased to basal levels and maintained until the end of the experiments (Fig. 3A).
Fig. 3.

Renal function and survival rate of two-stage unilateral ischemia-reperfusion injury (uIRI) model. Renal function of the animals in the two-stage uIRI groups was determined by plasma creatinine (PCr; A) and glomerular filtration rate (GFR; B). The survival rate of the mice in each group was scored and presented as a percentage (C). After removal of the intact right kidneys at day 14, the survival rate was >80% in the two-stage uIRI-21 min group and 63% in the two-stage uIRI-24 min group. All mice in two-stage uIRI-27 min and two-stage uIRI-30 min groups died. All mice in the two-stage uIRI-15 min and two-stage uIRI-18 min group survived. After removal of the intact right kidneys, animals in the two-stage uIRI-21 min and two-stage uIRI-24 min groups exhibited continuous increases in PCr as well as declines in GFR, whereas animals in the two-stage uIRI-15 min and two-stage uIRI-18 min groups exhibited similar levels in PCr and GFR compared with the two-stage uIRI-0 min group. n = 15–16/group. *P < 0.01 vs. the two-stage uIRI-0 min group. The urine albumin-to-creatinine ratio (ACR) was significantly increased in the two-stage uIRI-21 min and two-stage uIRI-24 min groups, whereas it was not significantly changed in the two-stage uIRI-15 min and two-stage uIRI-18 min groups. n = 15–16/group. *P < 0.01 vs. the two-stage uIRI-0 min group.
The changes in GFR were in a similar pattern to PCr. After the original insult of IRI-induced AKI, GFR gradually declined from 133.3 ± 8.4 µl/min at 4 wk to 76.3 ± 8.1 µl/min at 12 wk in the two-stage uIRI-21 min group and from 113.7 ± 12.6 µl/min at 4 wk to 49.5 ± 11.3 µl/min at 12 wk in the two-stage uIRI-24 min group. GFR in the two-stage uIRI-15 min and two-stage uIRI-18 min groups restored to the level similar to the control group and maintained until the end of the experiments (Fig. 3B).
The survival rate was 84% in the two-stage uIRI-21 min group and 75% in the two-stage uIRI-24 min group at the end of the experiments. All mice in the two-stage uIRI-27 min and two-stage uIRI-30 min groups died within 7 days after removal of the right kidneys (Fig. 3C).
Urine ACR.
Urine ACR increased from a baseline level of 9.4 ± 8.2 to 729.1 ± 284.3 μg/mg in the two-stage uIRI-21 min group and from 16.1 ± 9.3 to 1,364.5 ± 180.7 μg/mg in the two-stage uIRI-24 min group at the end of the experiments. In contrast, urine ACR was not significantly changed in the two-stage uIRI-15 min, two-stage uIRI-18 min, and two-stage uIRI-0 min groups (Fig. 3D).
Kidney size and weight.
Kidney size and weight were assessed at the end of the experiments. Compared with the two-stage uIRI-0 min control group, the kidneys of the two-stage uIRI-21 min and two-stage uIRI-24 min groups were much smaller and lighter with granular appearances, whereas it was not significantly different in the two-stage uIRI-15 min or two-stage uIRI-18 min groups (Fig. 4A and Table 1).
Fig. 4.

Histopathological and transmission electron microscopy (TEM) ultrastructural analyses of the two-stage unilateral ischemia-reperfusion injury (uIRI) model. A: representative images of the left kidneys from each two-stage uIRI group. Kidney slices with Masson’s trichrome (B) or periodic acid-Schiff staining (C) showed the typical histopathological features of chronic kidney disease, including tubular atrophy (black star), interstitial fibrosis (yellow triangle), inflammatory cell infiltration (yellow star), glomerular sclerosis (green triangle), collapse of glomerular tufts (green star), and dilated Bowman’s capsule (red star). The TEM images (D) show the characteristic ultrastructural alterations of chronic kidney disease, including peritubular interstitial expansion (yellow line), collagen deposition (yellow star), cellular debris within Bowman’s space (green triangle), glomerular basement membrane thickening (red star), and podocyte foot process effacement (red triangle).
Table 1.
Characteristics of the two-stage uIRI model
| Compartment | Parameter | Two-Stage uIRI-0 Min Group | Two-Stage uIRI-15 Min Group | Two-Stage uIRI-18 Min Group | Two-Stage uIRI-21 Min Group | Two-Stage uIRI-24 Min Group |
|---|---|---|---|---|---|---|
| Tubule and interstitium | Tubular atrophy (0–3) | 0 | 0 | 0 | 3 | 2 |
| Interstitial fibrosis (0–3) | 0 | 0 | 0 | 2 | 3 | |
| Inflammatory cell invasion (0–3) | 0 | 0 | 0 | 1 | 3 | |
| Glomeruli | Glomerulosclerosis (present/absent) | 0/50 | 0/50 | 3/47 | 33/17 | 48/2 |
| Collapse of glomerular tufts (present/absent) | 0/50 | 0/50 | 0/50 | 11/39 | 37/13 | |
| Dilatation of Bowman’s capsules (present/absent) | 0/50 | 0/50 | 2/48 | 16/34 | 41/9 |
There was no significant difference in anesthetized mean arterial pressure among all groups at the end of the experiment. Compared with the two-stage unilateral ischemia-reperfusion (uIRI)-0 min group, body weight and kidney weight were lower in the two-stage uIRI-21 min and two-stage uIRI-24 min groups but was not significantly different in the two-stage uIRI-15 min or two-stage uIRI-18 min groups (n = 11–13).
Fig. 4.

—Continued
Histopathological analysis.
The histological analysis with Masson’s trichrome stain (Fig. 4B) and PAS stain (Fig. 4C) showed the typical morphological changes of CKD, including tubular atrophy, interstitial fibrosis, inflammatory cell infiltration, glomerulosclerosis, collapse of glomerular tufts, and dilated Bowman’s capsule, in the kidney slices of the two-stage uIRI-21 min and two-stage uIRI-24 min groups but not in the kidney slices of the two-stage uIRI-15 min, two-stage uIRI-18 min, or two-stage uIRI-0 min control groups (Table 2).
Table 2.
Histopathological analysis of the two-stage uIRI model
| Groups | Anesthetized Mean Arterial Pressure, mmHg | Body Weight, g | Kidney Weight, g |
|---|---|---|---|
| Two-stage uIRI-0 min | 91 ± 3.4 | 32.6 ± 2.2 | 0.271 ± 0.024 |
| Two-stage uIRI-15 min | 89 ± 4.3 | 31.8 ± 1.7 | 0.263 ± 0.022 |
| Two-stage uIRI-18 min | 88 ± 4.1 | 33.1 ± 1.9 | 0.246 ± 0.017 |
| Two-stage uIRI-21 min | 92 ± 5.4 | 28.4 ± 1.5 | 0.136 ± 0.025* |
| Two-stage uIRI-24 min | 90 ± 3.7 | 25.1 ± 2.1 | 0.083 ± 0.034* |
Tubular atrophy, interstitial fibrosis, inflammatory cell infiltration, glomerulosclerosis, collapse of glomerular tufts, and dilated Bowman’s capsule were observed in the two-stage unilateral ischemia-reperfusion injury (uIRI)-21 min and two-stage uIRI-24 min groups but not in two-stage uIRI-15 min, two-stage uIRI-1 8 min, or two-stage uIRI-0 min control groups. Tubular atrophy, interstitial fibrosis, and inflammatory cell invasion were scored using the following criteria: 0, affecting 0–5% of the renal area; 1, 6–25%; 2, 26–50%; and 3, >50% (n = 10 fields/mice, 11–13 mice/group). Glomerulosclerosis, collapse of glomerular tufts, and dilatation of Bowman’s capsules were graded as present or absent (n = 50 glomeruli/group).
P < 0.01 vs. the two-stage uIRI-0 min group.
TEM ultrastructural analysis.
The TEM experiments showed the characteristic ultrastructural alterations of CKD, including peritubular interstitial expansion, collagen deposition, cellular debris within Bowman’s space, glomerular basement membrane thickening, and podocyte foot process effacement, in the kidneys of the two-stage uIRI-21 min and two-stage uIRI-24 min groups but not in the kidneys of the two-stage uIRI-15 min, two-stage uIRI-18 min, or two-stage uIRI-0 min control groups (Fig. 4D and Table 3).
Table 3.
Transmission electron microscopy ultrastructural analysis of the two-stage uIRI model
| Parameter | Two-Stage uIRI-0 Min Group | Two-Stage uIRI-15 Min Group | Two-Stage uIRI-18 Min Group | Two-Stage uIRI-21 Min Group | Two-Stage uIRI-24 Min Group |
|---|---|---|---|---|---|
| Peritubular interstitium | Normal | Normal | Normal | Wide | Wide |
| Collagen | − | − | − | + | + |
| Cellular debris in Bowman’s space | − | − | − | + | + |
| Glomerular basement membrane thickness, nm | 131 ± 37 | 164 ± 52 | 148 ± 33 | 286 ± 61* | 362 ± 89* |
| Foot process width, nm | 136 ± 44 | 132 ± 35 | 182 ± 52 | 624 ± 78* | 962 ± 113* |
Peritubular interstitium expansion, collagen deposition, cellular debris within Bowman’s space, glomerular basement membrane thickening, and podocyte foot process effacement were detected in the two-stage unilateral ischemia-reperfusion injury (uIRI)-21 min and two-stage uIRI-24 min groups but not in two-stage uIRI-15 min, two-stage uIRI-18 min, or two-stage uIRI-0 min control groups (n = 5 fields/mice, 11–13 mice/group).
P < 0.01 vs. the two-stage uIRI-0 min group.
DISCUSSION
In the present study, we generated a new model of AKI to CKD transition in C57BL/6 mice. In these animals, renal two-stage unilateral IRI resulted in AKI and subsequent development of CKD with the typical characteristics, including a progressive decline in kidney function, worsening of proteinuria, and deleterious histopathological changes.
The animal models of renal IRI-induced AKI generated by temporary unilateral (14, 27, 34) or bilateral (4, 26, 51, 53) clamping of renal pedicles or renal arteries have been well established and broadly used in experimental studies. Several experimental animal models for AKI to CKD transition have been generated, which are widely used and have advanced our understanding of the pathophysiological mechanisms for the transition from AKI to CKD. However, none of them exhibit the typical changes in GFR and PCr, the hallmarks of AKI to CKD transition (3, 13, 56).
In the present study, IRI was performed at 37°C of body temperature in C57BL/6 mice and kidney function was monitored for over 12 wk. We found an almost complete restoration of kidney function in animals with unilateral clamping of <15 min and bilateral clamping of <18 min. All animals were dead within 72 h when unilateral clamping was >18 min and bilateral clamping was >21 min. As shown in Fig. 5, A and B, based on the severity of AKI, bilateral IRI models and unilateral IRI with contralateral nephrectomy models resulted in either an almost full recovery or death but not a subsequent transition to CKD. In renal IRI models, ischemic duration and kidney temperature are two key determinants for the severity of AKI. The time length of renal pedicle clamping in mice varied from 14 to 90 min, with plasma creatinine values from 1.4 to 2.8 mg/dl, in different laboratories (13, 22, 28, 39, 53). We think that the huge variation in ischemic time and renal injury is mainly because of the kidney temperature during ischemia. A few laboratories controlled body temperature; some just controlled the temperature of the heating pad and others did not control temperature at all. In all renal IRI experiments of our laboratory, we maintained body temperature at 37°C during the entire surgical procedure (49, 51).
Fig. 5.
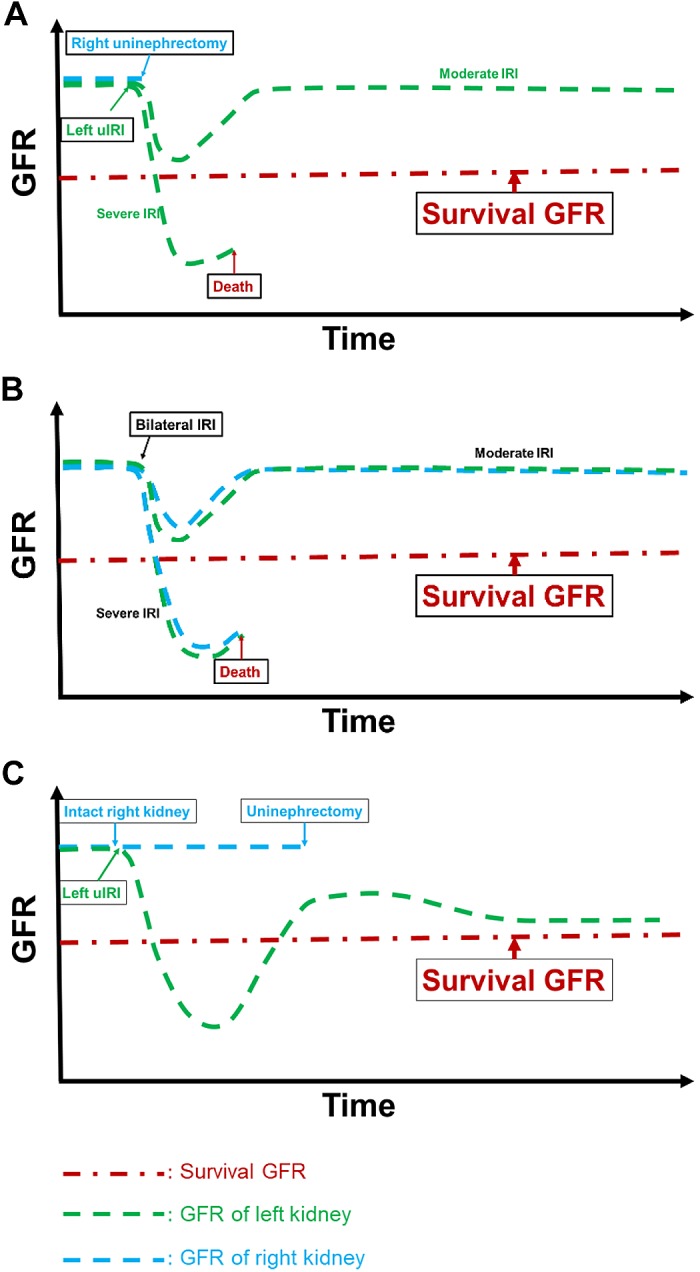
Rational for the acute kidney injury to chronic kidney disease transition in the two-stage unilateral ischemia-reperfusion injury (uIRI) model. The red line suggests a threshold of the glomerular filtration rate (GFR) for the animal to survive. The green line and blue line represent the projected GFR changes for the left and right kidneys. In the uIRI (A) and bilateral IRI (B) models, animals would either exhibit a full recovery in GFR if the injury is moderate or die within a few days if the injury is too severe. In the two-stage uIRI model (C), fatal IRI was induced in the left kidney while the right kidney was kept intact. Even though the GFR of the injured left kidney decreases below the survival threshold, the nonischemic right kidney could still maintain the animal to survive. After the GFR of the injured left kidney recovers above the survival threshold, the intact right kidney was removed. The animals would exhibit a partial recovery followed by a continuous decline in GFR.
Why does the AKI to CKD transition occur in patients but not in these experimental animals? Although not all results from experimental animal models can be extrapolated into clinics, and vice versa, the disease history and systemic responses are quite different between humans and animal models. For example, patients with AKI usually have underlying diseases and complications, whereas animals used for experiments are healthy. An additional reason might be dialysis treatment for those patients who suffer fatal AKI (16, 18, 54). The United States nationwide incidence of hospitalizations with life-threatening (dialysis-requiring) AKI was over 164,000 cases, but the fatality rate for these patients was only 23.5% (18). With the help of medical treatment, like hemodialysis, the majority of patients with deadly AKI could survive. Moreover, these survivors from an episode of dialysis-requiring AKI was associated with a much higher prevalence of subsequent CKD, end-stage renal disease, and long-term mortality (7, 40, 55). Accordingly, the experimental model of unilateral IRI without contralateral nephrectomy has been used to study the AKI to CKD transition (10, 11, 25, 57). In this model, because of the intact contralateral kidneys, animals are able to survive with severe unilateral ischemic AKI and develop to CKD in the IRI kidney. However, systemic biomarkers for renal functions, such as GFR and PCr, cannot be continuously assessed because of the presence of an intact kidney.
We speculate, as shown in Fig. 5C, that GFR of the kidney with severe IRI declines below the survival threshold, whereas the intact contralateral kidney maintains the animal survival, functioning like dialysis in patients with severe AKI. Then, after a period of recovery, when GFR of the injured kidney restores above the survival level, the contralateral intact kidney is removed. Thus, in the present study, we developed a two-stage unilateral IRI model, in which life-threatening IRI was performed in one kidney, whereas the contralateral kidney was kept intact and removed 2 wk later. To figure out the optimal condition that causes the transition to CKD, the severity of unilateral IRI was determined with different ischemic durations at 37°C of body temperature. We found that all mice in the two-stage uIRI-27 min and uIRI-30 min models died within a few days after the removal of the contralateral kidney, suggesting that the renal injury was too severe to recover, whereas the two-stage uIRI-15 min and uIRI-18 min models survived well after the contralateral nephrectomy and the IRI kidney almost completely restored in clearance function and histopathology. Only the animals of the two-stage uIRI-21 min and uIRI-24 min models developed into CKD with the characteristic features after contralateral nephrectomy, including a continuous decline in GFR, elevation in PCr, and exacerbation of proteinuria. Moreover, the IRI kidney exhibited the typical morphological changes of CKD, including contracted size with granular appearance, tubulointerstitial fibrosis, and glomerular and tubular damage. These phenotypes demonstrated that the two-stage uIRI-21 min and uIRI-24 min models induced an incomplete recovery of the IRI kidney from ischemic AKI and subsequent transition to CKD. A similar approach was performed in the studies of Skrypnyk et al. (42, 43), in which BALB/c mice were subjected to unilateral renal ischemia for 30−31 min on a heating platform at 38°C followed by contralateral nephrectomy 8 days later. However, a gradual decrease in GFR or increase in PCr or proteinuria, the hallmarks of CKD, were not observed in these animals, despite the significant expansion in renal fibrosis.
In conclusion, we generated and characterized a novel model in C57BL/6 mice exhibiting the transition from AKI to CKD. In this model, renal two-stage unilateral IRI resulted in an incomplete recovery from ischemic AKI and the subsequent progression of CKD with the characteristics, including progressive decline in kidney function, worsening of proteinuria, and typical morphological changes. The successful development of this experimental model provides an additional tool to investigate the underlying mechanisms and explore the potential therapeutic targets for the AKI to CKD transition.
GRANTS
This work was supported by an American Society of Nephrology Ben J. Lipps Research Fellowship Award (to J. Wei), American Society of Nephrology Ben J. Lipps Research Fellowship Award (to J. Zhang), American Heart Association Career Development Award 18CDA34110441 (to L. Wang), and National Institutes of Health Grants DK-099276, DK-098582, and HL-137987 (to R. Liu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.W. and R.L. conceived and designed research; J.W. and J.Z. performed experiments; J.W. and J.Z. analyzed data; J.Z. interpreted results of experiments; J.W. and J.Z. prepared figures; J.W. and J.Z. drafted manuscript; L.W., S.J., L.F., J.B., and R.L. edited and revised manuscript; J.W., J.Z., L.W., S.J., L.F., J.B., and R.L. approved final version of manuscript.
REFERENCES
- 1.Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med 357: 797–805, 2007. doi: 10.1056/NEJMra064398. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, Kellum JA, Ronco C; Acute Dialysis Quality Initiative XIII Working Group . Cellular and molecular mechanisms of AKI. J Am Soc Nephrol 27: 1288–1299, 2016. doi: 10.1681/ASN.2015070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281: F887–F899, 2001. doi: 10.1152/ajprenal.00050.2001. [DOI] [PubMed] [Google Scholar]
- 4.Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Challiner R, Ritchie JP, Fullwood C, Loughnan P, Hutchison AJ. Incidence and consequence of acute kidney injury in unselected emergency admissions to a large acute UK hospital trust. BMC Nephrol 15: 84, 2014. doi: 10.1186/1471-2369-15-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chawla LS, Bellomo R, Bihorac A, Goldstein SL, Siew ED, Bagshaw SM, Bittleman D, Cruz D, Endre Z, Fitzgerald RL, Forni L, Kane-Gill SL, Hoste E, Koyner J, Liu KD, Macedo E, Mehta R, Murray P, Nadim M, Ostermann M, Palevsky PM, Pannu N, Rosner M, Wald R, Zarbock A, Ronco C, Kellum JA; Acute Disease Quality Initiative Workgroup 16. . Acute kidney disease and renal recovery: consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat Rev Nephrol 13: 241–257, 2017. doi: 10.1038/nrneph.2017.2. [DOI] [PubMed] [Google Scholar]
- 8.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 82: 516–524, 2012. doi: 10.1038/ki.2012.208. [DOI] [PubMed] [Google Scholar]
- 9.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53: 961–973, 2009. doi: 10.1053/j.ajkd.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colombaro V, Jadot I, Declèves AE, Voisin V, Giordano L, Habsch I, Malaisse J, Flamion B, Caron N. Lack of hyaluronidases exacerbates renal post-ischemic injury, inflammation, and fibrosis. Kidney Int 88: 61–71, 2015. doi: 10.1038/ki.2015.53. [DOI] [PubMed] [Google Scholar]
- 11.Danelli L, Madjene LC, Madera-Salcedo I, Gautier G, Pacreau E, Ben Mkaddem S, Charles N, Daugas E, Launay P, Blank U. Early phase mast cell activation determines the chronic outcome of renal ischemia-reperfusion injury. J Immunol 198: 2374–2382, 2017. doi: 10.4049/jimmunol.1601282. [DOI] [PubMed] [Google Scholar]
- 12.Desai AA, Baras J, Berk BB, Nakajima A, Garber AM, Owens D, Chertow GM. Management of acute kidney injury in the intensive care unit: a cost-effectiveness analysis of daily vs alternate-day hemodialysis. Arch Intern Med 168: 1761–1767, 2008. doi: 10.1001/archinte.168.16.1761. [DOI] [PubMed] [Google Scholar]
- 13.Fu Y, Tang C, Cai J, Chen G, Zhang D, Dong Z. Rodent models of AKI-CKD transition. Am J Physiol Renal Physiol 315: F1098–F1106, 2018. doi: 10.1152/ajprenal.00199.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gall JM, Wong V, Pimental DR, Havasi A, Wang Z, Pastorino JG, Bonegio RG, Schwartz JH, Borkan SC. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int 79: 1207–1216, 2011. doi: 10.1038/ki.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, Salvadori M, Haynes RB, Clark WF. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA 290: 1360–1370, 2003. doi: 10.1001/jama.290.10.1360. [DOI] [PubMed] [Google Scholar]
- 16.Heung M, Faubel S, Watnick S, Cruz DN, Koyner JL, Mour G, Liu KD, Cerda J, Okusa MD, Lukaszewski M, Vijayan A; American Society of Nephrology Acute Kidney Injury Advisory Group . Outpatient dialysis for patients with AKI: a policy approach to improving care. Clin J Am Soc Nephrol 10: 1868–1874, 2015. doi: 10.2215/CJN.02290215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O’Connor C, Yang Y, Meadowbrooke C, Chowdhury M, Kikuchi M, Wiggins JE, Wiggins RC. Glomerular aging and focal global glomerulosclerosis: a podometric perspective. J Am Soc Nephrol 26: 3162–3178, 2015. doi: 10.1681/ASN.2014080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol 24: 37–42, 2013. doi: 10.1681/ASN.2012080800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol 80: 309–326, 2018. doi: 10.1146/annurev-physiol-022516-034227. [DOI] [PubMed] [Google Scholar]
- 20.Ishani A, Nelson D, Clothier B, Schult T, Nugent S, Greer N, Slinin Y, Ensrud KE. The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med 171: 226–233, 2011. doi: 10.1001/archinternmed.2010.514. [DOI] [PubMed] [Google Scholar]
- 21.James MT, Wald R, Bell CM, Tonelli M, Hemmelgarn BR, Waikar SS, Chertow GM. Weekend hospital admission, acute kidney injury, and mortality. J Am Soc Nephrol 21: 845–851, 2010. doi: 10.1681/ASN.2009070682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jankowski J, Perry HM, Medina CB, Huang L, Yao J, Bajwa A, Lorenz UM, Rosin DL, Ravichandran KS, Isakson BE, Okusa MD. Epithelial and endothelial pannexin1 channels mediate AKI. J Am Soc Nephrol 29: 1887–1899, 2018. doi: 10.1681/ASN.2017121306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia T, Olauson H, Lindberg K, Amin R, Edvardsson K, Lindholm B, Andersson G, Wernerson A, Sabbagh Y, Schiavi S, Larsson TE. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol 14: 116, 2013. doi: 10.1186/1471-2369-14-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kjellstrand CM, Ebben J, Davin T. Time of death, recovery of renal function, development of chronic renal failure and need for chronic hemodialysis in patients with acute tubular necrosis. Trans Am Soc Artif Intern Organs 27: 45–50, 1981. [PubMed] [Google Scholar]
- 25.Lech M, Gröbmayr R, Ryu M, Lorenz G, Hartter I, Mulay SR, Susanti HE, Kobayashi KS, Flavell RA, Anders HJ. Macrophage phenotype controls long-term AKI outcomes−kidney regeneration versus atrophy. J Am Soc Nephrol 25: 292–304, 2014. doi: 10.1681/ASN.2013020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HT, Park SW, Kim M, Ham A, Anderson LJ, Brown KM, D’Agati VD, Cox GN. Interleukin-11 protects against renal ischemia and reperfusion injury. Am J Physiol Renal Physiol 303: F1216–F1224, 2012. doi: 10.1152/ajprenal.00220.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Liu M, Bedja D, Thoburn C, Gabrielson K, Racusen L, Rabb H. Acute renal venous obstruction is more detrimental to the kidney than arterial occlusion: implication for murine models of acute kidney injury. Am J Physiol Renal Physiol 302: F519–F525, 2012. doi: 10.1152/ajprenal.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol 25: 2689–2701, 2014. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12: 976–998, 2016. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordoñez JD, Hsu CY. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int 76: 893–899, 2009. doi: 10.1038/ki.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowe KG. The late prognosis in acute tubular necrosis; an interim follow-up report on 14 patients. Lancet 259: 1086–1088, 1952. doi: 10.1016/S0140-6736(52)90744-7. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Wei J, Stec DE, Roman RJ, Ge Y, Cheng L, Liu EY, Zhang J, Hansen PB, Fan F, Juncos LA, Wang L, Pollock J, Huang PL, Fu Y, Wang S, Liu R. Macula densa nitric oxide synthase 1β protects against salt-sensitive hypertension. J Am Soc Nephrol 27: 2346–2356, 2016. doi: 10.1681/ASN.2015050515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuda H, Lavoie JL, Gaboury L, Hamet P, Tremblay J. HCaRG accelerates tubular repair after ischemic kidney injury. J Am Soc Nephrol 22: 2077–2089, 2011. doi: 10.1681/ASN.2010121265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A; Acute Kidney Injury Network . Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11: R31, 2007. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murugan R, Kellum JA. Acute kidney injury: what’s the prognosis? Nat Rev Nephrol 7: 209–217, 2011. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peres LA, Wandeur V, Matsuo T. Predictors of acute kidney injury and mortality in an Intensive Care Unit. J Bras Nefrol 37: 38–46, 2015. doi: 10.5935/0101-2800.20150007. [DOI] [PubMed] [Google Scholar]
- 38.Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol 130: 141–155, 2008. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao S, Walters KB, Wilson L, Chen B, Bolisetty S, Graves D, Barnes S, Agarwal A, Kabarowski JH. Early lipid changes in acute kidney injury using SWATH lipidomics coupled with MALDI tissue imaging. Am J Physiol Renal Physiol 310: F1136–F1147, 2016. doi: 10.1152/ajprenal.00100.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schiffl H, Lang SM, Fischer R. Long-term outcomes of survivors of ICU acute kidney injury requiring renal replacement therapy: a 10-year prospective cohort study. Clin Kidney J 5: 297–302, 2012. doi: 10.1093/ckj/sfs070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 7: 189–200, 2011. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 42.Skrypnyk NI, Harris RC, de Caestecker MP. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J Vis Exp 2013: e50495, 2013. doi: 10.3791/50495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skrypnyk NI, Voziyan P, Yang H, de Caestecker CR, Theberge MC, Drouin M, Hudson B, Harris RC, de Caestecker MP. Pyridoxamine reduces postinjury fibrosis and improves functional recovery after acute kidney injury. Am J Physiol Renal Physiol 311: F268–F277, 2016. doi: 10.1152/ajprenal.00056.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K, Haga H, Sharma K, Yanagita M. Severity and Frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol 27: 2393–2406, 2016. doi: 10.1681/ASN.2015060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 334: 1448–1460, 1996. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 46.Thakar CV, Christianson A, Himmelfarb J, Leonard AC. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin J Am Soc Nephrol 6: 2567–2572, 2011. doi: 10.2215/CJN.01120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 26: 1765–1776, 2015. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Song J, Buggs J, Wei J, Wang S, Zhang J, Zhang G, Lu Y, Yip KP, Liu R. A new mouse model of hemorrhagic shock-induced acute kidney injury. Am J Physiol Renal Physiol 312: F134–F142, 2017. doi: 10.1152/ajprenal.00347.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Wei J, Jiang S, Li HH, Fu L, Zhang J, Liu R. Effects of different storage solutions on renal ischemia tolerance after kidney transplantation in mice. Am J Physiol Renal Physiol 314: F381–F387, 2018. doi: 10.1152/ajprenal.00475.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei J, Song J, Jiang S, Zhang G, Wheeler D, Zhang J, Wang S, Lai EY, Wang L, Buggs J, Liu R. Role of intratubular pressure during the ischemic phase in acute kidney injury. Am J Physiol Renal Physiol 312: F1158–F1165, 2017. doi: 10.1152/ajprenal.00527.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei J, Zhang J, Wang L, Cha BJ, Jiang S, Liu R. A new low-nephron CKD model with hypertension, progressive decline of renal function, and enhanced inflammation in C57BL/6 mice. Am J Physiol Renal Physiol 314: F1008–F1019, 2018. doi: 10.1152/ajprenal.00574.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei Q, Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol Renal Physiol 303: F1487–F1494, 2012. doi: 10.1152/ajprenal.00352.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson FP, Yang W, Feldman HI. Predictors of death and dialysis in severe AKI: the UPHS-AKI cohort. Clin J Am Soc Nephrol 8: 527–537, 2013. doi: 10.2215/CJN.06450612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu VC, Shiao CC, Chang CH, Huang TM, Lai CF, Lin MC, Chiang WC, Chu TS, Wu KD, Ko WJ, Wang CY, Wang SM, Chen L. Long-term outcomes after dialysis-requiring acute kidney injury. BioMed Res Int 2014: 365186, 2014. doi: 10.1155/2014/365186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol 301: F1334–F1345, 2011. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Qu HY, Song J, Wei J, Jiang S, Wang L, Wang L, Buggs J, Liu R. Enhanced hemodynamic responses to angiotensin II in diabetes are associated with increased expression and activity of AT1 receptors in the afferent arteriole. Physiol Genomics 49: 531–540, 2017. doi: 10.1152/physiolgenomics.00025.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]