

Abstract
C‐to‐U RNA editing is the conversion from cytidine to uridine at RNA level. In plants, the genes undergo C‐to‐U RNA modification are mainly chloroplast and mitochondrial genes. Case studies have identified the roles of C‐to‐U editing in various biological processes, but the functional consequence of the majority of C‐to‐U editing events is still undiscovered. We retrieved the deep sequenced transcriptome data in roots and shoots of Arabidopsis thaliana and profiled their C‐to‐U RNA editomes and gene expression patterns. We investigated the editing level and conservation pattern of these C‐to‐U editing sites. The levels of nonsynonymous C‐to‐U editing events are higher than levels of synonymous events. The fraction of nonsynonymous editing sites is higher than neutral expectation. Highly edited cytidines are more conserved at DNA level, and the gene expression levels are correlated with C‐to‐U editing levels. Our results demonstrate that the global C‐to‐U editome is shaped by natural selection and that many nonsynonymous C‐to‐U editing events are adaptive. The editing mechanism might be positively selected and maintained and could have profound effects on the modified RNAs.
Keywords: adaptive, Arabidopsis thaliana, conservation, C‐to‐U RNA editing, expression, positive selection
1. INTRODUCTION
Most types of RNA modifications do not change the base‐pairing property of the modified nucleotides, with the exceptions of A‐to‐I and C‐to‐U RNA editing. A‐to‐I RNA editing is prevalent and well studied in animal species while in the plant kingdom, C‐to‐U RNA editing is enriched in the chloroplast and mitochondrial genes (Gray, 2009; Gray & Covello, 1993; Mulligan, Williams, & Shanahan, 1999; Rajasekhar & Mulligan, 1993; Tang & Luo, 2018; Yu & Schuster, 1995). Other types of potentially recoding modifications include the opposite U‐to‐C substitution (Takenaka, Zehrmann, Verbitskiy, Hartel, & Brennicke, 2013), which is much less frequent than C‐to‐U editing. The factor responsible for C‐to‐U RNA editing is PPR (pentatricopeptide repeat) protein, which recognizes single‐strand RNAs (Yin et al., 2013).
The biological functions of particular C‐to‐U editing events were consecutively discovered (Takenaka, 2010; Takenaka, Neuwirt, & Brennicke, 2004; Takenaka, Verbitskly, Merwe, Zehrmann, & Brennicke, 2008; Tang, Kobayashi, Suzuki, Matsumoto, & Muranaka, 2010), and systematic identification of these editing sites appeared in recent years with the help of next‐generation sequencing (NGS) technique. Bioinformatic tools are also developed to facilitate researchers to accurately identify C‐to‐U RNA editing sites from the big data (Du, Jia, & Li, 2009; Li et al., 2019; Lo Giudice, Hernandez, Ceci, Pesole, & Picardi, 2019; Picardi & Pesole, 2013), among which REDItools (Picardi & Pesole, 2013) is not restricted to plant species since it has been broadly cited by the animal A‐to‐I RNA editing studies. With increasing numbers of editing sites identified in plants, database is needed to store the editing sites found by different studies. Up to now, REDIdb collected over 25,000 RNA editing sites among 281 species (Lo Giudice, Pesole, & Picardi, 2018), and database PED even contained the editing sites in 1,621 plant species (Li et al., 2019). All these tools and databases give convenience to people of the C‐to‐U RNA editing community.
Researchers have conducted expression and RNA editing analyses on Arabidopsis plastid genes and found that the editing efficiency varied moderately across different tissues (Tseng, Lee, Chung, Sung, & Hsieh, 2013). While most editing sites showed similar levels between green and non‐green seedlings, some other sites were lost in non‐green tissues. Given this observation that many editing sites were shared across different tissues while a few other sites showed tissue specificity (Tseng et al., 2013), it is still unclear whether the steadily edited sites are more essential and beneficial and therefore exhibit higher editing levels. Moreover, it remains to be tested whether the global editome is adaptive.
Recently another study profiled the landscape of C‐to‐U RNA editome in 17 diverse angiosperm species (Edera, Gandini, & Sanchez‐Puerta, 2018). The authors found that nonsynonymous editing sites (at the 1st and 2nd codon positions) showed higher conservation levels as well as editing levels than synonymous editing sites (at the 3rd codon position). This finding, based on edited positions, likely shows that nonsynonymous editing is selectively advantageous and maintained by natural selection. However, further analysis is needed to verify this assumption because the genome‐wide nonsynonymous sites naturally undergo stronger constraint than synonymous sites (the substitution rate dN is usually smaller than dS). Thus, it would be helpful to compare the edited versus unedited sites to exclude the effect from genomic background. Besides, combining editing analysis with gene expression analysis would also be helpful. It is established that highly expressed genes are usually more conserved (and likely to be functionally more essential) and this idea prompts us to test whether these genes undergo stronger editing.
In this study, we retrieved the deep sequenced transcriptome data in roots and shoots of Arabidopsis thaliana and profiled their C‐to‐U RNA editomes and gene expression patterns. We draw to the conclusion that the global C‐to‐U editome is favored by natural selection. The editing mechanism might be positively selected and maintained.
2. METHODS
2.1. Data collection
The reference genome and CDS sequences of Arabidopsis thaliana were downloaded from TAIR database. The TAIR 9 version of annotation was used. The mRNA‐seq data of Arabidopsis thaliana (three replicates for roots and shoots) were retrieved from a previous study (Hsu et al., 2016). As described by the paper, the Arabidopsis Columbia‐0 (abbr. Col‐0) seeds (around 1,500 seeds per vessel) were surface sterilized and imbibed at 4°C for 2 days. They were grown hydroponically on fine nylon mesh supported by a customized rack in Magenta vessel GA‐7‐3 (Sigma; V8380) with filtered sterile liquid media and shaken at 85 rpm under 16‐hr light (110–115 µmol m−2 s−1 from cool white fluorescent bulbs) and 8‐hr dark at 22°C (Hsu et al., 2016). The tissues were collected from 4‐day old seedlings.
2.2. NGS data processing
We mapped the mRNA‐seq reads to the reference CDS sequence using Bowtie2 (version 2.2) (Langmead & Salzberg, 2012). The uniquely mapped reads were extracted using SAMtools (version 1.5) (Li et al., 2009). The variant calling process is accomplished by SAMtools “mpileup” with default parameters. The output file of mpileup is “vcf” format, which contains one variation site per line. The information for each variation site includes total depth on each site, the reference allele count and each alternative allele count. Sites with more than one variation type are discarded. Variation level = alternative allele count/ total depth. Take C‐to‐U RNA editing sites for instance, editing level = T/(C + T). To avoid the false‐positive variants caused by technical limitations and polymorphism at DNA level, the variant sites with levels between 0.05 and 0.95 and appear in all six samples were regarded as RNA editing events (extremely low level is possibly caused by sequencing errors and extremely high level is likely caused by mutations at DNA level) (Peng et al., 2012; Ramaswami et al., 2013; Yu et al., 2016). Gene expression levels are measured by RPKM (reads per kilobase per million mapped reads). The total number of (unique) reads mapped to each gene is counted. We conducting gene count, the longest isoform of each gene is chosen. If two isoforms have the same length, they are sorted alphabetically. Htseq‐count (Anders, Pyl, & Huber, 2015) is used to count the reads for each gene.
2.3. Conservation and divergence analysis
The CDS sequences of Arabidopsis thaliana and Arabidopsis lyrata (http://plants.ensembl.org/) were translated into protein sequences. We aligned these protein sequences (blastp, version 2.2) (Camacho et al., 2009) between two sibling species to define the best‐matched pair of genes (orthologous gene) according to the lowest E‐value (E‐value < 1e−5 by default) and high identity. By this step, we obtained the orthologous gene list between the two species. Next, the CDS sequences were aligned (tranalign, version 6.6) (Rice, Longden, & Bleasby, 2000) within each pair of orthologous genes. The orthologous sites are split manually according to the CDS alignment files. Given the aligned CDS of each pair of Arabidopsis thaliana and Arabidopsis lyrata genes, the nonsynonymous and synonymous substitution rates are calculated by yn00 (Xu & Yang, 2013).
2.4. Differential and non‐differential editing sites
For each C‐to‐U editing site, we calculated the standard deviation (SD) of editing levels across all samples. We ranked the sites with increasing SD and divided them into two halves, termed non‐differential sites and differential sites. “sd” was performed in R language with function “sd().” The same idea is applied to the dN dS comparison. We have divided the editing sites into two halves according to the dN values of host genes, or divided them into three equal bins with increasing dN/dS values.
2.5. Statistical analysis and code availability
All statistical analyses and the graphic work were conducted in R environment (http://www.R-project.org/) (version 3.3.3). The Wilcoxon rank sum tests were performed with command “wilcox.test()” and Fisher's exact tests were performed with command “fisher.test().”
3. RESULTS
3.1. The profiles of C‐to‐U RNA editome in Arabidopsis thaliana
The mRNA‐seq data of Arabidopsis thaliana were retrieved from a previous study (Hsu et al., 2016). After variant calling process (METHODS), in total, 174 variation sites in CDS were identified and 130 of them (75%) are C‐to‐T variations (Figure 1a), indicating that the C‐to‐U RNA editing events are reliably detected. The majority (123, 94.6%, Data S1) of these 130 C‐to‐U editing sites are located in chloroplast or mitochondrial genes (Figure 1b). Interestingly, the context flanking the focal editing sites shows considerable differences with the background (Figure 1c), with guanosine (G) strongly avoided upstream and slightly favored downstream the focal editing sites. When we look at the C‐to‐U editing level (edited allele divided by total coverage on a site), we found no remarkable differences between chloroplast and mitochondrial editing sites (Figure 1d).
Figure 1.
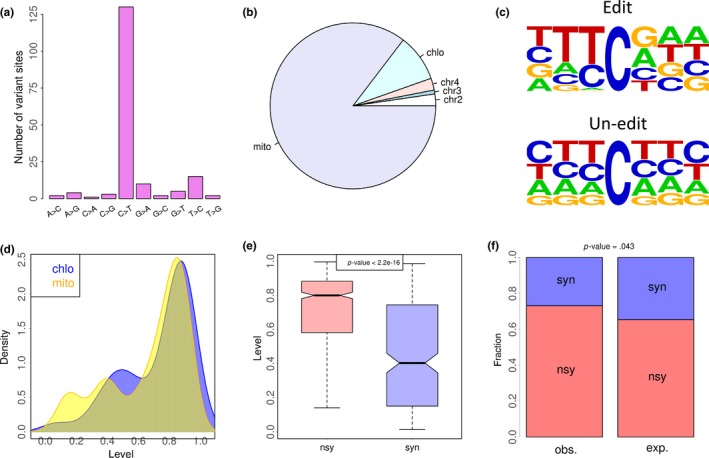
C‐to‐U RNA editome in CDS of Arabidopsis thaliana and the signals of adaptation. (a) Fractions of different types of variation sites. The variation sites in CDS region are displayed. One hundred and thirty (75%) out of 174 variants are C‐to‐T changes. (b) Genomic locations of the 130 coding C‐to‐U editing sites. One hundred and twenty‐three of them are located in mitochondria or chloroplast. Chromosome names are shown in the pie chart. mito, mitochondrion; chlo, chloroplast. (c) Context flanking the focal C‐to‐U editing sites (123 sites) or the background (unedited) cytidines. Only the cytidine sites in the CDS of mitochondria and chloroplast genes are considered. (d) C‐to‐U editing levels of the 123 mitochondrial and chloroplast editing events. The editing sites in three biological replicates (roots and shoots) are pooled. (e) C‐to‐U editing levels of 90 nonsynonymous (nsy) and 33 synonymous (syn) editing events. The editing sites in three biological replicates (roots and shoots) are pooled. p‐value is calculated by Wilcoxon rank sum test. (f) Fractions of nonsynonymous and synonymous editing sites. Observed (obs.) and expected (exp.) values are shown. The number of expected nonsynonymous and synonymous sites is defined by assuming all cytidines in the CDS in mitochondria or chloroplast genes to be mutated to thymidine. p‐value is calculated by Fisher's exact test
3.2. Signals of adaptation
Among these 123 C‐to‐U editing sites in chloroplast or mitochondrial genes (Data S1), 90 were nonsynonymous (cause AA change) and 33 were synonymous (AA is unchanged). Interestingly, the levels of nonsynonymous C‐to‐U events are significantly higher than the levels of synonymous sites (Figure 1e, p < 2.2e−16, Wilcoxon rank sum test), suggesting that high level of nonsynonymous C‐to‐U editing is favored by natural selection. Moreover, given the fact that 73% (90/123) of C‐to‐U editing sites are nonsynonymous, if we mutate all cytidines to thymidines in chloroplast and mitochondrial genes, the expected fraction of nonsynonymous mutations is significantly lower (Figure 1f, p < .05, Fisher's exact test). This pattern again indicates that many nonsynonymous C‐to‐U editing events are adaptive and maintained by positive selection.
We next investigated the DNA‐level conservation of the edited cytidines. We aligned the CDS between Arabidopsis thaliana and Arabidopsis lyrata to determine whether a cytidine is conserved in the two species (METHODS). We divided the 90 nonsynonymous editing sites into conserved and non‐conserved groups according to whether the orthologous site in Arabidopsis lyrata is cytidine. The result shows that conserved editing sites have higher editing levels (Figure 2a, p < .001, Wilcoxon rank sum test). This trend informs us that the highly edited cytidine sites should be selectively advantageous so that the evolution on their DNA sites is slowed down in order to maintain the editing status.
Figure 2.
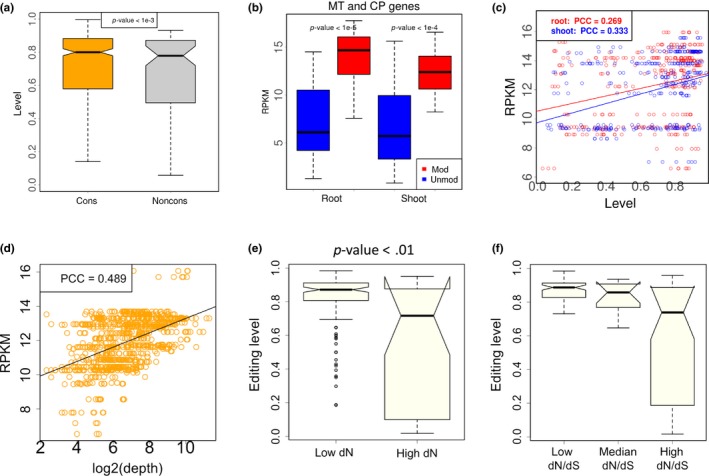
Levels of C‐to‐U RNA editing in CDS of Arabidopsis thaliana and the correlation with gene expression level. (a) C‐to‐U editing levels of the nonsynonymous editing sites. These editing sites are classified into two categories according to the conservation at DNA level. If the orthologous site in Arabidopsis lyrata is cytidine, then this editing site is conserved (cons). The other editing sites are non conserved (noncons). In this plot, the nonsynonymous editing sites in three biological replicates (roots and shoots) are pooled. p‐value is calculated by Wilcoxon rank sum test. (b) Gene expression levels of the C‐to‐U edited genes and the unedited genes in mitochondria and chloroplast. RPKM (reads per kilobase per million mapped reads) is used to measure the gene expression level. Three biological replicates (roots or shoots) are pooled. MT, mitochondria; CP, chloroplast; mod, modified (edited) genes; unmod, unmodified (unedited) genes. p‐value is calculated by Wilcoxon rank sum test. (c) Pearson's correlation between editing level of each site and the expression level (RPKM) of its host gene. PCC, Pearson's correlation coefficient. (d) Pearson's correlation between sequencing depth (coverage) of each editing site and the expression level (RPKM) of its host gene. (e) Comparison between editing levels in conserved genes (low dN) and non‐conserved genes (high dN). Editing sites are divided into two halves according to the dN values of host genes. p‐value is calculated by Wilcoxon rank sum test. (f) Comparison of editing levels in different categories. Editing sites are divided into three equal bins according to the dN/dS values of host genes
When we link C‐to‐U editing events with gene expression levels, we found that edited genes have higher expression levels than the unedited genes in chloroplast and mitochondria (Figure 2b, p < 1e−4 in both cases, Wilcoxon rank sum tests) and that the editing level of a site is positively correlated with the expression level of the host gene (Figure 2c). Since highly expressed genes are usually more conserved and functionally more important, our observation indicates that the C‐to‐U editing events might be evolutionarily adaptive so that they are favored by highly expressed genes.
A potential trick here is that editing events are more likely to be detected in highly expressed genes, so that the result in Figure 2b might be due to detection bias. However, we intend to solve this problem by using the editing level as a variable instead of the occurrence of editing events. The C‐to‐U editing level is positively correlated with gene expression level (Figure 2c). Editing level = edited reads/ total reads covering a site (depth). The number of total reads (depth) is intuitively positively correlated with gene expression level (Figure 2d), so in theory editing level should have a negative correlation with expression level. However, we observed the opposite trend (Figure 2c). This result excludes the potential bias of gene expression in the editing level analysis.
We also investigated the relationship between editing level and the gene conservation level (dN and dN/dS) (METHODS). Even the gene expression level suffers from detection bias, the dN and dS values are purely calculated from genomic sequences and are unaffected by the NGS reads. We divided the editing sites into two halves according to the dN values of host genes. We observed that editing level is generally higher in genes with higher conservation level (Figure 2e, p < .01, Wilcoxon rank sum test). Considering that dN is often negatively correlated with expression level, which makes it difficult to tease apart what factors might contribute to differences in editing between genes. We next divided the editing sites into three equal bins according to the dN/dS values of host genes. We found that the pattern observed for dN also holds for dN/dS (Figure 2f), suggesting that C‐to‐U editing occurs in more conserved genes.
We also stress the reason for discarding the sites with extremely high levels. Here, we display the variation profile between level 0.95 ~ 1 (Figure 3a). The variation types do not enrich C‐to‐T mutations and the variant sites are not enriched in mitochondrial or chloroplast genes. This mutation profile resembles the landscape of DNA mutations, suggesting that these sites are not bona fide RNA modification sites and should not be included in our analyses.
Figure 3.
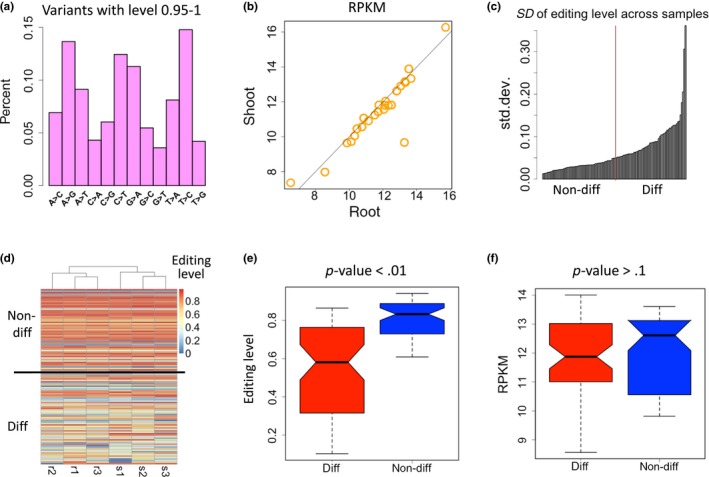
Editing levels of highly edited sites are less variable. (a) Percentages of each mutation type of the variation with level 0.95 ~ 1. (b) Correlation between the expression level (RPKM) of edited genes in roots and shoots. (c) Standard deviation (SD) of editing level across the samples. The lower half is termed non‐differential sites and the higher half is termed differential sites. (d) Heatmap showing the editing level of each site in all six samples (r, root; s, shoot). The sites are sorted by the level difference between roots and shoots. Differential sites (diff) have larger difference of editing levels between roots and shoots, and vice versa. (e) Editing levels of differential and non‐differential sites. p‐value is calculated by Wilcoxon rank sum test. (f) Expression levels (RPKM) of the host genes of differential and non‐differential sites. p‐value is calculated by Wilcoxon rank sum test
3.3. Highly edited sites have less variable editing levels
It is conceivable that the editing events and levels could be tissue‐specific as previous study revealed (Tseng et al., 2013). The tissue‐specific sites could be caused by the tissue‐specific expressed genes. Our strict pipeline only included the editing sites appearing in all samples (three root samples and three shoot samples), which consequently discarded the root‐ or shoot‐specific genes or sites. We ask that if highly edited sites are more likely to be beneficial and functional, then they should exhibit consistent editing level to assure their proper function.
Given the fact that the edited genes show highly correlated expression levels between roots and shoots (Figure 3b), our editing level comparison would not suffer from detection bias across different tissues. We rank the editing sites by the editing level difference between the root and shoot samples (standard deviation across the samples, Figure 3c, also see METHODS). Differential sites (diff) have larger difference of editing levels across the samples, and vice versa (Figure 3c,d). We find that the less variable half of sites have significantly higher editing levels than the more variable half of editing sites (Figure 3e, p < .001, Wilcoxon rank sum test). It is possible that the highly edited sites (mostly nonsynonymous sites) are more essential and functional so that their editing levels tend to be “fixed.” We further tested that the host genes of differential or non‐differential sites have similar expression levels (Figure 3f), indicating that the variation observed in editing level might not be caused by gene expression difference.
4. DISCUSSION
According to Edera et al. (2018, their findings could draw to a conclusion that the nonsynonymous editing events are selectively favored, which is similar to ours. However, their multiple angiosperm study revealed another interesting trend that highly edited cytidines were likely to be replaced with thymidines during genome evolution. Although other study also discovered that the loss of editing events in kin species might be due to the changed editing factor (Kawabe et al., 2019), the C‐to‐T DNA switch found by Edera et al. caught our interest. If the advantage of RNA editing is increasing the protein diversity, then the C‐to‐T‐mutated DNA would actually abolish this diversity. An alternative explanation for the C‐to‐T DNA switch is, the edited isoform (U/T version) is more advantageous than the unedited isoform (C version), so that those extremely highly edited sites are almost equivalent to a permanent DNA mutation. This idea could be supported by our observation that highly edited sites have less variable editing levels (Figure 3), which possibly reflects the necessity for steady and consistent editing level for these sites. At this stage, why these edited genes do not directly change their DNA sequences before the occurrence of RNA editing remains unknown.
Here, we show what patterns of editing would look like if C‐to‐U editing is generally selectively neutral, deleterious, or beneficial. Positive selection: in a previous paper studying A‐to‐I RNA editing sites in cephalopods (Liscovitch‐Brauer et al., 2017), the authors proved the positive selection on RNA editing events by showing that (a) The fraction of nonsynonymous editing events is higher than the genomic background; (b) The nonsynonymous editing levels are generally higher than the synonymous editing levels. In our study, we observed exactly the same pattern in Arabidopsis C‐to‐U RNA editome. Slightly deleterious or neutral: the literatures on C‐to‐U and A‐to‐I RNA editing in human (Liu & Zhang, 2018; Xu & Zhang, 2014) proved the non‐adaptive feature of the RNA editing events by showing that (a) the fraction of nonsynonymous editing events is lower than the fraction of synonymous editing; (b) the nonsynonymous editing levels are no higher than the synonymous editing levels. The author concludes that these editing events are slightly deleterious or neutral under the observed patterns. We surmise that if C‐to‐U editing in Arabidopsis is slightly deleterious or neutral, it should exhibit those patterns as described above.
Our study systematically demonstrates that the C‐to‐U editing events in Arabidopsis thaliana are overall adaptive and favored by natural selection. Together with previous literatures, the advantage of this editing mechanism might be the amino acid changes conferred by the high‐level nonsynonymous editing events. Our work analyzed the C‐to‐U RNA editome from novel aspects, which broadened our horizon in the Arabidopsis community and should also be appealing to the evolutionary phytologists.
CONFLICT OF INTEREST
The authors declare they have no conflict of interest.
AUTHOR CONTRIBUTIONS
Lai Wei designed and supervised this research. Duan Chu and Lai Wei analyzed the data. Duan Chu and Lai Wei wrote this article.
Supporting information
ACKNOWLEDGMENTS
We thank all members in Wei Lab for their constructive suggestions to this project.
Chu D, Wei L. The chloroplast and mitochondrial C‐to‐U RNA editing in Arabidopsis thaliana shows signals of adaptation. Plant Direct. 2019;3:1–7. 10.1002/pld3.169
Funding information
This research was financially supported by the National Natural Science Foundation of China (Grant no. 31770213). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- Anders, S. , Pyl, P. T. , & Huber, W. (2015). HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10, 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, P. F. , Jia, L. Y. , & Li, Y. D. (2009). CURE‐chloroplast: A chloroplast C‐to‐U RNA editing predictor for seed plants. Bioinformatics, 10(1), 135 10.1186/1471-2105-10-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edera, A. A. , Gandini, C. L. , & Sanchez‐Puerta, M. V. (2018). Towards a comprehensive picture of C‐to‐U RNA editing sites in angiosperm mitochondria. Plant Molecular Biology, 97, 215–231. 10.1007/s11103-018-0734-9 [DOI] [PubMed] [Google Scholar]
- Gray, M. W. (2009). RNA editing in plant mitochondria: 20 years later. IUBMB Life, 61, 1101–1104. 10.1002/iub.272 [DOI] [PubMed] [Google Scholar]
- Gray, M. W. , & Covello, P. S. (1993). RNA editing in plant‐mitochondria and chloroplasts. The FASEB Journal, 7, 64–71. 10.1096/fasebj.7.1.8422976 [DOI] [PubMed] [Google Scholar]
- Hsu, P. Y. , Calviello, L. , Wu, H. Y. L. , Li, F. W. , Rothfels, C. J. , Ohler, U. , & Benfey, P. N. (2016). Super‐resolution ribosome profiling reveals unannotated translation events in Arabidopsis . Proceedings of the National Academy of Sciences of the United States of America, 113, E7126–E7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe, A. , Furihata, H. Y. , Tsujino, Y. , Kawanabe, T. , Fujii, S. , & Yoshida, T. (2019). Divergence of RNA editing among Arabidopsis species. Plant Science, 280, 241–247. 10.1016/j.plantsci.2018.12.009 [DOI] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–354. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , … 1000 Genome Project Data Processing Subgroup . (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Xia, L. , Zhang, Y. , Niu, G. , Li, M. , Wang, P. , … Zhang, Z. (2019). Plant editosome database: A curated database of RNA editosome in plants. Nucleic Acids Research, 47, D170–D174. 10.1093/nar/gky1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch‐Brauer, N. , Alon, S. , Porath, H. T. , Elstein, B. , Unger, R. , Ziv, T. , … Eisenberg, E. (2017). Trade‐off between transcriptome plasticity and genome evolution in cephalopods. Cell, 169(2), 191–202.e11. 10.1016/j.cell.2017.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , & Zhang, J. (2018). Human C‐to‐U coding RNA editing is largely nonadaptive. Molecular Biology and Evolution, 35(4), 963–969. 10.1093/molbev/msy011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Giudice, C. , Hernandez, I. , Ceci, L. R. , Pesole, G. , & Picardi, E. (2019). RNA editing in plants: A comprehensive survey of bioinformatics tools and databases. Plant Physiology and Biochemistry, 137, 53–61. 10.1016/j.plaphy.2019.02.001 [DOI] [PubMed] [Google Scholar]
- Lo Giudice, C. , Pesole, G. , & Picardi, E. (2018). REDIdb 3.0: A comprehensive collection of RNA editing events in plant organellar genomes. Frontiers in Plant Science, 9:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan, R. M. , Williams, M. A. , & Shanahan, M. T. (1999). RNA editing site recognition in higher plant mitochondria. Journal of Heredity, 90, 338–344. 10.1093/jhered/90.3.338 [DOI] [PubMed] [Google Scholar]
- Peng, Z. , Cheng, Y. , Tan, B.‐M. , Kang, L. , Tian, Z. , Zhu, Y. , … Wang, J. (2012). Comprehensive analysis of RNA‐Seq data reveals extensive RNA editing in a human transcriptome. Nature Biotechnology, 30, 253–260. 10.1038/nbt.2122 [DOI] [PubMed] [Google Scholar]
- Picardi, E. , & Pesole, G. (2013). REDItools: High‐throughput RNA editing detection made easy. Bioinformatics, 29, 1813–1814. 10.1093/bioinformatics/btt287 [DOI] [PubMed] [Google Scholar]
- Rajasekhar, V. K. , & Mulligan, R. M. (1993). RNA editing in plant‐mitochondria ‐ alpha‐phosphate is retained during C‐to‐U conversion in messenger‐RNAs. The Plant Cell, 5, 1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami, G. , Zhang, R. , Piskol, R. , Keegan, L. P. , Deng, P. , O'Connell, M. A. , & Li, J. B. (2013). Identifying RNA editing sites using RNA sequencing data alone. Nature Methods, 10, 128–132. 10.1038/nmeth.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, P. , Longden, I. , & Bleasby, A. (2000). EMBOSS: The European molecular biology open software suite. Trends in Genetics, 16, 276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- Takenaka, M. (2010). MEF9, an E‐subclass pentatricopeptide repeat protein, is required for an RNA editing event in the nad7 transcript in mitochondria of Arabidopsis . Plant Physiology, 152, 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka, M. , Neuwirt, J. , & Brennicke, A. (2004). Complex cis‐elements determine an RNA editing site in pea mitochondria. Nucleic Acids Research, 32, 4137–4144. 10.1093/nar/gkh763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka, M. , Verbitskly, D. , van der Merwe, J. A. , Zehrmann, A. , & Brennicke, A. (2008). The process of RNA editing in plant mitochondria. Mitochondrion, 8, 35–46. 10.1016/j.mito.2007.09.004 [DOI] [PubMed] [Google Scholar]
- Takenaka, M. , Zehrmann, A. , Verbitskiy, D. , Hartel, B. , & Brennicke, A. (2013). RNA editing in plants and its evolution. Annual Review of Genetics, 47, 335–352. 10.1146/annurev-genet-111212-133519 [DOI] [PubMed] [Google Scholar]
- Tang, J. W. , Kobayashi, K. , Suzuki, M. , Matsumoto, S. , & Muranaka, T. (2010). The mitochondrial PPR protein LOVASTATIN INSENSITIVE 1 plays regulatory roles in cytosolic and plastidial isoprenoid biosynthesis through RNA editing. The Plant Journal, 61, 456–466. 10.1111/j.1365-313X.2009.04082.x [DOI] [PubMed] [Google Scholar]
- Tang, W. , & Luo, C. (2018). Molecular and functional diversity of RNA editing in plant mitochondria. Molecular Biotechnology, 60, 935–945. 10.1007/s12033-018-0126-z [DOI] [PubMed] [Google Scholar]
- Tseng, C. C. , Lee, C. J. , Chung, Y. T. , Sung, T. Y. , & Hsieh, M. H. (2013). Differential regulation of Arabidopsis plastid gene expression and RNA editing in non‐photosynthetic tissues. Plant Molecular Biology, 82, 375–392. 10.1007/s11103-013-0069-5 [DOI] [PubMed] [Google Scholar]
- Xu, B. , & Yang, Z. (2013). PAMLX: A graphical user interface for PAML. Molecular Biology and Evolution, 30, 2723–2724. 10.1093/molbev/mst179 [DOI] [PubMed] [Google Scholar]
- Xu, G. , & Zhang, J. (2014). Human coding RNA editing is generally nonadaptive. Proceedings of the National Academy of Sciences of the United States of America, 111, 3769–3774. 10.1073/pnas.1321745111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, P. , Li, Q. , Yan, C. , Liu, Y. , Liu, J. , Yu, F. , … Yan, N. (2013). Structural basis for the modular recognition of single‐stranded RNA by PPR proteins. Nature, 504, 168–171. 10.1038/nature12651 [DOI] [PubMed] [Google Scholar]
- Yu, W. , & Schuster, W. (1995). Evidence for a site‐specific cytidine deamination reaction involved in C to U RNA editing of plant‐mitochondria. Journal of Biological Chemistry, 270, 18227–18233. 10.1074/jbc.270.31.18227 [DOI] [PubMed] [Google Scholar]
- Yu, Y. , Zhou, H. , Kong, Y. , Pan, B. , Chen, L. , Wang, H. , … Li, X. (2016). The landscape of A‐to‐I RNA editome is shaped by both positive and purifying selection. PLoS Genetics, 12, e1006191 10.1371/journal.pgen.1006191 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials