

Abstract
Hypereosinophilic syndrome is characterized by sustained and marked eosinophilia leading to tissue damage and organ dysfunction. Morbidity and mortality occur primarily due to cardiac and thromboembolic complications. Understanding the cause and mechanism of disease would aid in the development of targeted therapies with greater efficacy and fewer side effects. We discovered a spontaneous mouse mutant in our colony with a hypereosinophilic phenotype. Mice develop peripheral blood eosinophilia; infiltration of lungs, spleen, and heart by eosinophils; and extensive myocardial damage and remodeling. This ultimately leads to heart failure and premature death. Histopathological assessment of the hearts revealed a robust inflammatory infiltrate composed primarily of eosinophils and B-lymphocytes, associated with myocardial damage and replacement fibrosis, consistent with eosinophilic myocarditis. In many cases, hearts showed dilatation and thinning of the right ventricular wall, suggestive of an inflammatory dilated cardiomyopathy. Most mice showed atrial thrombi, which often filled the chamber. Protein expression analysis revealed overexpression of chemokines and cytokines involved in innate and adaptive immunity including IL-4, eotaxin, and RANTES. Disease could be transferred to wild-type mice by adoptive transfer of splenocytes from affected mice, suggesting a role for the immune system. In summary, the pathologies observed in the mutant lines are reminiscent of those seen in patients with hypereosinophilia, where cardiac-related morbidities, like congestive heart failure and thrombi, are the most common causes of death. As such, our model provides an opportunity to test mechanistic hypotheses and develop targeted therapies.
NEW & NOTEWORTHY This article describes a new model of heart disease in hypereosinophilia. The model developed as a spontaneous mouse mutant in the colony and is characterized by peripheral blood eosinophilia and infiltration of lungs, spleen, and heart by eosinophils. In the heart, there is extensive myocardial damage, remodeling, fibrosis, and thrombosis, leading to heart failure and death. The immune microenvironment is one of increased innate and adaptive immunity, including Th1 and Th2 cytokines/chemokines. Finally, adoptive transfer of splenocytes transfers disease to recipient mice. In summary, this model provides an opportunity to test mechanistic hypotheses and develop targeted therapies for this rare but devastating disease.
INTRODUCTION
Eosinophils are bone marrow-derived cells that under normal physiological states represent a small subset of circulating white blood cells (<5% and <0.5 × 109/l), while the majority reside in lymphatic organs and mucosal linings of the intestinal tract (20). Eosinophils contain multiple biologically active molecules, including granule proteins [major basic protein (MBP), eosinophil peroxidase, eosinophil-derived neurotoxin, and eosinophil cationic protein] and cytokines. When activated, eosinophils release these mediators, which can lead to tissue damage resulting in impaired organ function. Hypereosinophilic syndrome (HES) is defined as blood and/or tissue hypereosinophilia for more than 1 mo with evidence of organ damage or dysfunction (23, 24). HES is classified as primary neoplastic (clonal), secondary reactive, and idiopathic HES. Eosinophils in primary neoplastic HES are clonal and typically derived from eosinophil progenitors containing genetic alterations in oncogenic tyrosine kinase receptors such as platelet-derived growth factor receptor-α (PDGFRA), platelet-derived growth factor receptor-β (PDGFRB), and fibroblast growth factor receptor-1 (FGFR1). Secondary reactive HES occurs in a variety of disease states including parasitic and fungal infection, hypersensitivity reactions, collagen vascular disease, and select malignancies that elicit an eosinophilic response but wherein eosinophils are not part of the clonal process. The final idiopathic HES category is a diagnosis of exclusion once reactive and neoplastic etiologies have been excluded. Idiopathic HES is the largest category, comprising nearly 50% of cases (26).
Some patients with sustained eosinophilia are at risk of developing cardiac complications (25). A chief question is to understand which patients with eosinophilia are at risk for these complications. For example, patients with primary neoplastic HES as well as patients with eosinophilic granulomatosis with polyangiitis (previously known as Churg-Strauss syndrome) have a high rate of cardiac morbidity, whereas most patients with eosinophilia secondary to atopy or drug reactions do not have this risk. Thus the presence of eosinophils in the peripheral blood is not sufficient for tissue damage, and additional factors leading to their recruitment and/or activation in the heart are involved. Genetic factors (e.g., HLA-Bw44) and subtype of disease (neoplastic versus reactive or idiopathic) influence the likelihood of heart disease in patients with HES (10). Eosinophil-mediated heart damage involves early endocardial damage and myocardial infiltration by eosinophils and lymphocytes with histopathological evidence of myocardiocyte necrosis, which progresses to ventricular and/or atrial thrombosis and later fibrosis (6, 17, 19, 21, 25).
The National Institutes of Health Taskforce on the Research needs of Eosinophil-Associated Diseases (TREAD) and recent RE-TREAD conferences identified an “urgent need for preclinical models and a mechanistic understanding of eosinophil-mediated cardiac damage” (2, 14). Attempts to model these findings in mice include spontaneous eosinophilic myocarditis (EM) in DBA/2 mice (11) and a model of antigen (cardiac myosin)-induced autoimmune EM elicited in interleukin-5 (IL-5) transgenic mice (7, 18). The DBA/2 model is limited in that it lacks the endocardial thrombosis, fibrosis, and systemic aspects typical of HES; furthermore, this model develops at very early ages and spontaneously resolves by ~3 mo old (12). Studies in myosin-challenged IL-5 transgenic mice identified eosinophils as activated (higher expression of Siglec-F and CD11b) and pathogenic, primarily via production of IL-4. Although informative, this model is driven by antigenic cardiac proteins, which is not a prominent feature of HES. In summary, there is a paucity of animal models that adequately replicate cardiac disease in hypereosinophilia, and development of such models would enable mechanistic studies aiming to identify new treatment strategies or refine current targeted therapy.
To address this unmet need, we have advanced a recently discovered mouse mutant that models hypereosinophilia with prominent heart disease. Herein, we describe the phenotype of two distinct mouse lines established from this founder mutant, which is highly reminiscent of the human disease in patients with hypereosinophilia involving the heart. Furthermore, we begin mechanistic dissection of this trait by demonstrating that the disease can be transferred to wild-type mice by adoptive transfer of splenocytes from affected mice, highlighting the central role for immune cells in the disease pathogenesis.
MATERIALS AND METHODS
Mice.
This work was reviewed and approved by the Cincinnati Children's Hospital Institutional Animal Care and Use Committee. From a spontaneous mutant founder mouse, we have established a novel mutant mouse line that naturally develops EM, which manifests itself by sudden death (27). The mutant lines are checked daily by Veterinary Services staff, in addition to routine husbandry by laboratory personnel. When mice are found moribund (e.g., panting, ungroomed, inactive, or cyanotic), investigators are promptly notified by Veterinary Services staff for their immediate use or euthanasia. This mutant line is maintained on a pure A/J inbred strain, with demonstrated heritability of these eosinophilic traits over many generations; herein, this line is referred to as AJ.EM. Mutant mice usually die by ~15 wk old of heart disease. For line maintenance, we use breeder pairs that together are predicted to have the alleles needed for disease. However, because the alleles necessary for disease are unknown and still segregating, only a proportion of mice in each litter develop a disease, necessitating us to retain many mating pairs. Depending on the alleles carried between the breeder mates, the percentage of affected offspring has ranged from none to considerably >50%. With this strategy, we have preserved the mutant line for over 6 yr.
We have also constructed a mixed-strain background from the A/J mutant and SJL/J (SJ) inbred strains that develops the mutant-derived EM phenotype and is referred to in this report as the SJ.EM line. This line was initiated from mating pairs of F2 mice generated for ongoing mapping studies and is currently at F6–7 in the inbreeding (brother-sister matings) process. The SJ.EM line has higher fecundity, thus making it easier than the AJ.EM line to maintain and easier to generate more affected offspring. Importantly, because its major histocompatibility region was derived from the founder A/J mutant line, this mixed-strain line is histocompatible with the A/J control and A/J-inbred mutant lines. The detailed genetic analysis to determine the loci involved in this complex phenotype is the subject of a separate manuscript in preparation.
For splenocyte transfer experiments, we used A/J mice (purchased from The Jackson Laboratory) as recipients and the spleen from moribund (confirmed heart disease by post mortem histology) SJ.EM mutant mice and unaffected littermates as donors. Initially, we performed an intravenous dose response study using 1–20 × 106 splenocytes in PBS, and in subsequent experiments 10 × 106 splenocytes were used. Recipients were sublethally irradiated (700 rad) 4 h before intravenous tail injection of splenocytes. Mice were followed by weekly determination of eosinophil count in peripheral blood by flow cytometry (see below) and euthanized when the first recipient mouse of the group was found dead or moribund. Organs (heart, lung, spleen, liver, kidney, bone, bone marrow, and skeletal muscle) were collected for histopathological assessment, as described below.
Histopathological assessment.
At necropsy, organs were fixed with 10% neutral buffered formalin, processed, and embedded into paraffin. The heart was transected in a four-chamber coronal plane. Sections were stained with hematoxylin and eosin (H&E), Trichrome or immunohistochemistry (IHC) for CD3, B220, MBP, F4/80, Ly6G, smooth muscle actin, and cardiac myosin heavy chain (MHC). For details on antibodies, please see Table 1. Guidelines for antibody use in physiology were followed (4). IHC was performed with a Benchmark Ultra (Roche) automated IHC stainer. H&E and IHC slides were evaluated by a pathologist (N. Zimmermann). Working positive and negative controls were verified in each batch of IHC staining performed.
Table 1.
Antibodies
| Antibody | Manufacturer, Cat. No., RRID | Dilution | Antigen Retrieval |
|---|---|---|---|
| CD3 | Roche, 790-4341, AB_2335978 | None | EDTA |
| B220 | Abcam, ab10558 | 1:200 | Citrate |
| MBP | J. J. and N. Lee, Mayo Clinic, Scottsdale, AZ | 1:2,000 | Proteinase K |
| Ly6G | Biolegend, 127602, AB_1089180 | 1:25 | EDTA |
| Smooth muscle actin | LSBio, LS-B4742, AB_10973744 | 1:25 | EDTA |
| Cardiac myosin heavy chain | Abcam, ab185967 | 1:1,000 | Citrate |
| CD68 | Abcam, ab125212, AB_10975465 | 1:25 | EDTA |
RRID, research resource identifier; MBP, major basic protein.
Quantification of peripheral blood eosinophilia.
Cohorts (entire litters) of mice were bled weekly from 4 to 10 wk of age, and eosinophils were quantified by Discombe’s staining and/or flow cytometry. Blood was diluted (1:10) in Discombe’s solution, and eosinophils were counted in a hemocytometer (3, 8). The percentage of eosinophils in the peripheral blood was determined by flow cytometry labeling eosinophils with CD11b and Siglec-F. Specifically, peripheral blood was lysed using lysis buffer (in mM: 155 NH4Cl, 12 NaHCO3, and 0.1 EDTA) and stained with CD11b and Siglec-F (PE anti-Siglec-F and PE-Cy7 CD11b; nos. 552126 and 552850 respectively; BD) antibodies for 30–60 min at 4°C in the dark. Cells were washed with FACS buffer (1× PBS + 1% FBS), subjected to flow cytometry on FACSCanto instrument, and analyzed using BD FACSDiva software.
Chemokine and cytokine expression in heart extracts.
Protein was extracted from heart tissue of affected and control mice and submitted to Eve Technologies (Calgary, Canada; https://www.evetechnologies.com). The levels of 44 cytokine and chemokine proteins (44-Plex; MD44) in 1,500 μg/ml total proteins were measured by multiplex analysis. IL-5 level was measured using ELISA 88-7054-22; eBioscience). Samples were diluted 1:4, and assay was performed according to manufacturer’s instructions.
IgE level measurement.
Serum and heart homogenate IgE level was measured using ELISA (88-50460-88; eBioscience). Samples were diluted 1:25 and assay performed according to the manufacturer’s instructions.
Statistical analysis.
Prism (GraphPad Software) was used to determine statistical significance. Data in figures represent mean values with error bars indicating SD of replicates within an experiment. The number of mice within experiment and independent experiments is denoted in figure legends. P < 0.05 by unpaired t-test (if 2 groups) or ANOVA (if >2 groups) was regarded as statistically significant.
RESULTS
EM mice exhibit sudden, premature death.
The penetrance of the disease trait is markedly reduced in the mutant lines and varies significantly among the breeder pairs, ranging from no affected offspring to one to several affected offspring for most litters and up to 50% or more affected offspring for a few breeder pairs. Such a diverse range of disease occurrence is consistent with the involvement of multiple genes. Our long-term maintenance of these lines, which includes hundreds of mating pairs over 6 yr, suggests that approximately three genes are segregating for the trait. This genetic complexity explains the unpredictable and low penetrance of the overall trait and is the focus of ongoing mapping studies. Regardless of the number of genes involved, mice of the AJ.EM and SJ.EM mutant lines that do carry the correct combination of variant alleles die suddenly and prematurely, often without visible signs of distress. About 74% of affected mice in the AJ.EM line die by 15 wk old, with median and mean survival ages of 79 and 101 days, respectively. A later onset disease makes up the remaining (~25%) of cases, but the full range of survival age is unknown since we routinely euthanize nonbreeders by 20 wk old (a time at which ~85% of all affected mice have died). Figure 1 portrays the age at death for all AJ.EM mutant mice that have died of disease (n = 279).
Fig. 1.
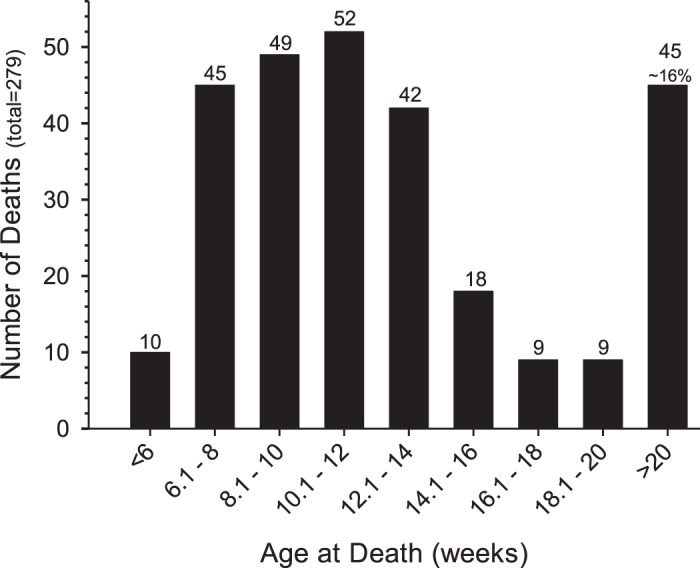
Survival times of AJ.EM-affected mice. A summary of the “age at death” (in wk) of all AJ.EM mutant mice with disease. Ages are grouped as <6 wk old, >20 wk old, and 2 wk age bins between 6 and 20 wk old. For convenience, the total number of mice dying in each group is provided at the top of each bar.
Affected mice develop EM.
To understand the reason for sudden death, we visually and microscopically analyzed collected organs from moribund or recently deceased mice. Affected mice exhibited cardiomegaly with tan foci visible on the pericardial surface, significant expansion of the right ventricle with translucency of the wall, and/or expansion of the atria (Fig. 2). Some mice exhibited generalized subcutaneous edema.
Fig. 2.

Gross findings of the heart. Images of hearts from a control (AJ) mouse and representative affected mice (AJ.EM, including age at death; d, days) taken at same magnification are shown.
Histopathological assessment of the hearts of affected mice revealed an inflammatory infiltrate of the biventricular myocardium, ranging from focal in mild cases to full thickness in severe cases (Fig. 3). Immunohistochemistry confirmed the infiltrate to be primarily composed of eosinophils (MBP+ cells) and B lymphocytes (B220+ cells), with a minor component of T lymphocytes (CD3+ cells). There was minimal infiltration of monocytes/macrophages (F4/80+ cells) or neutrophils (Ly6G+ cells). There was clear myocardial damage and replacement fibrosis, confirmed by trichrome staining. A subset of mice (37%) revealed dilatation and thinning of the ventricular wall, suggestive of inflammatory dilated cardiomyopathy. A group of mice (n = 5) was analyzed by two-dimensional echocardiography, and two of the mice that went on to die within hours after testing were found to have increased left ventricular internal dimension, mass, and volume and decreased ejection fraction and fractional shortening (data not shown), supporting the macroscopic findings of cardiomegaly and heart failure.
Fig. 3.

Histopathology of the heart. Heart from a representative mutant mouse stained with hematoxylin and eosin (A, D, E, G, and H), anti-major basic protein (MBP; B and F), anti-B220 (C), anti-Ly6G (J), anti-F4/80 (K), anti-CD3 (L), and trichrome (I). Photomicrographs taken with 2× (A–C), 10× (D and J–L), and 40× (E–I) magnification objective. Mouse shown is of the SJ.EM strain; however, there was no significant difference in histological findings between the SJ.EM and AJ.EM strains.
The majority of mice (64%) revealed atrial thrombi, which were associated with the chamber wall and often filled the entire or majority of chamber. There was no difference in the prevalence of chamber dilatation, thrombi formation, or histopathological findings between the AJ.EM and SJ.EM mice (data not shown). Furthermore, there was no difference in rate of development of heart disease or histopathological findings between mice of different gender (data not shown). In summary, the hearts of affected mice showed evidence of EM leading to inflammatory dilated cardiomyopathy or thrombotic chamber occlusion.
In some experiments, mice were euthanized before exhibiting symptoms of disease, which allowed us to see early disease in a subset of these mice (presumably those that were going to develop full-blown disease over time). These mice showed milder EM in both atrium and ventricles (Fig. 4). Interestingly, at that early time point the atrial inflammation was centered on the endocardium and no thrombosis was yet evident.
Fig. 4.

Early disease. In several instances we were able to observe early disease in mice that were not yet displaying signs of overt heart failure but already had peripheral blood eosinophilia. Ventricular (A and B) and atrial (C and D) wall in mouse with peripheral blood hypereosinophilia. Photomicrographs taken with 4× (A and C) or 20× (B and D) magnification objective. Mouse shown is of the SJ.EM strain; however, there was no significant difference in histological findings between the SJ.EM and AJ.EM strains.
The histopathology of lungs from affected mice demonstrated an inflammatory infiltrate around large and medium-sized vessels, primarily veins. This infiltrate was composed of lymphocytes and eosinophils (Fig. 5). Immunohistochemistry confirmed the perivascular infiltrate to be primarily composed of eosinophils (MBP+ cells), B lymphocytes (B220+ cells), with a minor component of T lymphocytes (CD3+ cells). There was no appreciable infiltration (beyond control mice) by monocytes/macrophages (F4/80+ cells) or neutrophils (Ly6G+ cells). While eosinophils were seen in the walls of vessels, no convincing evidence of vascular damage was observed (data not shown). The venous wall in some of the mice (53%) was surrounded by inflammatory cells and had an amorphous appearance on H&E-stained sections. To discern if this is due to smooth muscle hypertrophy, fibrosis, or deposition of foreign material, sections were stained with trichrome, anti-smooth muscle actin, and anti-cardiac MHC. As demonstrated in Fig. 5, the smooth muscle layer was not thickened, but the wall was positive for cardiac MHC, consistent with mouse lung veins containing a layer of cardiomyocytes (15) and raising the possibility that the inflammatory process seen in the lungs is an extension of the inflammatory process in the heart. Review of other tissues revealed heart failure-related changes in the liver (centrilobular congestion and necrosis) and increased eosinophilic precursors in the spleen (along with extramedullary hematopoiesis; the latter is also seen in control mice) and bone marrow (increased eosinophils and eosinophil precursors by anti-MBP staining, data not shown), and unremarkable kidney histology.
Fig. 5.

Histopathology of the lungs. Lungs from a representative affected mouse, stained with hematoxylin and eosin (H&E), trichrome, anti-major basic protein (MBP), anti-B220, anti-Ly6G, anti-F4/80, anti-CD3 anti-smooth muscle actin (SMA), and anti-myosin heavy chain (MHC). Mouse shown is of the AJ.EM strain; however, there was no significant difference in histological findings between the SJ.EM and AJ.EM strains.
Inflammatory microenvironment in the heart of affected mice.
To understand the signals that lead to accumulation and activation of eosinophils and other inflammatory cells in the hearts of affected mice, we assessed the protein expression of cytokines and chemokines in heart homogenates of affected mice compared with those of control mice. Expression analysis by protein multiplex assay in heart homogenates revealed overexpression of Th1 and Th2 cytokines and chemokines, including IL-4, IL-6, LIF, IL-16, IP-10/Cxcl10, MIG/Cxcl9, eotaxin-1/Ccl11, RANTES/Ccl5, MIP-1β/Ccl4, MIP-3β/Ccl19, MCP-1/Ccl-2, and MDC/Ccl22 (Fig. 6A). IL-5 level was measured separately by ELISA and also showed increased levels in heart homogenates of affected compared with control mice (9.3 ± 9.5 and 68 ± 36 pg/ml, P = 0.01, n = 5 control and 6 SJ.EM mice, Fig. 6B).
Fig. 6.

Immune microenvironment in the heart: protein expression. A: levels of cytokines and chemokines in heart homogenates were determined by multiplex array in A/J (n = 5) and AJ.EM (n = 6) and SJ.EM (n = 6). Those with statistical significance (ANOVA *P < 0.05) are shown; pairwise comparisons of the individual groups were performed. B: levels of IL-5 were measured in heart homogenates by ELISA (n = 5 control and 6 SJ.EM mice). C: levels of IgE in serum (n = 6 each group; AJ.EM strain) were determined by ELISA. EM, eosinophilic myocarditis.
The overexpression of IL-4 led us to hypothesize that immunoglobulin class switching would be skewed to IgE. Thus, we assessed levels of IgE in serum of AJ.EM mice compared with controls (Fig. 6C). The level of IgE was increased (22 ± 16 vs. 175 ± 68 ng/ml, P = 0.05, n = 6 in each group). Together, these findings support our histological findings and suggest a role for innate and adaptive humoral immunity.
Peripheral blood eosinophilia as biomarker of disease.
Since only a proportion of each litter develops disease, understanding the natural progression of disease is difficult, as any mice euthanized at younger ages would be hard to know if they would have developed disease or not. Thus we set out to identify biomarkers of disease that would predict which mice would eventually be affected. Peripheral blood eosinophilia was initially assessed by Discombe’s staining and showed that peripheral blood eosinophil count is increased in mice that eventually went on to develop EM: at 6 wk of age, there were 87 ± 73 and 222 ± 117 eosinophils/μl in unaffected and affected mice, respectively (P = 0.05, Fig. 7A). In later experiments, we compared Discombe’s staining and flow cytometry for assessment of peripheral blood eosinophilia. While there was correlation between the two methods (R2 = 0.197, P = 0.01), receiver operating characteristic analysis demonstrated that flow cytometry is superior for predicting which mice will develop disease (Fig. 7B), with the area under the curve = 0.87 and P = 0.01. At a cutoff of 8.95% eosinophils in the peripheral blood, the sensitivity of peripheral blood eosinophilia by flow cytometry as a test for predicting EM phenotype is 80% and specificity is 82%. Analysis of flow cytometry data for other white blood cells (neutrophils, lymphocytes and monocytes) found that eosinophils were the only cell type that was consistently different between EM and control mice. In summary, peripheral blood eosinophilia occurs before sudden death only in affected mice and can be used as a predictor of impending death due to EM.
Fig. 7.

Peripheral blood eosinophil level by Discombe’s staining and flow cytometry as biomarker of eosinophilic myocarditis (EM) development. A: peripheral blood eosinophil level was initially determined by Discombe’s staining. A representative experiment with SJ.EM mice at 6 wk of age is shown (n = 5 control and n = 6 EM mice). B: similar trends were seen with AJ.EM mice; however, fewer mice were analyzed. In later experiments, eosinophil percent was determined by flow cytometric analysis and its ability to predict development of eosinophilic myocarditis analyzed using a receiver operating characteristic curve. Representative of 2 experiments is shown (n = 45 controls and 5 cases). Area under the curve = 0.87 and P = 0.01.
Transfer of disease by splenocytes from affected mice to wild-type mice.
To test the role of immunity in the model, we transferred splenocytes (initially dose response: 1, 5, 10, and 20 × 106 splenocytes/recipient mouse, later experiments with 10 × 106) from one moribund EM mouse and one unaffected control to wild-type A/J mice. Peripheral blood eosinophilia was already evident by 2 wk after transfer in mice that received splenocytes from EM donor, and peaked at 3 and 4 wk (representative experiment at week 3 shown in Fig. 8A). Mice that received splenocytes (doses 5–20 × 106) from the EM donor were moribund (and thus euthanized) 4 wk after transfer, while mice that received splenocytes from the unaffected donor did not display any signs of illness. Histological assessment revealed early EM in mice that received the highest doses of splenocytes from the mutant donor (doses 10–20 × 106). However, all mice that received splenocytes from the mutant donor (doses 1–20 × 106) showed rhabdomyositis with extensive infiltration of skeletal muscle by eosinophils accompanied by necrosis of myofibers (Fig. 8B). This was seen in muscles of the forelimb, hindlimb, as well as diaphragm. In summary, while the phenotype of the disease changed, the finding that an eosinophilia-associated disease is transferrable by splenocytes supports the role for the immune system in the EM model.
Fig. 8.

Splenocytes from affected mice transfer hypereosinophilia with early death. A: splenocytes (10 × 106) from an eosinophilic myocarditis (EM) and control mouse were transferred to A/J inbred mice. Peripheral blood eosinophilia was determined by flow cytometry analysis 3 wk posttransfer. Data shown are mean and standard deviation from a representative experiment with 9 control (Ctrl) and 7 EM mice. B: images of skeletal muscles from mouse receiving control (a) or EM splenocytes stained with hematoxylin and eosin (a–c) or anti-major basic protein (d) are shown. Photomicrographs taken with ×10 (a and b), ×20 (d), and ×40 (c) magnification objective. Representative experiment of 3 experiments with 7–10 mice in each group per experiment (total of 26 control and 24 EM).
DISCUSSION
Although therapies specifically targeting eosinophils have begun addressing the unmet need for effective therapies for eosinophil-associated diseases, additional targets are needed as not all patients and not all conditions respond to current therapies. This is especially true in HES, where heart involvement provides significant morbidity and mortality. For novel targets to be developed, robust preclinical models are needed. In this study, we describe a novel mouse model of hypereosinophilia with cardiac tissue damage and organ dysfunction, which mimics heart pathology observed in patients with HES.
There are several advantages of our model, over those currently available. The EM mouse lines naturally develop EM, without the need for antigen challenge and without genomic manipulation. This is more in line with human disease than that seen in IL-5 transgenic mice with experimental autoimmune eosinophilia (7) and IL-5/eotaxin-2 double transgenic (16) mouse models. In contrast to DBA/2 mice (11, 12), which feature EM alone, the disease in AJ.EM and SJ.EM mice is systemic: characterized by peripheral blood eosinophilia as well as involvement of heart, lungs, spleen, and in some instances skeletal muscle. Furthermore, the disease is progressive leading to early death if left untreated, while DBA/2 mice recover without intervention. Most importantly, while EM in DBA/2 mice is primarily subpericardial and lacks thrombosis and fibrosis, the cardiac pathology in our EM models is highly reminiscent of what is seen in human disease. Eosinophil-mediated heart disease in humans evolves in three stages (25). The first stage is an acute necrotic stage, in which there is damage to the endocardium and infiltration of the myocardium by eosinophils and lymphocytes, accompanied by evidence of myocardiocyte necrosis. The second stage is a thrombotic stage with formation of thrombi along the damaged endocardium. The third stage is the fibrotic stage characterized by replacement fibrosis and scarring. In our mouse model, early disease is characterized by endomyocardial damage with myocardiocyte necrosis, with later stages showing thrombosis and replacement fibrosis. Altogether, the disease pathology in our model is highly reminiscent of the human disease based on histopathological findings and cytokine milieu and thus is predicted to provide an accurate model to study the pathophysiology of human disease.
There are however limitations to our model. Because the disease variants are not known and still segregating, many mating pairs of both lines are maintained to keep the complete set of causal alleles in the breeding population; therefore, penetrance of the disease can be limited and unpredictable among the mating pairs. This also hampers our ability to perform mechanistic studies using knockout and transgenic approaches. However, our ongoing genetic studies are seeking to identify loci required for the phenotype. Once these loci are identified, we will be able to implement efficient breeding strategies and expand experimental options.
Our mechanistic studies revealed that the disease is transferrable into A/J inbred mice by adoptive transfer of splenocytes from affected mice. This supports a hematopoietic origin of the phenotype; however, the exact nature of the specific cell type that is sufficient to transfer disease is not known, as the spleen contains a mixture of T and B lymphocytes, as well as eosinophils and their precursors. Thus future studies will need to determine whether it is lymphocytes (and if so, which subset) or eosinophils and/or their precursors that mediate the disease transfer. The other interesting aspect of the transfer experiments is the finding that the recipient mice had prominent myositis and only early myocarditis. Why in the transfer model there is reactivity against skeletal muscle in addition to cardiac muscle is unclear. Skeletal muscle is a target for eosinophilic inflammation in patients and is seen in 12% of patients with HES (9) and in eosinophilia-myalgia syndrome in persons taking l-tryptophan dietary supplement (5). Eosinophilic infiltration of muscle is also seen in a spectrum of hereditary myopathies, including dysferlinopathies (22). Importantly, A/J (13) and SJ (1) mice have a dysferlin mutation and develop mild myopathy with aging. We speculate that either the radiation or transfer of activated splenocytes precipitated inflammation in the skeletal muscle in these susceptible hosts.
In summary, we have developed and characterized a mouse model of hypereosinophilia with extensive cardiac damage that is highly reminiscent of clinical cases of HES. This model will enable mechanistic characterization of the disease pathogenesis, which may lead to novel targeted therapies for this disease.
GRANTS
This project was funded by National Heart, Lung, and Blood Institute Grant HL-135507 (to D. R. Prows and N. Zimmmermann), University of Cincinnati Center for Environmental Genetics through the National Institute of Environmental Health Sciences Grant P30-ES-006096 (to D. R. Prows), in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant P30-DK-078392 (Pathology Core, Flow Cytometry Core) of the Digestive Diseases Research Core Center in Cincinnati, and Campaign Urging Research on Eosinophilic Diseases (CURED).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.R.P. and N.Z. conceived and designed research A.K., W.J.G.J., and S.M.H. performed experiments; A.K., W.J.G.J., S.M.H., and N.Z. analyzed data; D.R.P. and N.Z. interpreted results of experiments; N.Z. prepared figures; N.Z. drafted manuscript; D.R.P. and N.Z. edited and revised manuscript; D.R.P. and N.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Marc Rothenberg for suggestions and critical review of the manuscript, Jeff Bailey and Victoria Summey of the Comprehensive Mouse and Cancer Core for assistance with irradiation of animals and adoptive transfer experiments, and Chris Woods (Cincinnati Children’s Hospital Medical Center, Pathology) for assistance with DSL photography images.
Portions of this article were presented at the American Society for Hematology annual conference in 2018 (27).
REFERENCES
- 1.Bittner RE, Anderson LV, Burkhardt E, Bashir R, Vafiadaki E, Ivanova S, Raffelsberger T, Maerk I, Höger H, Jung M, Karbasiyan M, Storch M, Lassmann H, Moss JA, Davison K, Harrison R, Bushby KM, Reis A. Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat Genet 23: 141–142, 1999. doi: 10.1038/13770. [DOI] [PubMed] [Google Scholar]
- 2.Bochner BS, Book W, Busse WW, Butterfield J, Furuta GT, Gleich GJ, Klion AD, Lee JJ, Leiferman KM, Minnicozzi M, Moqbel R, Rothenberg ME, Schwartz LB, Simon HU, Wechsler ME, Weller PF. Workshop report from the National Institutes of Health Taskforce on the Research Needs of Eosinophil-Associated Diseases (TREAD). J Allergy Clin Immunol 130: 587–596, 2012. doi: 10.1016/j.jaci.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt EB, Strait RT, Hershko D, Wang Q, Muntel EE, Scribner TA, Zimmermann N, Finkelman FD, Rothenberg ME. Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest 112: 1666–1677, 2003. doi: 10.1172/JCI19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks HL, Lindsey ML. Guidelines for authors and reviewers on antibody use in physiology studies. Am J Physiol Heart Circ Physiol 314: H724–H732, 2018. doi: 10.1152/ajpheart.00512.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control (CDC) Update: eosinophilia-myalgia syndrome associated with ingestion of L-tryptophan—United States. MMWR Morb Mortal Wkly Rep 38: 842–843, 1989. [PubMed] [Google Scholar]
- 6.Cheung CC, Constantine M, Ahmadi A, Shiau C, Chen LY. Eosinophilic myocarditis. Am J Med Sci 354: 486–492, 2017. doi: 10.1016/j.amjms.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Diny NL, Baldeviano GC, Talor MV, Barin JG, Ong S, Bedja D, Hays AG, Gilotra NA, Coppens I, Rose NR, Čiháková D. Eosinophil-derived IL-4 drives progression of myocarditis to inflammatory dilated cardiomyopathy. J Exp Med 214: 943–957, 2017. doi: 10.1084/jem.20161702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Discombe G. Criteria of eosinophilia. Lancet 247: 195–196, 1946. doi: 10.1016/S0140-6736(46)91306-2. [DOI] [PubMed] [Google Scholar]
- 9.Fauci AS, Harley JB, Roberts WC, Ferrans VJ, Gralnick HR, Bjornson BH. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med 97: 78–92, 1982. doi: 10.7326/0003-4819-97-1-78. [DOI] [PubMed] [Google Scholar]
- 10.Harley JB, Fauc AS, Gralnick HR. Noncardiovascular findings associated with heart disease in the idiopathic hypereosinophilic syndrome. Am J Cardiol 52: 321–324, 1983. doi: 10.1016/0002-9149(83)90131-5. [DOI] [PubMed] [Google Scholar]
- 11.Hirasawa M, Deguchi H, Ukimura A, Kitaura Y. Immunologic interaction between infiltrating eosinophils and T lymphocytes in murine spontaneous eosinophilic myocarditis. Int Arch Allergy Immunol 130: 73–81, 2003. doi: 10.1159/000068371. [DOI] [PubMed] [Google Scholar]
- 12.Hirasawa M, Kitaura Y, Deguchi H, Ukimura A, Kawamura K. Spontaneous myocarditis in DBA/2 mice. Light microscopic study with transmission and X-ray analytical electron microscopic studies. Virchows Arch 432: 461–468, 1998. doi: 10.1007/s004280050192. [DOI] [PubMed] [Google Scholar]
- 13.Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, Watkins SC, Cox GA, Brown RH Jr. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet 13: 1999–2010, 2004. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- 14.Khoury P, Akuthota P, Ackerman SJ, Arron JR, Bochner BS, Collins MH, Kahn JE, Fulkerson PC, Gleich GJ, Gopal-Srivastava R, Jacobsen EA, Leiferman KM, Francesca LS, Mathur SK, Minnicozzi M, Prussin C, Rothenberg ME, Roufosse F, Sable K, Simon D, Simon HU, Spencer LA, Steinfeld J, Wardlaw AJ, Wechsler ME, Weller PF, Klion AD. Revisiting the NIH Taskforce on the Research needs of Eosinophil-Associated Diseases (RE-TREAD). J Leukoc Biol 104: 69–83, 2018. doi: 10.1002/JLB.5MR0118-028R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer AW Jr, Marks LS. The occurrence of cardiac muscle in the pulmonary veins of Rodenita. J Morphol 117: 135–149, 1965. doi: 10.1002/jmor.1051170202. [DOI] [PubMed] [Google Scholar]
- 16.Ochkur SI, Jacobsen EA, Protheroe CA, Biechele TL, Pero RS, McGarry MP, Wang H, O’Neill KR, Colbert DC, Colby TV, Shen H, Blackburn MR, Irvin CC, Lee JJ, Lee NA. Coexpression of IL-5 and eotaxin-2 in mice creates an eosinophil-dependent model of respiratory inflammation with characteristics of severe asthma. J Immunol 178: 7879–7889, 2007. doi: 10.4049/jimmunol.178.12.7879. [DOI] [PubMed] [Google Scholar]
- 17.Ogbogu PU, Rosing DR, Horne MK 3rd. Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin North Am 27: 457–475, 2007. doi: 10.1016/j.iac.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N, Fontes JA, Gebremariam E, Kass DA, Rose NR, Čiháková D. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol 185: 847–861, 2015. doi: 10.1016/j.ajpath.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parrillo JE, Borer JS, Henry WL, Wolff SM, Fauci AS. The cardiovascular manifestations of the hypereosinophilic syndrome. Prospective study of 26 patients, with review of the literature. Am J Med 67: 572–582, 1979. doi: 10.1016/0002-9343(79)90227-4. [DOI] [PubMed] [Google Scholar]
- 20.Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol 24: 147–174, 2006. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 21.Sasano H, Virmani R, Patterson RH, Robinowitz M, Guccion JG. Eosinophilic products lead to myocardial damage. Hum Pathol 20: 850–857, 1989. doi: 10.1016/0046-8177(89)90096-8. [DOI] [PubMed] [Google Scholar]
- 22.Schröder T, Fuchss J, Schneider I, Stoltenburg-Didinger G, Hanisch F. Eosinophils in hereditary and inflammatory myopathies. Acta Myol 32: 148–153, 2013. [PMC free article] [PubMed] [Google Scholar]
- 23.Valent P, Gleich GJ, Reiter A, Roufosse F, Weller PF, Hellmann A, Metzgeroth G, Leiferman KM, Arock M, Sotlar K, Butterfield JH, Cerny-Reiterer S, Mayerhofer M, Vandenberghe P, Haferlach T, Bochner BS, Gotlib J, Horny HP, Simon HU, Klion AD. Pathogenesis and classification of eosinophil disorders: a review of recent developments in the field. Expert Rev Hematol 5: 157–176, 2012. doi: 10.1586/ehm.11.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, Hellmann A, Metzgeroth G, Leiferman KM, Arock M, Butterfield JH, Sperr WR, Sotlar K, Vandenberghe P, Haferlach T, Simon HU, Reiter A, Gleich GJ. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 130: 607–612.e9, 2012. doi: 10.1016/j.jaci.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood 83: 2759–2779, 1994. [PubMed] [Google Scholar]
- 26.Williams KW, Ware J, Abiodun A, Holland-Thomas NC, Khoury P, Klion AD. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract 4: 941–947.e1, 2016. doi: 10.1016/j.jaip.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmermann N, Gibbons WJ, Homan SM, Klingler A, Prows DR. Characterization of a mouse model of hypereosinophilic syndrome. Blood 132: 3686, 2018. doi: 10.1182/blood-2018-99-113909. [DOI] [Google Scholar]