

Dear Editor,
Sex in mammals is genetically determined and defined at the cellular level by the sex chromosome constitution (XY males and XX females) and at the phenotypic level by the development of gender-specific anatomy, physiology, and behavior.1 46, XY complete gonadal dysgenesis (46, XY CGD), first reported by Swyer in 1955,2 is a rare congenital condition with completely or partially disordered gonadal development, leading to discordance between the genetic, gonadal, and phenotypic sex. Affected patients have a 46, XY karyotype, a female phenotype, normal female external genitalia, and CGD (“streak gonads”) without sperm production and follicular or steroid function.3 Moreover, the streak gonads in individuals with 46, XY CGD have a high propensity toward malignancy, and especially dysgerminoma, which occurs in about 20%–50% of patients.1 Most cases of 46, XY CGD are sporadic, but familial cases have also been reported. It is estimated that mutations in sex-determining region Y (SRY) gene were found in 30% of 46, XY CGD cases, and other cases have unknown genetic origins,4 although mutations in several genes involved in testis differentiation may be causative, such as nuclear receptorsubfamily 5 group A member 1 (NR5A1), SRY-related HMG box genes 9 (SOX9), Wilms’ tumor-4 (WNT4), dosage-sensitive sex reversal (DAX1), and desert hedgehog (DHH).5 Here, we describe a de novo frameshift mutation leading to a truncated and dysfunctional form of the SRY protein.
A 35-year-old female, married for 7 years, was referred to the urology clinic because of concerns about gonadal tumors. She presented for evaluation at 29 years due to the absence of menarche. Physical examination showed a patient 1.64-m tall and weighing 57.5 kg with a female appearance and voice and with no beard or laryngeal prominence. Tanner Stage II breast development and Tanner Stage I pubic hairs were noted. Gynecological examination confirmed normal female external genitalia, and the vagina was well canalized. No palpable mass was identified in the groin. Cytogenetic analysis revealed a karyotype of 46, XY with no evidence of mosaicism, and C-banding analysis further confirmed the existence of a Y chromosome. Y-chromosome microdeletions detected by multiplex polymerase chain reaction (PCR) analysis showed the presence of SRY gene. Pelvis imaging with ultrasonography and computed tomography (CT) scanning revealed a hypoplastic uterus with a thin endometrium and bilateral fallopian tubes, while no ovaries were visualized. Laparoscopy was performed to detect the ovaries and evaluate the risk of tumor development. The bilateral streak gonads were noted (Figure 1a) and then resected. Histological examination of the fallopian tube and streak gonad showed nonneoplastic changes. Secretory cells and ciliated cells could be found in the oviduct (Figure 1b). Primordial follicles were detected in ovarian tissue (Figure 1c).
Figure 1.
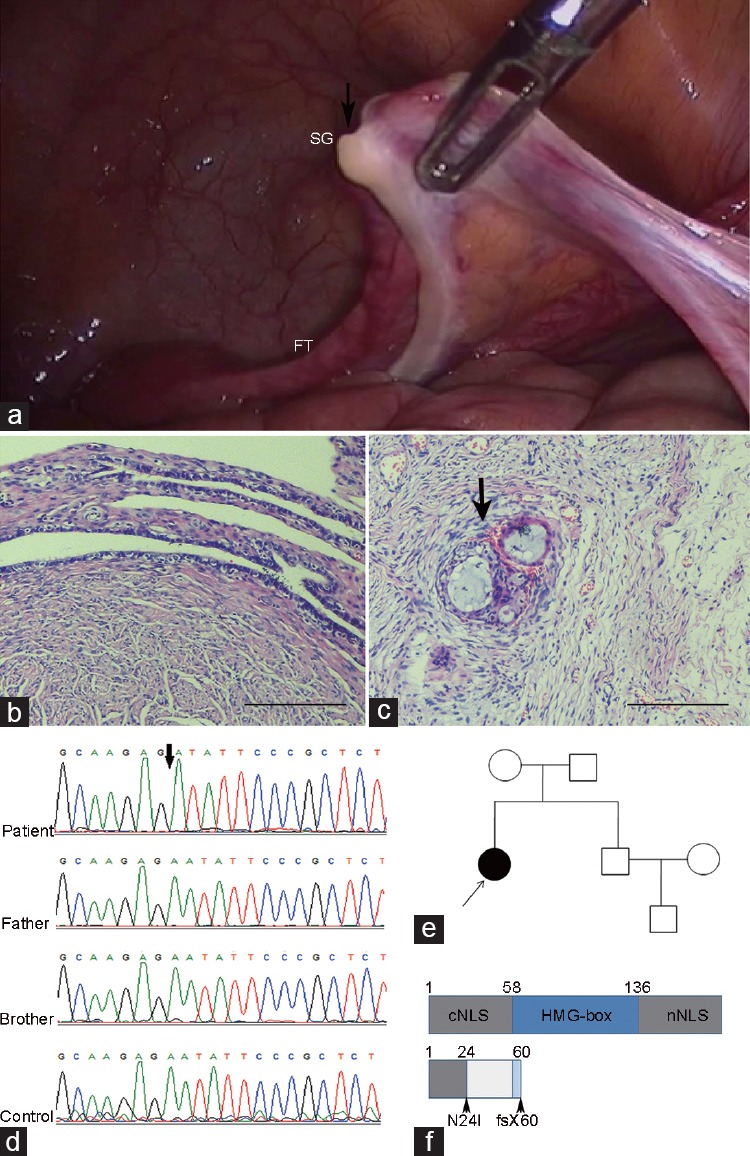
Laparoscopic, histopathological, and genetical examination of the patient with poor pubertal development. (a) Gonad imaging by laparoscopy. (b) Secretory cells and ciliated cells could be found in the oviduct. (c) Primordial follicles could be seen in the streak gonad (marked by arrow). (d) The deletion nucleotide indicated by the arrows was found in the patient, which was not shared by her father, brother or control. (e) According to the family tree, the SRY mutation is de novo. (f) Schematic description of the full-length and truncated SRY proteins. The p.Asn24Ile (N24I) mutation resides in the nNLS domain, and the fsTer60 resides in the HMG box. Scale bars = 100 mm. SG: streak gonad (marked by arrow); FT: fallopian tube; SRY: sex-determining region Y; NLS: nuclear localization; HMG: high mobility group.
On admission, she was asked for a familial history to identify possible causes for the disorder. The patient was born at term by normal delivery to nonconsanguineous parents, and her mother denied any use of hormone drugs or exposure to radioactive substances during her pregnancy. The patient has a younger brother and an 8-year-old nephew. There was no other family history of delayed puberty or infertility.
Genomic DNA was isolated from the blood leukocytes of the patient, her parents, her sibling, and five fertile control males. The study was approved by the Ethics Committee of Shanghai General Hospital (Shanghai, China). Written informed consent was obtained from each participant. The human SRY gene (reference sequence: NM_003140.2) was amplified by the following two sets of primers, which were designed using Primer 5.0 (PREMIER Biosoft International, Palo Alto, CA, USA): SRY-forward 01: 5’-gaatacattgtcagggtactagggg-3’; SRY-reverse 01: 5’-caggctcacttctggatgtctta-3’; SRY-forward 02: 5’-gggcaagtagtcaacgttactga-3’; and SRY-reverse 02: 5’-ggctcacttctggatgtcttatttctt-3’. The PCR products were resolved on a 2.0% agarose gel, and then purified with gel (Nest, Wuxi, China). The resulting purified DNA was sequenced. A SRY gene mutation was identified between the patient and the reference genome (reference sequence: NM_003140.2), and then screened against the NCBI known mutation database (www.ncbi.nlm.nih.gov/SNP). Direct screening of the SRY coding region revealed a deletion of adenine (A) at nucleotide position 70 (c.70delA) in the patient (Figure 1d). The mutation was not found in her family members or control individuals, which revealed that the SRY mutation occurred de novo (Figure 1e). The mutation (c.70delA, p. Asn24Ile fsTer60; GenBank: AFG33955.1) led to a frameshift and introduced a premature termination codon at position in place of Glu, resulting in a truncated form of the SRY protein (Figure 1f). The mutation was confirmed by bidirectional sequencing.
Human bipotential gonads can differentiate into either testes or ovaries, which is depending on the presence or absence of the SRY gene. Expression of SRY gene above a critical threshold at the appropriate time and place drives the expression of the downstream target gene SOX9 and eventually leads to the formation of pre-Sertoli cells. This further orchestrates the formation of functional testes, which determines the development of male primary and secondary sex characteristics. With lack or dysfunction of SRY, the bipotential gonads instead develop as ovaries, and then female primary and secondary sex characteristics occur. The human SRY gene encodes a protein with 204 amino acids that consists of three domains, a central part of 79 amino acids known as a high mobility group (HMG) box and a C-terminal and an N-terminal domain.6 Most mutations occur within the HMG-box as a single amino acid substitution, which can alter DNA binding or prevent nuclear import of the SRY protein.7 Moreover, recent studies of the three-dimensional structure of the human sex-determining region Y (hSRY)-DNA complex in solution show that some residues of the HMG box domain are involved in nuclear localization (NLS) as well as DNA binding.7,8 The point mutation identified in the patient modified the residues of the N-terminal NLS (nNLS) and removed the C-terminal NLS (cNLS) residues and the HMG box domain, which abolished the function of the HMG box domain as a transcriptional regulator of the SOX9 gene in the sex determination cascade.9
The SRY gene with this frameshift mutation identified in the patient is unable to induce SOX9 expression and the differentiation of pre-Sertoli cells, which is essential for testis cord formation. The abnormal gonads cannot drive the development of Wolffian ducts and repress the development of fallopian tubes in the early stages of embryogenesis. However, the ovary does not develop normally, and only primordial follicles can be detected in the streak gonad. This is consistent with previous reports on the relationship between mutations in the SRY gene and the 46, XY gonadal dysgenesis.10,11 We conclude that the SRY gene mutation is a pathogenic factor for the patient.
Although the same point mutation resulting in premature termination and a dysfunctional SRY protein has previously been described,10 we are the first to describe it occurring de novo, as opposed to the previous report where it was inherited from a mosaic father.12 Our data also suggest that a mutation hot spot might exist in this region of the SRY gene.
AUTHOR CONTRIBUTIONS
XBW and YLL identified the case, conducted the genetic studies, and drafted the manuscript. QYZ and JPC carried out the laparoscopy. ZJZ, YZ, and PL participated in the genetic analysis and PL coordinated to draft the manuscript. QL and ZL conceived of the study and reviewed and edited the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declared no competing interests.
ACKNOWLEDGMENT
This work is supported by the National Key Research and Development Program (Grand No. 2017YFC1002003), the National High-tech Research and Development Program (863) of China (Grand No. 2015AA020404), the National Natural Science Foundation of China (Grand No. 81771637, 81571488), and the Frontier Technology Project of Shanghai (Grand No. SHDC12015122)
REFERENCES
- 1.Doherty LF, Rackow BW. Abnormal streak gonads in 46, XY complete gonadal dysgenesis. Fertil Steril. 2011;96:1415–6. doi: 10.1016/j.fertnstert.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 2.Swyer GI. Male pseudohermaphroditism: a hitherto undescribed form. Br Med J. 1955;2:709. doi: 10.1136/bmj.2.4941.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorgensen PB, Kjartansdóttir KR, Fedder J. Care of women with XY karyotype: a clinical practice guideline. Fertil Steril. 2010;94:105. doi: 10.1016/j.fertnstert.2009.02.087. [DOI] [PubMed] [Google Scholar]
- 4.Salehi LB, Scarciolla O, Vanni GF, Nardone AM, Frajese G, et al. Identification of a novel mutation in the SRY gene in a 46, XY female patient. Eur J Med Genet. 2006;49:494–8. doi: 10.1016/j.ejmg.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Otake T, Kuroiwa A. Molecular mechanism of male differentiation is conserved in the SRY-absent mammal, Tokudaia osimensis. Sci Rep. 2016;6:32874. doi: 10.1038/srep32874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bastian C, Muller JB, Lortatjacob S, Bignontopalovic J, Mcelreavey K, et al. Genetic mutations and somatic anomalies in association with 46, XY gonadal dysgenesis. Fertil Steril. 2015;103:1297–304. doi: 10.1016/j.fertnstert.2015.01.043. [DOI] [PubMed] [Google Scholar]
- 7.Shahid M, Dhillon VS, Khalil HS, Haque S, Batra S, et al. A SRY-HMG box frame shift mutation inherited from a mosaic father with a mild form of testicular dysgenesis syndrome in Turner syndrome patient. BMC Med Genet. 2010;11:131. doi: 10.1186/1471-2350-11-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helszer Z, Dmochowska A, Szemraj J, Słowikowska-Hilczer J, Wieczorek M, et al. A novel mutation (c.341A>G) in the SRY gene in a 46, XY female patient with gonadal dysgenesis. Gene. 2013;526:467–70. doi: 10.1016/j.gene.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 9.Ait Benkhali J, Coppin E, Brun S, Peraza-Reyes L, Martin T, et al. A network of HMG-box transcription factors regulates sexual cycle in the fungus Podospora anserina. PLoS Genet. 2013;9:e1003642. doi: 10.1371/journal.pgen.1003642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andonova S, Robeva R, Sirakov M, Mainhard K, Tomova A, et al. A novel SRY gene mutation p.F109L in a 46, XY female with complete gonadal dysgenesis. Sex Dev. 2015;9:333–7. doi: 10.1159/000443807. [DOI] [PubMed] [Google Scholar]
- 11.De Sousa SM, Kassahn KS, McIntyre LC, Chong CE, Scott H, et al. Case report of whole genome sequencing in the XY female: identification of a novel SRY mutation and revision of a misdiagnosis of androgen insensitivity syndrome. BMC Endocr Disord. 2016;16:58. doi: 10.1186/s12902-016-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isidor B, Capito C, Paris F, Baron S, Corradini N, et al. Familial frameshift SRYmutation inherited from a mosaic father with testicular dysgenesis syndrome. J Clin Endocrinol Metab. 2009;94:3467–71. doi: 10.1210/jc.2009-0226. [DOI] [PubMed] [Google Scholar]