

Abstract
Bile acids are involved in the emulsification and absorption of dietary fats, as well as acting as signaling molecules. Recently, bile acid signaling through farnesoid X receptor and G protein-coupled bile acid receptor (TGR5) has been reported to elicit changes in not only bile acid synthesis but also metabolic processes, including the alteration of gluconeogenic gene expression and energy expenditure. A role for bile acids in glucose metabolism is also supported by a correlation between changes in the metabolic state of patients (i.e., obesity or postbariatric surgery) and altered serum bile acid levels. However, despite evidence for a role for bile acids during metabolically challenging settings, the direct effect of elevated bile acids on insulin action in the absence of metabolic disease has yet to be investigated. The present study examines the impact of acutely elevated plasma bile acid levels on insulin sensitivity using hyperinsulinemic-euglycemic clamps. In wild-type mice, elevated bile acids impair hepatic insulin sensitivity by blunting the insulin suppression of hepatic glucose production. The impaired hepatic insulin sensitivity could not be attributed to TGR5 signaling, as TGR5 knockout mice exhibited a similar inhibition of insulin suppression of hepatic glucose production. Canonical insulin signaling pathways, such as hepatic PKB (or Akt) activation, were not perturbed in these animals. Interestingly, bile acid infusion directly into the portal vein did not result in an impairment in hepatic insulin sensitivity. Overall, the data indicate that acute increases in circulating bile acids in lean mice impair hepatic insulin sensitivity via an indirect mechanism.
Keywords: bile acid, insulin resistance, TGR5
INTRODUCTION
Bile acids have a well-established role in the solubilization and absorption of dietary fats and in the regulation of their own synthesis (13, 41). The physiological effects attributed to bile acid receptor signaling have become more well defined over the last few decades (13, 30, 51, 66). Most recently, with the increased prevalence of bariatric surgery, bile acids have been propelled into focus as potential mediators of metabolic signaling events within major metabolically active organs, such as the liver (2, 43, 46, 51, 61, 63). Bariatric surgery, which is currently one of the most effective treatments for metabolic disorders (i.e., obesity and type 2 diabetes), results in improvements in metabolic parameters, such as insulin sensitivity, which correlate with increases in plasma bile acid levels in both humans and rodents (2, 10, 29, 33, 45, 48, 54), leading to the hypothesis that elevated bile acids may contribute to the improvements in metabolic parameters.
The association between metabolic outcomes and bile acids is complex and paradoxical, as both metabolic disease and bariatric surgery can cause alterations in both the concentration and composition of the bile acid pool (10, 21, 57). The bile acid pool is composed of diverse bile acid species that can be categorized into subgroups based on several characteristics, including processing by intestinal bacteria (primary or secondary bile acids), amino acid conjugation status (conjugated or unconjugated bile acids), as well as their hydroxylation status (12α-hydroxylated or non-12α-hydroxylated bile acids). Elevations in fasting serum total bile acid levels (12, 50, 57) and alterations in the composition of the bile acid pool (1, 22, 23, 34, 48, 54, 60) have been reported in obese and insulin-resistant individuals and rodents irrespective of diabetes status (51, 60). Interestingly, the altered bile acid pool is often further altered in obese patients and rodents that have undergone bariatric surgery (3, 10, 21, 48, 57). Furthermore, sequestration of bile acids and the dietary addition of bile acids both have positive metabolic benefits (58). These presumably contradictory observations make the relationship between bile acids and metabolic alterations difficult to interpret. The alterations in the composition and concentration of total bile acids in serum have been disputed in a few studies that report no difference in total levels (3, 22) or conjugation status (9) after bariatric surgery. To complicate the biology further, circulating bile acid levels are not constant. The levels change throughout the feeding fasting cycle (17). Bile acids increase after a meal, the magnitude of which is dependent on the fatty acid composition and nutritional content (5, 15, 56). Overall, there is a complex and paradoxical relationship between bile acids and metabolism.
Bile acids can exert their effects by acting through bile acid receptors. Expression of both farnesoid X receptor (FXR) and G protein-coupled bile acid receptor (TGR5), major bile acid receptors, has been detected in multiple tissues, including highly metabolically active tissues, such as adipose tissue, skeletal muscle, liver, and the central nervous system (20, 25, 26). Given the differential affinities of the receptors for different bile acid species, it follows that an alteration in the composition of species within the bile acid pool would lead to activation of different signaling pathways (20, 42). Whereas there is extensive evidence that bile acid signaling and subsequent endocrine responses within the gastrointestinal compartment have profound metabolic effects, including insulin sensitivity, the specific impact that increases in circulating bile acids alone have on insulin action has not been thoroughly examined (35, 58). To understand the mechanism by which increased bile acids induce their metabolic effects in the presence of metabolic disease, we first must understand the effect of elevated bile acids in healthy subjects. Thus in this study, we have investigated the effects of increased circulating bile acids on insulin sensitivity in the absence of metabolic disease or surgery. We discovered that acute increases in bile acids, independent of the conjugation status of the bile acid species, impair hepatic insulin action in lean mice. This impairment is not dependent on TGR5 and is achieved through an indirect mechanism, as direct delivery of bile acids to the liver via the portal vein does not result in impaired hepatic insulin action.
MATERIALS AND METHODS
Mice.
Adult C57BL/6 (The Jackson Laboratory, Bar Harbor, ME) wild-type (WT) and TGR5 (Gpbar1)-deficient [TGR5 knockout (TGR5KO)] mice were used for these studies. TGR5 heterozygous breeder mice were obtained from Dr. David Wasserman (Vanderbilt University School of Medicine, Nashville, TN) and generated as described in Vassileva et al. (64). TGR5KO mice are on a 129S3/SvImJ × C57BL/6 background. Mice were bred in house and housed within Vanderbilt University under a 12:12-h light/dark cycle with free access to food and water. All experiments were performed on male mice (8 weeks of age or older) on a standard chow diet. Procedures were approved and monitored by the Vanderbilt University Institutional Animal Care and Use Committee.
Surgical procedures.
Chronic catheters (carotid artery and jugular vein) were surgically implanted 5 days before the study, as previously described (8). Briefly, mice were anesthetized with isoflurane. The left carotid artery was catheterized for arterial blood sampling. The right jugular vein was catheterized for bile acid, insulin, glucose, or tracer infusions. In some studies, an additional catheter was placed into the portal vein (14). The free ends of the catheters were tunneled under the skin to the back of the neck where the loose ends of the catheters were attached via stainless-steel connectors to tubing made of Micro-Renathane. The tubes were exteriorized and sealed with stainless-steel plugs. Animals were individually housed after surgery.
In vivo metabolic experiments.
All metabolic experiments were performed following a 5-day postoperative recovery. For metabolic studies, conscious, unrestrained, catheterized mice were examined, as previously described (8). Briefly, mice were placed in a plastic container lined with bedding and fasted at 7 AM. Mice were then immediately connected to a dual-channel, stainless-steel swivel (Instech Laboratories, Plymouth Meeting, PA) to allow simultaneous jugular vein infusion and sampling of arterial blood. Three different metabolic studies were performed to assess the in vivo impact of bile acids on the following: 1) insulin action using the hyperinsulinemic-euglycemic clamp, 2) hepatic metabolic flux using stable isotopes, or 3) hepatic signaling.
Hyperinsulinemic-euglycemic clamp.
On the morning of the study, a primed (1.0-µCi), continuous (0.05-µCi/min) infusion of [3-3H]glucose was initiated into the jugular vein catheter at 10 AM (t = −120 min). At t = −10 and 0 min, blood samples were taken from the arterial catheter to assess arterial glucose, insulin, and glucose-specific activity, after which, an infusion of insulin (2 mU · kg−1 · min−1) was initiated along with red blood cells (4.5 µl/min) to replace blood collected during the study. A variable glucose infusion containing [3-3H]glucose was also initiated to maintain euglycemia.
At t = 0 min, mice received a constant infusion of saline, deoxycholic acid (DCA; 0.496 µmol·kg−1·min−1; Sigma-Aldrich, St. Louis, MO), or taurocholic acid (TCA; 0.496 µmol·kg−1·min−1; Sigma-Aldrich) into the jugular vein catheter for the duration of the study. In some studies, DCA was infused into the portal vein catheter instead of the jugular vein. Blood glucose was monitored every 10 min for the duration of the study. At t = 80, 100, 110, and 120 min, blood was obtained from the arterial catheter to assess glucose-specific activity and plasma insulin.
In WT mice, after t = 120 min, a bolus of [2-14C]deoxyglucose (DG; 12 µCi) was given into the jugular vein catheter, followed by a 20-µl saline flush. Samples were taken from the arterial catheter at t = 122, 225, 300, 400, and 155 min. After t = 155 min, animals were anesthetized with Nembutal (Hospira, Lake Forest, IL), and the following tissues were collected: soleus muscle, gastrocnemius (gastroc) muscle, vastus lateralis (Vastus L) muscle, white adipose tissue (WAT), liver, heart, and brain.
Bile acid measurements.
Bile acids were measured by liquid chromatography–mass spectrometry, as previously described (3, 21, 52).
Muscle and plasma sample analysis.
Plasma insulin was assayed using radioimmunoassay in the Vanderbilt Hormone Assay and Analytical Services Core (Nashville, TN). To measure [3-3H]DG and [2-14C]DG in the plasma, samples were deproteinized with barium hydroxide and zinc sulfate and dried, and radioactivity was determined using liquid scintillation counting (Tri-Carb liquid scintillation analyzer; PerkinElmer Life and Analytical Sciences, Downers Grove, IL). Excised soleus, gastroc, superficial Vastus L, gonadal adipose tissue (WAT), heart, and brain were deproteinized with perchloric acid and subsequently neutralized to a pH of ∼7.5. A portion of the sample was counted {[2-14C]DG and [2-14C]DG-phosphate (DGP)}, while a portion was treated with Ba(OH)2 and ZnSO4, and the supernatant was counted ([2-14C]DG). Both [2-14C]DG and [2-14C]DGP radioactivity levels were determined using liquid scintillation counting. Rate of glucose appearance (Ra) and disappearance (Rd; i.e., whole-body glucose uptake) were determined using nonsteady-state equations (59). Endogenous glucose production (EndoRa; mg·kg−1·min−1) was determined by subtraction of glucose infusion rate (GIR) from total Ra. Tissue-specific clearance (Kg) of [2-14C]DG and glucose uptake (Rg) were calculated as previously described (24):
where [2-14C]DGPtissue is the [2-14C]DGP radioactivity (disintegrations per minute/gram) in the tissue, AUC [2-14C]DGplasma is the area under the plasma [2-14C]DG disappearance curve (disintegrations per minute·milliliter−1·minute−1), and [glucose]plasma is the average blood glucose (microgram/microliter) during the experimental period.
Hepatic metabolic flux analysis.
Five days after surgery, on the day of the study, mice were placed in bedded containers (at 7 AM), and food was removed. Infusion lines were connected through a swivel system to the catheters of unrestrained, conscious mice to allow freedom of movement. At 10 AM (t = −150 min), a bolus of [6,6-D2]glucose (80 mg/kg) and D2O (1.5 mg/kg) was given over a 40-min period. This was followed by a constant infusion of [6,6-D2]glucose (0.8 mg·kg−1·min−1), diluted in saline, containing 4.5% D2O, which was maintained for the duration of the study. At t = −20 and 0 min, blood samples were taken to assess glucose concentration and glucose isotopomer distribution. At t = 0 min, an infusion of saline or DCA was given via the jugular vein catheter. Blood samples were taken every 10 min for 30 min. Reconstituted red blood cells from a donor mouse were continuously infused (4 μl/min) for the duration of the study.
Glucose isotopomer distribution in arterial plasma was determined in the Metabolic Flux Analysis Subcore of the Analytical Resources Core of the Mouse Metabolic Phenotyping Center using an Agilent 5977A mass spectrometric detector GC-MS, according to the method of Antoniewicz et al. (6) and analyzed using isotopomer computational analysis software (67). Glucose fluxes were assessed using steady-state equations from plasma [6,6-D2]glucose enrichment. The contribution of gluconeogenesis to glucose flux was assessed as the enrichment ratio of C5/C2 (6, 11, 24).
Bile acid infusion.
Five days after surgery, mice were placed in bedded containers (at 7 AM), and food was removed. Infusion lines were connected through a swivel system to the jugular catheter of unrestrained, conscious mice. DCA was infused into the jugular vein at a constant rate (0.496 µmol·kg−1·min−1) for 30 min. After t = 30 min, animals were anesthetized with Nembutal, and the liver was collected.
HepG2 cell stimulation.
Hepatocellular carcinoma (HepG2) cells were incubated in DMEM with 10% FBS, penicillin, and streptomycin for 2 days at 37°C. Cells were then plated to ~50% confluence in six-well plates. After overnight incubation at 37°C, the media were switched to DMEM (no serum) with 4.5 g/l glucose and l-glutamine for 3 h. Cells were then washed with serum-free media two times and incubated in serum-free media for 3 h. DCA (10, 50, or 100 µM) or 50 µl PBS was added to the media and incubated for 20 min at 37°C. Cells were then washed in PBS, two times. Cells were then harvested and lysed using 150 µl radioimmunoprecipitation buffer with phosphatase and protease inhibitors (Roche, Basel, Switzerland), and the plates were incubated on ice. The plates were then scraped, and lysates were collected and immediately snap frozen in liquid nitrogen.
Primary hepatocyte isolation and stimulation.
Primary hepatocyte isolation from ~16-week-old mice was performed, as previously described (53a). Hepatocytes were prepared from whole liver with some modifications of that previously reported. After anesthesia with sodium pentobarbital (70 mg/kg ip), the portal vein was cannulated, and the liver was perfused at 37°C in flowthrough mode at 5 ml/min of flow rate using oxygenated Ca2+-free Krebs bicarbonate buffer containing 0.5 mM EGTA for 15 min, after which, the liver was perfused at 37°C in recirculating mode at 5 ml/min with 50 ml for mice of the liver-digestion medium (Gibco Life Technologies, Waltham, MA) for 30 min. The liver was then dispersed in hepatocyte wash medium (Gibco Life Technologies) and filtered through nylon mesh (100 µm), and the suspension was centrifuged at 50 g for 2 min. The precipitated cells were washed four times by resuspension in fresh wash media. The supernatant was centrifuged at 50 g again to remove remaining hepatocytes, and the supernatant was centrifuged at 500 g for 5 min to collect nonparenchymal cells. The precipitated nonparenchymal cells were washed two times by resuspension in fresh wash media. Hepatocytes and nonparenchymal cells were incubated overnight in DMEM-based attachment media with 20 mM glucose on plates coated with collagen I. The cells were washed with PBS, and the media were changed to DMEM growth media with 20 mM glucose for 16 h.
After 16 h incubation at 37°C, the media were switched to DMEM (no serum) with 4.5 g/l glucose and l-glutamine for 3 h. Cells were then washed with serum-free media, two times, and incubated in serum-free media for 3 h. DCA (100 µM), 1 µM of the TGR5 agonist INT-777, or 50 µl PBS was added to the media, and the cells were incubated for 20 min at 37°C. The plates were then placed on ice to inhibit further reactions. The cells were harvested in radioimmunoprecipitation buffer, as described above.
Gene-expression analysis.
Mouse livers were harvested after the hyperinsulinemic-euglycemic clamp and frozen in liquid nitrogen. Tissues and primary hepatocytes were then homogenized in TRIzol (RNA; Ambion, Carlsbad, CA), and a chloroform extraction was performed. The aqueous phase was used to extract mRNA with the RNeasy Mini kit (Qiagen, Hilden, Germany). cDNA was obtained using a cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) and stored at −20°C. PCR amplification via Applied Biosystems and TaqMan probes from Thermo Fisher Scientific (Waltham, MA) for phosphoenolpyruvate carboxykinase 1 (Pck1; Mm01247058_m1), phosphoenolpyruvate carboxykinase 2 (Pck2; Mm00551411_m1), glucose-6-phosphatase (G6pc; Mm00839363_m1), and Gpbar1 (Mm04212121_s1) allowed mRNA quantification using the comparative threshold cycle (or ΔΔCt) method. Gene expression was normalized using Gapdh (Mm99999915_g1) as a housekeeping gene.
Tissue protein extraction.
Mouse livers were harvested after the bile acid infusion experiments described above. Liver protein extraction was performed as in previous studies (44). Briefly, tissues frozen at −80°C were homogenized on ice in a 50-nM Tris buffer containing 1 mM EDTA, 1 mM EGTA, 10% glycerol, and 1% Triton X-100, at pH 7.5, and then, 1 mM DTT, 1 mM PMSF, 5 μg/ml protease inhibitor cocktail, 10 mg/ml trypsin inhibitor, 50 mM NaF, and 5 mM NaPP were added the day of extraction. Protein in tissue extract was quantified using the protein assay (Bio-Rad, Hercules, CA).
Western blot analysis.
Protein (~30 μg) from liver, hepatocyte, or HepG2 cell lysates and Precision Plus Protein Kaleidoscope Standard (Bio-Rad) were size fractionated in 10% SDS-PAGE. Proteins and standard molecular weight ladder were transferred to polyvinylidene difluoride membranes. Membranes were rinsed with 1.0% Tween in Tris-buffered saline (TBS-T) and blocked with 5% nonfat dried milk in TBS-T. Membranes were incubated overnight at 4°C with primary antibodies: phosphorylated cAMP response element-binding protein (P-CREB), CREB, phosphorylated PKB [P-Akt (Ser473)], AKT, phosphorylated STAT3 [P-STAT3 (Tyr705)], STAT3, and β-tubulin. After the TBS-T wash, membranes were incubated with corresponding secondary anti-rabbit or anti-mouse peroxidase-conjugated antibody. The proteins were detected with a chemiluminescence system and the ChemiDoc MP (Bio-Rad). All antibodies used were obtained from Cell Signaling Technology (Danvers, MA). Band intensity was quantified using ImageJ software.
Statistics.
Mouse data were analyzed using a Student’s t-test, one-way nonrepeated measures ANOVA, or two-way repeated-measures ANOVA, assuming normal distribution and equal variance. Post hoc analyses were performed using the Bonferroni correction for multiple comparisons. P < 0.05 was considered significant. Data are presented as means ± SE.
RESULTS
Bile acids acutely blunt insulin-dependent suppression of endogenous glucose production.
Recent studies have demonstrated that bile acids are elevated in the circulation of obese individuals and patients after receiving bariatric surgery (1). It has been hypothesized that the increase in bile acids is associated with the improved metabolic state of these patients postsurgery; however, the effect of increased bile acids in the absence of metabolic dysfunction or surgery is still unknown. In this study, we investigated whether circulating bile acids directly affect glucose metabolism by examining alterations in insulin action in response to exogenous bile acid species infused peripherally into lean animals. Insulin action was assessed using the hyperinsulinemic-euglycemic clamp in chronically catheterized, conscious WT mice. During the 2-h insulin-infusion period, mice also received a continuous intravenous infusion of saline vehicle (control); an unconjugated, hydrophobic bile acid (DCA); or a conjugated, hydrophilic bile acid (TCA). DCA and TCA were chosen to represent the extremes of the bile acid species spectrum, as they are highly hydrophobic and hydrophilic, respectively (49). The mice had slightly different body weights at the start of the study but similar arterial blood glucose (Fig. 1A), plasma insulin (Fig. 1B), rate of Rd (Fig. 1C), and rate of EndoRa (Fig. 1D) in the basal period. During the clamp period (t = 80–120), the blood glucose was clamped to the same level (~120 mg/dl) in all three treatment groups (Fig. 1A), and arterial insulin was raised to similar concentrations during the clamp period (Fig. 1B). To ensure that the level of bile acids increased after the infusion, DCA and TCA concentrations were assessed in DCA- and TCA-treated mice before and after the infusion. As expected, continuous infusion of either bile acid species significantly elevated bile acid concentrations in circulation (Fig. 1, E and F). Despite equivalent infusion rates of DCA and TCA, absolute levels of TCA were less than DCA. Infusion of either bile acid species, however, resulted in significantly decreased GIRs compared with the saline-treated controls (Fig. 1G). Remarkably, differences in the GIR of bile acid-treated and control mice begin to appear within 30 min of the start of infusion, suggesting that circulating bile acids have a rapid effect on glucose metabolism. The decrease in GIR suggests that both conjugated and unconjugated bile acids acutely impair insulin action in mice.
Fig. 1.

Hyperinsulinemic-euglycemic clamp of wild-type chronically catheterized, conscious mice with jugular vein infusion of saline or bile acid. A: blood glucose concentrations during clamp. B: plasma insulin concentrations during basal period and during the clamp. C: rate of glucose disappearance [whole-body glucose uptake (Rd)]. D: endogenous glucose production (EndoRa). E: plasma deoxycholic acid (DCA) concentration during basal period and during the clamp. F: plasma taurocholic acid (TCA) concentration during basal period and during the clamp. G: glucose infusion rate during clamp. H and I: tissue-specific glucose uptake (Rg). Data are expressed as means ± SE. aP < 0.05 DCA vs. saline treated; bP < 0.05 TCA vs. saline treated; cP < 0.05 DCA vs. TCA treated; dP < 0.05 vs. basal of same treatment. Gastroc, gastrocnemius; Vastus L., vastus lateralis; WAT, white adipose tissue.
The incorporation of [3-3H]glucose in the hyperinsulinemic-euglycemic clamp allowed assessment of the mechanism by which bile acids impair insulin action by measuring the EndoRa and Rd. In saline-infused mice, insulin suppressed EndoRa by 70%, whereas the infusion of DCA or TCA impaired insulin suppression of EndoRa (Fig. 1D). As seen in Fig. 1C, insulin increased Rd (approximately twofold) in saline-infused mice. This increase was attenuated in mice infused with DCA, suggesting that circulating, unconjugated bile acids impair whole-body Rg in conscious mice. To determine which tissues were affected, we assessed tissue-specific Rg in the soleus, gastroc, Vastus L, WAT, heart, and brain. We did not observe significant differences in Rg in most tissues (Fig. 1, H and I); however, a small but significant decrease in Rg in the soleus of mice treated with DCA compared with TCA and saline was detected (Fig. 1H). There was also a significant increase in the Rg of the heart in TCA-treated mice compared with both DCA and control mice (Fig. 1I). Overall, infusion of bile acids—DCA more potently than TCA—into the systemic circulation via the jugular vein of WT mice impaired insulin action by the blunting of insulin-dependent suppression of EndoRa during a hyperinsulinemic-euglycemic clamp.
Classical regulation of gluconeogenic signaling does not play a role in the blunted insulin-dependent suppression of EndoRa.
The liver is the major source of EndoRa. Therefore, we hypothesized that circulating bile acids affect signaling events in the liver that are important for the regulation of hepatic glucose production. Given the greater effect that DCA treatment had on insulin action in WT mice (Fig. 1), only DCA treatment was used in subsequent experiments. Samples were probed for activation of the transcription factor Creb by phosphorylation, which initiates the transcription of genes in hepatocytes involved in hepatic gluconeogenesis. We hypothesized that increased levels of P-Creb would be detected in hepatocytes treated with DCA.
HepG2 cells, a surrogate for hepatocytes, were treated with 10, 50, and 100 µM DCA in culture for 20 min and then collected for analysis. P-Creb was significantly elevated in HepG2 cells that were treated with DCA compared with saline-treated controls (Fig. 2, A and B), suggesting that bile acids can act directly on hepatocytes to elevate P-Creb and ultimately increase expression of gluconeogenic genes. However, there was no difference in P-Creb levels in primary hepatocytes isolated from WT mice treated with saline or DCA (Fig. 2, C and D). Primary hepatocytes that do not replicate the effect seen in HepG2 cells are not surprising, given that differences between the HepG2 cell line and primary hepatocytes are well documented. Specifically, there are differences in the number of chromosomes and the concentration of transporters and metabolic enzymes between HepG2 cells and hepatocytes, which has been investigated in depth previously (65). Furthermore, there was no difference in P-Creb levels in whole-liver lysates harvested from mice that received a 30-min continuous infusion of insulin, with or without infusion of DCA (Fig. 2, E and F). Therefore, the observed blunting of insulin suppression of EndoRa, which occurs acutely in the presence of elevated systemic bile acids, is not dependent on the activation of Creb.
Fig. 2.

Western blot analysis of gluconeogenic regulators. A: immunoblot of phosphorylated cAMP response element-binding protein (P-CREB) and total CREB from hepatocellular carcinoma (HepG2) cells after exposure to PBS or 10, 50, or 100 µM deoxycholic acid (DCA) for 30 min. B: quantification of P-CREB/total CREB ratio from HepG2 immunoblot. C: immunoblot of P-CREB and total CREB from wild-type (WT) primary hepatocytes after exposure to PBS or DCA for 30 min. D: quantification of P-CREB/total CREB ratio from primary hepatocyte immunoblot. E: immunoblot of P-CREB and total CREB from liver lysates of WT mice after saline or DCA infusion during clamp. F: quantification of P-CREB/total CREB ratio from liver lysate immunoblot. Data are expressed as means ± SE. aP < 0.05 vs. PBS treated.
Pck1 and G6pc, two key genes involved in the regulation of hepatic gluconeogenesis, are regulated by P-CREB. Consistent with the P-Creb data described above, we did not observe increased expression of Pck1 (Fig. 3A) or G6pc (Fig. 3B) in the whole-liver lysates of mice treated with DCA compared with control-treated mice. However, a second isoform of Pck (Pck2) was also analyzed, and a significant increase in Pck2 expression in the livers of DCA-treated mice compared with control mice was observed (Fig. 3C).
Fig. 3.

RNA expression of gluconeogenic genes in wild-type mice after saline or deoxycholic acid (DCA) infusion. RNA expression of phosphoenolpyruvate carboxykinase 1 (Pck1; A), glucose-6-phosphatase (G6pc; B), and phosphoenolpyruvate carboxykinase 2 (Pck2; C). Data are expressed as means ± SE. aP < 0.05 vs. saline treated.
Gluconeogenic genes are also regulated by Akt. P-Akt negatively regulates the expression of Pck1 and G6pc (47). We did not detect the decreased expression of Pck1 or G6pc that would be expected in the presence of blunted suppression of hepatic glucose production. Therefore, we hypothesized that there would be decreased P-Akt detected in liver lysates from DCA-treated mice compared with saline-treated mice, leading to diminished suppression of Pck1 and G6pc expression. However, there was no difference in P-Akt protein expression between the two groups (Fig. 4, A and B). Overall, these results suggest that circulating bile acids blunt insulin-dependent suppression of hepatic glucose production, in part, by opposing insulin suppression of gluconeogenic genes, such as Pck2. Nonetheless, given the rapid onset of impairments in insulin action, it is unlikely that transcriptional-mediated effects alone can explain the bile acid-induced hepatic insulin resistance.
Fig. 4.
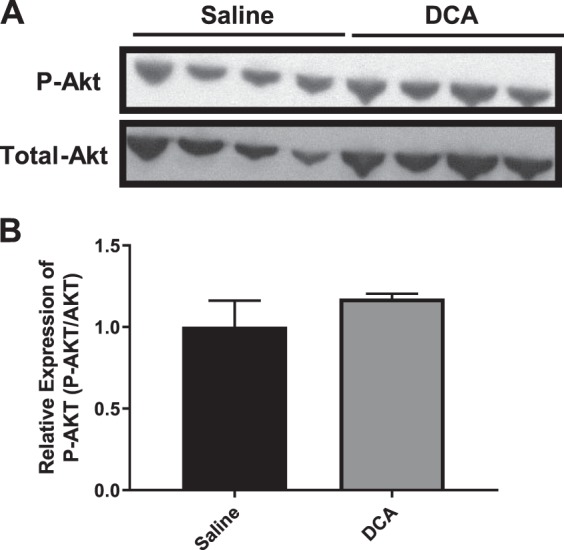
Western blot analysis of gluconeogenic regulators. A: immunoblot of phosphorylated PKB (P-Akt) and total Akt from liver lysates of wild-type mice after saline or deoxycholic acid (DCA) infusion during clamp. B: quantification of P-Akt/total Akt ratio from liver lysate immunoblot. Data are expressed as means ± SE.
It was possible that the failure of insulin to suppress EndoRa during DCA infusion was because DCA directly stimulated EndoRa by augmenting hepatic glycogenolysis and/or gluconeogenesis. This would be interpreted as an impairment in hepatic insulin action. With the use of stable isotopes, we monitored the acute effect of DCA on hepatic glucose production, gluconeogenesis, and glycogenolysis. However, we found that the acute infusion of DCA did not alter arterial glucose levels (Fig. 5A), hepatic glucose production (Fig. 5B), or the contribution of glycogenolysis to total EndoRa (Fig. 5C). Thus DCA infusion does not augment hepatic glucose flux, but rather, it opposes the ability of insulin to suppress endogenous hepatic glucose production.
Fig. 5.

Stable isotope analysis of hepatic glucose production and glycogenolysis. A: blood glucose concentrations during stable isotope and deoxycholic acid (DCA) infusion. B: total hepatic glucose production during stable isotope and DCA infusion. C: fractional glycogenolysis of total hepatic glucose production during stable isotope and DCA infusion. Data are expressed as means ± SE.
Inhibition of insulin induction of gluconeogenic gene expression is TGR5 dependent.
Previous work has demonstrated that signaling through TGR5 is able to mediate insulin action, specifically EndoRa in mice (62). Therefore, we hypothesized that systemic bile acid signaling through TGR5 leads to inhibition of the insulin suppression of hepatic glucose production. We wanted to determine if increased expression of Pck2 would be detected in TGR5KO mice, as it is in WT mice. Whole livers collected from TGR5KO mice were analyzed, and no difference in expression of Pck1, G6pc, or Pck2 between TGR5KO mice with DCA or saline infusion was detected (Fig. 6, A–C). Therefore, we concluded that bile acid signaling through the TGR5 receptor attenuates the ability of insulin to suppress the transcription of the gluconeogenic gene Pck2. To determine the mechanism of action, we examined the expression of TGR5 in primary hepatocytes and determined that there is minimal expression of TGR5 in primary WT hepatocytes but significantly higher expression in primary, nonparenchymal cells (Fig. 7). These data indicate that signaling to hepatocytes through TGR5 would have to occur through indirect mechanisms.
Fig. 6.

RNA expression of gluconeogenic genes in G protein-coupled bile acid receptor knockout mice after saline or deoxycholic acid (DCA) infusion. RNA expression of phosphoenolpyruvate carboxykinase 1 (Pck1; A), glucose-6-phosphatase (G6pc; B), and phosphoenolpyruvate carboxykinase 2 (Pck2; C). Data are expressed as means ± SE.
Fig. 7.
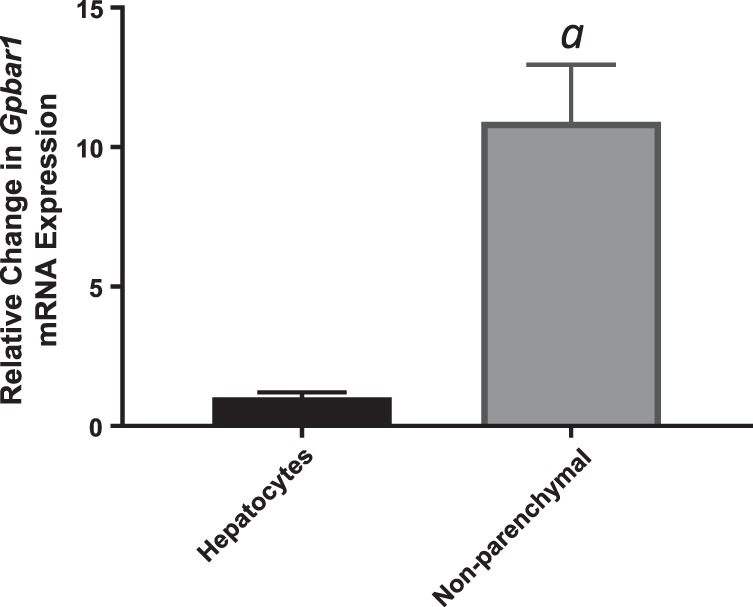
G Protein-coupled bile acid receptor (Gpbar1) expression in wild-type primary hepatocytes and primary nonparenchymal cells. Data are expressed as means ± SE. aP < 0.05 vs. hepatocytes.
Bile acids oppose insulin action independently of TGR5 signaling.
Given the TGR5-dependent induction of Pck2 expression, we hypothesized that TGR5KO mice would be protected against the impairment in insulin signaling at the liver in the presence of bile acids. Hyperinsulinemic-euglycemic clamps were performed on TGR5KO mice. DCA or saline was infused into the jugular vein of TGR5KO mice for 2 h, as was done in WT mice. The body weight of both groups was ~25 g, and plasma insulin levels of both DCA- and saline-treated mice were raised to the same level during the steady-state period (clamp; Fig. 8A). Similar to our studies in WT mice, the blood glucose levels of both groups were clamped at the same level (Fig. 8B). Surprisingly, in the absence of TGR5, a systemic increase of bile acids still acutely reduced the GIR of TGR5KO mice (Fig. 8C). In contrast to WT mice, bile acid infusion did not result in differences in Rd (Fig. 8D) compared with saline-treated TGR5KO mice. DCA infusion led to impaired insulin suppression of EndoRa in TGR5KO mice, as was detected in WT mice (Fig. 8E). This supports the hypothesis that infusion of bile acids leads to an impairment of insulin suppression of hepatic glucose production, but it occurs through a TGR5-independent mechanism. Furthermore, the TGR5 signaling cascade leading to the induction of Pck2 is not necessary for the bile acid-induced impairment in hepatic insulin action.
Fig. 8.

Hyperinsulinemic-euglycemic clamp of G protein-coupled bile acid receptor knockout chronically catheterized, conscious mice with jugular vein infusion of saline or deoxycholic acid (DCA). A: plasma insulin concentrations during basal period and during the clamp. B: blood glucose concentrations during clamp. C: glucose infusion rate during clamp. D: rate of glucose disappearance [whole-body glucose uptake (Rd)]. E: endogenous glucose production (EndoRa). Data are expressed as means ± SE. aP < 0.05 vs. saline treated; dP < 0.05 vs. basal of same treatment.
Impaired hepatic insulin action is not conferred via brain-insulin STAT3 and IL-6 signaling.
The canonical and direct regulation of insulin signaling did not appear to be altered in WT or TGR5KO mice (Figs. 3, 4, and 6); signaling in the brain is a potential indirect mediator of hepatic glucose production (27). Specifically, insulin signaling in the brain leads to an increase in the proinflammatory IL-6 expression in the liver, which leads to activation of hepatic Stat3 via phosphorylation, and ultimately, results in hepatic insulin resistance (32). In TGR5KO mice that received peripheral DCA infusions, there was no difference in hepatic IL-6 expression (data not shown) or hepatic P-Stat3 levels (Fig. 9, A and B). Absence of alterations in this signaling cascade is confirmed by a lack of altered G6pc and Pck1 expression, which would be inhibited by Stat3 activation.
Fig. 9.
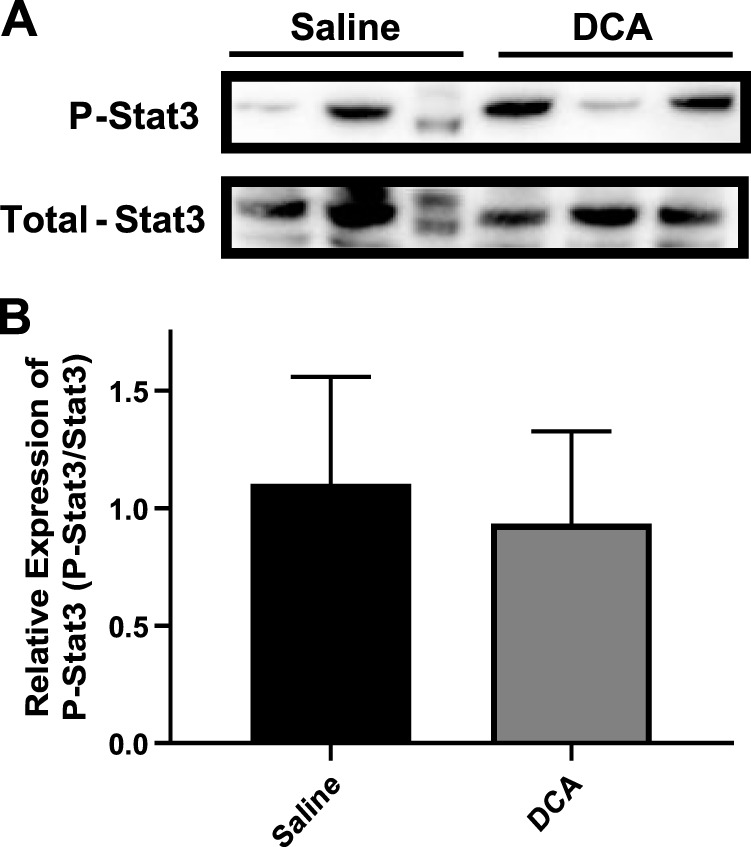
Western blot analysis of STAT3 activation in G protein-coupled bile acid receptor knockout mice. A: immunoblot of phosphorylated Stat3 (P-Stat3) and total Stat3 from liver lysates of wild-type mice after saline or deoxycholic acid (DCA) infusion during clamp. B: quantification of P-Stat3/total Stat3 ratio from liver lysate immunoblot. Data are expressed as means ± SE.
Increased systemic, but not portal vein, bile acid concentration impairs insulin action.
Finally, we have determined that insulin action in the liver is impaired via an increase in systemic bile acid levels from infusion directly into the jugular vein. However, physiologically, bile acids are delivered from the intestinal tract to the liver via the portal vein, and hepatic first-pass extraction of bile acids is high, which would limit the spillover of bile acids into the systemic circulation. Thus by the delivery of bile acids via the portal vein, we could determine if direct delivery of DCA to the liver, a more physiologically relevant mechanism, impairs hepatic insulin action. Hyperinsulinemic-euglycemic clamps were performed in conscious, chronically catheterized TGR5KO mice implanted with three catheters (carotid artery, jugular vein, and portal vein). DCA or saline was infused directly into the portal vein, whereas insulin and glucose were infused into the jugular vein during the clamp. There was no difference in body weight between mice receiving bile acids compared with saline control, and blood glucose was clamped to the same level (~120 mg/dl) in both groups (Fig. 10A). Serum DCA levels were significantly increased from basal after infusion (Fig. 10B). Interestingly, there was no difference in GIR between DCA- and saline-treated controls (Fig. 10C), and both treatment groups have a similar rise in plasma insulin during the clamp (Fig. 10D). Additionally, there were no detectable differences between mice treated with bile acid or saline in the Rd (Fig. 10E) or EndoRa (Fig. 10F). Together, these data demonstrate that an increase in bile acid levels, specifically in the systemic circulation, can result in impaired suppression of hepatic glucose production by insulin, but elevated bile acid levels directly to the liver via infusion into the portal vein do not affect insulin action at the liver.
Fig. 10.

Hyperinsulinemic-euglycemic clamp of G protein-coupled bile acid receptor knockout chronically catheterized, conscious mice with infusion of saline or deoxycholic acid (DCA) into the portal vein. A: blood glucose concentrations during clamp. B: plasma DCA concentration during basal period and during the clamp. C: glucose infusion rate during clamp. D: plasma insulin concentrations during basal period and during the clamp. E: rate of glucose disappearance [whole-body glucose uptake (Rd)]. F: endogenous glucose production (EndoRa). Data are expressed as means ± SE. aP < 0.05 vs. saline treated; dP < 0.05 vs. basal of same treatment.
DISCUSSION
Bile acids have gained much attention in the metabolic field since the observation that an association exists between systemic bile acid levels and the changes in the metabolic state of a patient. For example, after bariatric surgery, there is a consistent rise in serum bile acids, corresponding with the improvement in metabolic health, independent of weight loss (7, 55). A current hypothesis in the field is that this increase in systemic bile acid levels, specifically conjugated bile acids, leads to improvements in insulin signaling (3, 28). As bile acid levels also increase in response to a meal (5, 15, 56), it is essential first to establish the effects of an acute rise in bile acids on glucose metabolism in healthy animals. To our knowledge, this is the first study to investigate the direct effects of increases in conjugated and unconjugated bile acids in the systemic circulation on insulin sensitivity in the absence of metabolic disease. In contrast to the hypothesized association of increased bile acids with metabolic benefit, we demonstrated that in lean animals, systemic increases of both conjugated and unconjugated bile acid resulted in defects in insulin sensitivity, albeit to different extents. The primary target tissue is the liver, where it manifests as a failure of insulin to suppress EndoRa. Our data also suggest that the hepatic insulin resistance is via a peripheral, indirect mechanism, as direct delivery of bile acids into the portal vein does not induce hepatic insulin resistance. Furthermore, it occurs in a TGR5-independent manner, as TGR5KO mice develop hepatic insulin resistance in response to increases in systemic bile acids similar to WT mice. Thus whereas increases in systemic bile acid species may correlate with metabolic benefit, it is unlikely that the increased systemic bile acids alone are driving the metabolic benefit.
It is not surprising that bile acid signaling directly at the liver does not result in impaired insulin suppression of hepatic glucose production. In the normal postprandial state, bile acid levels in the portal vein are elevated, but there is no impairment in liver insulin signaling (5). Additionally, bile acid portal vein levels are higher than systemic levels after a meal, but the bile acid concentration differences between the portal vein and the peripheral circulation may be altered during obesity or after bariatric surgery compared with normal conditions that could potentially highlight an important physiological difference between metabolically healthy and unhealthy states. Under normal conditions, first-pass extraction of bile acids from the enterohepatic circulation at the liver is effective; ~90–95% of conjugated bile acids are transported from the portal vein into the hepatocytes (16, 35). Because of this high first-pass extraction rate, it is not unexpected that the portal vein infusion of bile acids yields a lower peripheral level than when bile acids are infused directly into the jugular vein. However, after bariatric surgery, specifically Roux-en-Y gastric bypass, in mini pigs, conjugated bile acids in the portal vein are decreased, whereas conjugated bile acids in the systemic circulation are significantly increased (12). This increase is associated with a decrease in hepatic bile acid transporter expression, similar to the decreased expression detected in obese patients, which could contribute to the increased spillover into the peripheral circulation (12, 23). Therefore, Roux-en-Y gastric bypass and obesity may not only change the composition of bile acid species but also the ability of the liver to take up and recycle the bile acids, leading to an elevation specifically in systemic bile acids.
As bile acid signaling directly at the liver did not alter insulin action, according to our portal vein delivery studies, bile acid signaling from peripheral tissue to the liver must impair insulin action indirectly. Established signaling cascades between the brain and gut can potentially be involved in the indirect signaling events that lead to altered insulin action in the liver. The signaling events in the liver leading to hepatic insulin resistance, in response to elevated systemic bile acids, are not clear. Inflammation does not appear to be playing a role in the response. We examined the expression of two proinflammatory cytokines—IL-6 and TNF-α—and did not detect a change in either after bile acid infusion (data not shown). Furthermore, the signaling events do not involve the classical PKA cascade, as P-Creb or P-Akt expression is not changed after bile acid infusion.
With specific focus on the FXR signaling, bile acids can activate FGF15/19 in the intestine, which leads to inhibition of agouti-related protein neurons in the arcuate nucleus (38). Modulation of the activity of these neurons has a well-established role in modulation of food-intake behavior, as well as alteration of hepatic insulin action (53). FXR is expressed in tissues other than the liver, which have established signaling cascades to the liver and can also play a role in the suppression of insulin action. Indeed, bile acid signaling through FXR, leading to decreased gluconeogenesis by induction of glycogen synthesis (51), complementary to insulin action, has been described, but this effect has only been established for signaling directly at the liver. Additionally, given the rapid effect of bile acids to impair hepatic insulin action, it is unlikely that FXR-driven transcriptional events alone can explain the effects of bile acids seen in this study.
Whereas not the specific focus of these studies, we did observe that insulin action in WT and TGR5KO was very similar. The average GIRs were 20 and 25 mg⋅kg−1⋅min−1 in WT and TGR5KO, respectively. Previous hyperinsulinemic-euglycemic clamps performed by Ding et al. (18) reported GIRs similar to those found in our studies, when the mice were compared directly. Furthermore, in our studies, the addition of bile acids, specifically DCA, to the clamp protocol resulted in a reduction in the GIRs to similar levels (~12) in WT and TGR5KO mice. Therefore, the absence of TGR5 alone does not appear to affect insulin action significantly in lean mice.
Our studies relied on DCA (hydrophilic, unconjugated) and TCA (hydrophobic, conjugated) to represent the wide spectrum of bile acid species. DCA and TCA are both natural ligands for TGR5, but DCA has a higher affinity for TGR5 based on a cAMP production assay (31). Given the greater effect of DCA on insulin sensitivity in WT animals and the higher affinity of DCA for TGR5, we used only DCA for hyperinsulinemic-euglycemic clamps in TGR5KO mice. Interestingly, although equal molar quantities of DCA and TCA were infused during the clamp, the concentration of DCA was ~10 times higher than the TCA concentration at the end of the clamp. Therefore, whereas the fold increase in the bile acids was similar, the clearance of bile acids is species specific, and so, the absolute levels of DCA and TCA do not represent total flux during the study but rather, a combination of species flux, interconversion, and clearance. The DCA concentration detected after peripheral bile acid infusion in our studies is similar to that detected in the streptozotocin-treated and ob/ob mouse models of diabetes (36), meaning that we induced systemic bile acid levels normally exhibited in the pathophysiological states. Furthermore, although insulin action is similar between WT and TGR5KO, in TGR5KO mice, the basal levels of DCA were lower than those in WT animals. Genetic manipulation has been reported to alter bile acid pool size and bile acid pool composition. Specifically, in TGR5KO mice, total bile acids are decreased (37, 39), and bile acid composition is altered (19, 37) compared with control mice. Additionally, after portal vein infusion, the total serum bile acid levels did not reach those of the WT mice after peripheral infusion of DCA (data not shown). The first-pass extraction of bile acids into the liver is efficient, so it is not unexpected that the total peripheral levels were lower.
Although humans also exhibit an increase in bile acids during the diabetic state and in response to a meal, similar to mice, there are several major differences between mouse and human bile acid physiology. Namely, the primary bile acid in humans is cholic acid, whereas in a mouse, it is muricholic acid (4). Moreover, the amino acid, to which the majority of bile acids are conjugated, differs between mice and humans and glycine and taurine, respectively. In this study, we focused on two bile acids—one conjugated and one unconjugated—which were highly hydrophilic or hydrophobic, respectively, to encompass the spectrum of bile acids. Less emphasis was placed on the specific bile acid species than on a total rise in serum levels. We believe our conclusions encompass the broad spectrum of bile acids, and therefore, the specific differences between mice and humans have little impact on the conclusions.
Finally, the metabolic state and route of bile acid delivery play a major role in the physiologic response to bile acids. These studies were completed in lean animals; however, bile acid signaling and regulation may differ in an obesogenic environment. It is possible that the response to bile acids could be modified by the presence of obesity and the accompanying insulin resistance. Additionally, in the presence of metabolic disorders, there is a chronic elevation in serum bile acids, whereas in these studies, the elevation was acute, as is seen after a meal. Finally, the delivery of bile acids intravenously to alter the bile acid pool rapidly does not mimic the physiological rise in bile acids. Therefore, although these studies unexpectedly determined that an acute elevation in systemic bile acid levels leads to impaired insulin action, future studies are necessary to determine the effects of chronic elevation in the lean setting, the indirect signaling responsible for the alteration in liver insulin action, and how these effects are modified in a metabolically challenging setting.
GRANTS
Support for this work was provided by the National Institute of Diabetes and Digestive and Kidney Diseases (Grants R01 DK105847 to C. R. Flynn and DK078188 to O. P. McGuinness) and Mouse Metabolic Phenotyping Center (MMPC) MICROMouse Program (Grant U24DK076169 to O. P. McGuinness). N. A. Mignemi and T. C. Beck were supported by Training Grants T32DK101003 and T32DK007563, respectively. The Diabetes Research Training Center is also acknowledged (Grant DK020593), as well as the Vanderbilt Hormone and Analytical Services Core, Mass Spectrometry Research Center Core, Digestive Disease Research Center (Grant P30DK058404) and MMPC (Grant U24DK059637).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.J.C., T.C.B., and O.P.M. conceived and designed research; K.E.S., T.J.C., T.C.B., and C.R.F. performed experiments; K.E.S., T.J.C., T.C.B., and O.P.M. analyzed data; K.E.S., T.J.C., T.C.B., N.A.M., and O.P.M. interpreted results of experiments; K.E.S. prepared figures; K.E.S. and T.C.B. drafted manuscript; K.E.S., T.J.C., T.C.B., C.R.F., N.A.M., and O.P.M. edited and revised manuscript; K.E.S., T.J.C., T.C.B., C.R.F., N.A.M., and O.P.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank C. Malabanan and A. Kellakaros for performing catheterization surgeries, L. Chen for managing the animal colony, M. Shiota for performing hepatocyte and nonparenchymal cell isolation, and Dr. Galya Vassilev, Merck, for providing TGR5KO (gpbar1−/−) mice. The authors gratefully acknowledge the use of services provided by the Vanderbilt Hormone and Analytical Services Core, Mass Spectrometry Research Center Core, Digestive Disease Research Center, and Mouse Metabolic Phenotyping Center (MMPC).
REFERENCES
- 1.Ahmad NN, Pfalzer A, Kaplan LM. Roux-en-Y gastric bypass normalizes the blunted postprandial bile acid excursion associated with obesity. Int J Obes 37: 1553–1559, 2013. doi: 10.1038/ijo.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albaugh VL, Banan B, Ajouz H, Abumrad NN, Flynn CR. Bile acids and bariatric surgery. Mol Aspects Med 56: 75–89, 2017. doi: 10.1016/j.mam.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albaugh VL, Flynn CR, Cai S, Xiao Y, Tamboli RA, Abumrad NN. Early increases in bile acids post Roux-en-Y gastric bypass are driven by insulin-sensitizing, secondary bile acids. J Clin Endocrinol Metab 100: E1225–E1233, 2015. doi: 10.1210/jc.2015-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alnouti Y, Csanaky IL, Klaassen CD. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 873: 209–217, 2008. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angelin B, Björkhem I, Einarsson K, Ewerth S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J Clin Invest 70: 724–731, 1982. doi: 10.1172/JCI110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Measuring deuterium enrichment of glucose hydrogen atoms by gas chromatography/mass spectrometry. Anal Chem 83: 3211–3216, 2011. doi: 10.1021/ac200012p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arble DM, Sandoval DA, Seeley RJ. Mechanisms underlying weight loss and metabolic improvements in rodent models of bariatric surgery. Diabetologia 58: 211–220, 2015. doi: 10.1007/s00125-014-3433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayala JE, Bracy DP, Malabanan C, James FD, Ansari T, Fueger PT, McGuinness OP, Wasserman DH. Hyperinsulinemic-euglycemic clamps in conscious, unrestrained mice. J Vis Exp pii: 3188, 2011. doi: 10.3791/3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belgaumkar AP, Vincent RP, Carswell KA, Hughes RD, Alaghband-Zadeh J, Mitry RR, le Roux CW, Patel AG. Changes in bile acid profile after laparoscopic sleeve gastrectomy are associated with improvements in metabolic profile and fatty liver disease. Obes Surg 26: 1195–1202, 2016. doi: 10.1007/s11695-015-1878-1. [DOI] [PubMed] [Google Scholar]
- 10.Bhutta HY, Rajpal N, White W, Freudenberg JM, Liu Y, Way J, Rajpal D, Cooper DC, Young A, Tavakkoli A, Chen L. Effect of Roux-en-Y gastric bypass surgery on bile acid metabolism in normal and obese diabetic rats. PLoS One 10: e0122273, 2015. doi: 10.1371/journal.pone.0122273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burgess SC, Hausler N, Merritt M, Jeffrey FM, Storey C, Milde A, Koshy S, Lindner J, Magnuson MA, Malloy CR, Sherry AD. Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J Biol Chem 279: 48941–48949, 2004. doi: 10.1074/jbc.M407120200. [DOI] [PubMed] [Google Scholar]
- 12.Chávez-Talavera O, Baud G, Spinelli V, Daoudi M, Kouach M, Goossens JF, Vallez E, Caiazzo R, Ghunaim M, Hubert T, Lestavel S, Tailleux A, Staels B, Pattou F. Roux-en-Y gastric bypass increases systemic but not portal bile acid concentrations by decreasing hepatic bile acid uptake in minipigs. Int J Obes 41: 664–668, 2017. doi: 10.1038/ijo.2017.7. [DOI] [PubMed] [Google Scholar]
- 13.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 50: 1955–1966, 2009. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chueh FY, Malabanan C, McGuinness OP. Impact of portal glucose delivery on glucose metabolism in conscious, unrestrained mice. Am J Physiol Endocrinol Metab 291: E1206–E1211, 2006. doi: 10.1152/ajpendo.00608.2005. [DOI] [PubMed] [Google Scholar]
- 15.Costarelli V, Sanders TA. Acute effects of dietary fat composition on postprandial plasma bile acid and cholecystokinin concentrations in healthy premenopausal women. Br J Nutr 86: 471–477, 2001. doi: 10.1079/BJN2001431. [DOI] [PubMed] [Google Scholar]
- 16.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res 50: 2340–2357, 2009. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Barros SG, Balistreri WF, Soloway RD, Weiss SG, Miller PC, Soper K. Response of total and individual serum bile acids to endogenous and exogenous bile acid input to the enterohepatic circulation. Gastroenterology 82: 647–652, 1982. [PubMed] [Google Scholar]
- 18.Ding L, Sousa KM, Jin L, Dong B, Kim BW, Ramirez R, Xiao Z, Gu Y, Yang Q, Wang J, Yu D, Pigazzi A, Schones D, Yang L, Moore D, Wang Z, Huang W. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 64: 760–773, 2016. doi: 10.1002/hep.28689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donepudi AC, Boehme S, Li F, Chiang JY. G-Protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology 65: 813–827, 2017. doi: 10.1002/hep.28707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis 46: 302–312, 2014. doi: 10.1016/j.dld.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flynn CR, Albaugh VL, Cai S, Cheung-Flynn J, Williams PE, Brucker RM, Bordenstein SR, Guo Y, Wasserman DH, Abumrad NN. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat Commun 6: 7715, 2015. doi: 10.1038/ncomms8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glicksman C, Pournaras DJ, Wright M, Roberts R, Mahon D, Welbourn R, Sherwood R, Alaghband-Zadeh J, le Roux CW. Postprandial plasma bile acid responses in normal weight and obese subjects. Ann Clin Biochem 47: 482–484, 2010. doi: 10.1258/acb.2010.010040. [DOI] [PubMed] [Google Scholar]
- 23.Haeusler RA, Camastra S, Nannipieri M, Astiarraga B, Castro-Perez J, Xie D, Wang L, Chakravarthy M, Ferrannini E. Increased bile acid synthesis and impaired bile acid transport in human obesity. J Clin Endocrinol Metab 101: 1935–1944, 2016. doi: 10.1210/jc.2015-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasenour CM, Wall ML, Ridley DE, Hughey CC, James FD, Wasserman DH, Young JD. Mass spectrometry-based microassay of (2)H and (13)C plasma glucose labeling to quantify liver metabolic fluxes in vivo. Am J Physiol Endocrinol Metab 309: E191–E203, 2015. doi: 10.1152/ajpendo.00003.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang C, Wang J, Hu W, Wang C, Lu X, Tong L, Wu F, Zhang W. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett 590: 3233–3242, 2016. doi: 10.1002/1873-3468.12373. [DOI] [PubMed] [Google Scholar]
- 26.Huber RM, Murphy K, Miao B, Link JR, Cunningham MR, Rupar MJ, Gunyuzlu PL Jr, Haws TF Jr, Kassam A, Powell F, Hollis GF, Young PR, Mukherjee R, Burn TC. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 290: 35–43, 2002. doi: 10.1016/S0378-1119(02)00557-7. [DOI] [PubMed] [Google Scholar]
- 27.Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, Teshigawara K, Matsuki Y, Watanabe E, Hiramatsu R, Notohara K, Katayose K, Okamura H, Kahn CR, Noda T, Takeda K, Akira S, Inui A, Kasuga M. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab 3: 267–275, 2006. doi: 10.1016/j.cmet.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Jahansouz C, Xu H, Hertzel AV, Serrot FJ, Kvalheim N, Cole A, Abraham A, Luthra G, Ewing K, Leslie DB, Bernlohr DA, Ikramuddin S. Bile acids increase independently from hypocaloric restriction after bariatric surgery. Ann Surg 264: 1022–1028, 2016. doi: 10.1097/SLA.0000000000001552. [DOI] [PubMed] [Google Scholar]
- 29.Jansen PL, van Werven J, Aarts E, Berends F, Janssen I, Stoker J, Schaap FG. Alterations of hormonally active fibroblast growth factors after Roux-en-Y gastric bypass surgery. Dig Dis 29: 48–51, 2011. doi: 10.1159/000324128. [DOI] [PubMed] [Google Scholar]
- 30.Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol 15: 111–128, 2018. doi: 10.1038/nrgastro.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. J Biol Chem 278: 9435–9440, 2003. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 32.Kim JH, Kim JE, Liu HY, Cao W, Chen J. Regulation of interleukin-6-induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3-SOCS3 pathway. J Biol Chem 283: 708–715, 2008. doi: 10.1074/jbc.M708568200. [DOI] [PubMed] [Google Scholar]
- 33.Kohli R, Bradley D, Setchell KD, Eagon JC, Abumrad N, Klein S. Weight loss induced by Roux-en-Y gastric bypass but not laparoscopic adjustable gastric banding increases circulating bile acids. J Clin Endocrinol Metab 98: E708–E712, 2013. doi: 10.1210/jc.2012-3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohli R, Kirby M, Setchell KD, Jha P, Klustaitis K, Woollett LA, Pfluger PT, Balistreri WF, Tso P, Jandacek RJ, Woods SC, Heubi JE, Tschoep MH, D’Alessio DA, Shroyer NF, Seeley RJ. Intestinal adaptation after ileal interposition surgery increases bile acid recycling and protects against obesity-related comorbidities. Am J Physiol Gastrointest Liver Physiol 299: G652–G660, 2010. doi: 10.1152/ajpgi.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 66: 948–983, 2014. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li T, Francl JM, Boehme S, Ochoa A, Zhang Y, Klaassen CD, Erickson SK, Chiang JY. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem 287: 1861–1873, 2012. doi: 10.1074/jbc.M111.305789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li T, Holmstrom SR, Kir S, Umetani M, Schmidt DR, Kliewer SA, Mangelsdorf DJ. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol 25: 1066–1071, 2011. doi: 10.1210/me.2010-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S, Marcelin G, Blouet C, Jeong JH, Jo YH, Schwartz GJ, Chua S Jr. A gut-brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Mol Metab 8: 37–50, 2018. doi: 10.1016/j.molmet.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, Miyamoto Y, Kanatani A, Tamai Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol 191: 197–205, 2006. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 41.Mertens KL, Kalsbeek A, Soeters MR, Eggink HM. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front Neurosci 11: 617, 2017. doi: 10.3389/fnins.2017.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal 8: e005, 2010. doi: 10.1621/nrs.08005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molinaro A, Wahlström A, Marschall HU. Role of bile acids in metabolic control. Trends Endocrinol Metab 29: 31–41, 2018. doi: 10.1016/j.tem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 44.Mulligan KX, Morris RT, Otero YF, Wasserman DH, McGuinness OP. Disassociation of muscle insulin signaling and insulin-stimulated glucose uptake during endotoxemia. PLoS One 7: e30160, 2012. doi: 10.1371/journal.pone.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakatani H, Kasama K, Oshiro T, Watanabe M, Hirose H, Itoh H. Serum bile acid along with plasma incretins and serum high-molecular weight adiponectin levels are increased after bariatric surgery. Metabolism 58: 1400–1407, 2009. doi: 10.1016/j.metabol.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Noel OF, Still CD, Argyropoulos G, Edwards M, Gerhard GS. Bile acids, FXR, and metabolic effects of bariatric surgery. J Obes 2016: 4390254, 2016. doi: 10.1155/2016/4390254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh K-J, Han H-S, Kim M-J, Koo S-H. CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep 46: 567–574, 2013. doi: 10.5483/BMBRep.2013.46.12.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, Badman MK, Maratos-Flier E, Mun EC, Pihlajamaki J, Auwerx J, Goldfine AB. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 17: 1671–1677, 2009. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powell AA, LaRue JM, Batta AK, Martinez JD. Bile acid hydrophobicity is correlated with induction of apoptosis and/or growth arrest in HCT116 cells. Biochem J 356: 481–486, 2001. doi: 10.1042/bj3560481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prinz P, Hofmann T, Ahnis A, Elbelt U, Goebel-Stengel M, Klapp BF, Rose M, Stengel A. Plasma bile acids show a positive correlation with body mass index and are negatively associated with cognitive restraint of eating in obese patients. Front Neurosci 9: 199, 2015. doi: 10.3389/fnins.2015.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shapiro H, Kolodziejczyk AA, Halstuch D, Elinav E. Bile acids in glucose metabolism in health and disease. J Exp Med 215: 383–396, 2018. doi: 10.1084/jem.20171965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shibao CA, Celedonio JE, Tamboli R, Sidani R, Love-Gregory L, Pietka T, Xiong Y, Wei Y, Abumrad NN, Abumrad NA, Flynn CR. CD36 modulates fasting and preabsorptive hormone and bile acid levels. J Clin Endocrinol Metab 103: 1856–1866, 2018. doi: 10.1210/jc.2017-01982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin AC, Filatova N, Lindtner C, Chi T, Degann S, Oberlin D, Buettner C. Insulin receptor signaling in POMC, but not AgRP, neurons controls adipose tissue insulin action. Diabetes 66: 1560–1571, 2017. doi: 10.2337/db16-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53a.Shiota M, Fujimoto Y, Inagami M, Hiramatsu M, Moriyama M, Kimura K, Ohta M, Sugano T. Adaptive changes in zonation for gluconeogenic capacity in liver lobules of cold-exposed rats. Am J Physiol Endocrinol Metab 265: E559–E564, 1993. doi: 10.1152/ajpendo.1993.265.4.E559. [DOI] [PubMed] [Google Scholar]
- 54.Simonen M, Dali-Youcef N, Kaminska D, Venesmaa S, Käkelä P, Pääkkönen M, Hallikainen M, Kolehmainen M, Uusitupa M, Moilanen L, Laakso M, Gylling H, Patti ME, Auwerx J, Pihlajamäki J. Conjugated bile acids associate with altered rates of glucose and lipid oxidation after Roux-en-Y gastric bypass. Obes Surg 22: 1473–1480, 2012. doi: 10.1007/s11695-012-0673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sinclair P, Docherty N, le Roux CW. Metabolic effects of bariatric surgery. Clin Chem 64: 72–81, 2018. doi: 10.1373/clinchem.2017.272336. [DOI] [PubMed] [Google Scholar]
- 56.Sonne DP, van Nierop FS, Kulik W, Soeters MR, Vilsbøll T, Knop FK. Postprandial plasma concentrations of individual bile acids and FGF-19 in patients with type 2 diabetes. J Clin Endocrinol Metab 101: 3002–3009, 2016. doi: 10.1210/jc.2016-1607. [DOI] [PubMed] [Google Scholar]
- 57.Spinelli V, Lalloyer F, Baud G, Osto E, Kouach M, Daoudi M, Vallez E, Raverdy V, Goossens JF, Descat A, Doytcheva P, Hubert T, Lutz TA, Lestavel S, Staels B, Pattou F, Tailleux A. Influence of Roux-en-Y gastric bypass on plasma bile acid profiles: a comparative study between rats, pigs and humans. Int J Obes 40: 1260–1267, 2016. doi: 10.1038/ijo.2016.46. [DOI] [PubMed] [Google Scholar]
- 58.Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care 32, Suppl 2: S237–S245, 2009. doi: 10.2337/dc09-S355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steele R, Wall JS, De Bodo RC, Altszuler N. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol 187: 15–24, 1956. doi: 10.1152/ajplegacy.1956.187.1.15. [DOI] [PubMed] [Google Scholar]
- 60.Sun W, Zhang D, Wang Z, Sun J, Xu B, Chen Y, Ding L, Huang X, Lv X, Lu J, Bi Y, Xu Q. Insulin resistance is associated with total bile acid level in type 2 diabetic and nondiabetic population: a cross-sectional study. Medicine (Baltimore) 95: e2778, 2016. doi: 10.1097/MD.0000000000002778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taoka H, Yokoyama Y, Morimoto K, Kitamura N, Tanigaki T, Takashina Y, Tsubota K, Watanabe M. Role of bile acids in the regulation of the metabolic pathways. World J Diabetes 7: 260–270, 2016. doi: 10.4239/wjd.v7.i13.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10: 167–177, 2009. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis 28: 220–224, 2010. doi: 10.1159/000282091. [DOI] [PubMed] [Google Scholar]
- 64.Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, Hoos L, Tetzloff G, Levitan D, Murgolo NJ, Keane K, Davis HR Jr, Hedrick J, Gustafson EL. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J 398: 423–430, 2006. doi: 10.1042/BJ20060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiśniewski JR, Vildhede A, Norén A, Artursson P. In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J Proteomics 136: 234–247, 2016. doi: 10.1016/j.jprot.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 66.Xu Y. Recent progress on bile acid receptor modulators for treatment of metabolic diseases. J Med Chem 59: 6553–6579, 2016. doi: 10.1021/acs.jmedchem.5b00342. [DOI] [PubMed] [Google Scholar]
- 67.Young JD. INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30: 1333–1335, 2014. doi: 10.1093/bioinformatics/btu015. [DOI] [PMC free article] [PubMed] [Google Scholar]