

Abstract
The pathogenesis of small cell lung cancer (SCLC), a highly metastatic malignant tumor, remains unclear. In the present study, important genes and pathways that are involved in the pathogenesis of SCLC were identified. The following four datasets were downloaded from the Gene Expression Omnibus: GSE60052, GSE43346, GSE15240 and GSE6044. The differentially expressed genes (DEGs) between the SCLC samples and the normal samples were analyzed using R software. The limma package was used for every dataset. The RobustRankAggreg package was used to integrate the DEGs from the four datasets. Functional and pathway enrichment analyses were conducted using the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes databases with FunRich software and R software, respectively. In addition, the protein-protein interaction (PPI) network of the DEGs was constructed using the STRING database and Cytoscape software. Hub genes and significant modules were identified using Molecular Complex Detection in Cytoscape software. Finally, the expression values of hub genes were determined using the Oncomine online database. In total, 412 DEGs were identified following the integration of the four datasets, with 146 upregulated genes and 266 downregulated genes. The upregulated DEGs were primarily enriched in the cell cycle, cell division and microtubule binding. The downregulated DEGs were primarily enriched in the complement and coagulation cascades, the cytokine-mediated signaling pathway and protein binding. Eight hub genes and 1 significant module correlated to the cell cycle pathway were identified based on a subset of the PPI network. Finally, five hub genes were identified as highly expressed in SCLC tissue compared with normal tissue. The cell cycle pathway may be the pathway most closely associated with the pathogenesis of SCLC. NDC80, BUB1B, PLK1, CDC20 and MAD2L1 should be the focus of follow-up studies regarding the diagnosis and treatment of SCLC.
Keywords: small cell lung cancer, microarray analysis, protein-protein interaction, hub gene, bioinformatics analysis
Introduction
Cancer has placed a great burden on human health and the economy worldwide. Lung cancer has the highest rate of mortality and morbidity among both sexes, making it one of the most significant types of cancer, according to a major global epidemiological survey in 2018 (1). The financial burden of cancer not only takes a toll on an individual's life, but it also leads to a further loss of productivity. When adolescents with cancer face unbearable economic costs, their drug treatment and subsequent treatment will be affected, forming a vicious circle and causing further losses to society (2). Small cell lung cancer (SCLC) accounts for approximately 15% of the total cases of lung cancer (1). Compared with other types of lung cancer, SCLC is usually more sensitive to radiotherapy and chemotherapy, but its diagnosis and treatment are more difficult (3). Although SCLC is sensitive to chemotherapy, it is more likely to develop drug resistance and is highly prone to systemic metastasis (4). Patients with late-stage SCLC who are treated with standard chemotherapy have a median survival time of only 9–10 months from diagnosis (5). Unlike non-small cell lung cancer (NSCLC), there are no driver genes yet identified that can be used for clinically targeted therapy, so the treatment of SCLC primarily relies on radiotherapy and chemotherapy.
The etiology of malignant tumors is not yet fully understood. In addition to smoking, environmental exposure (6,7) and genetics (8) have been linked to the risk of developing cancer. Genetic factors are currently recognized as one of the endogenous causes of cancer. In addition, genetic mutations in patients with cancer can lead to resistance to cancer drugs. As a result, it is very important to investigate the molecular mechanisms underlying the occurrence and development of SCLC and to identify effective biomarkers.
With the continuous development of microarray and high-throughput sequencing technologies, molecular biomarkers associated with the occurrence, development, diagnosis and treatment of tumors can be identified (9–12). Compared with research on NSCLC, fewer bioinformatics analyses have been conducted in SCLC. However, certain studies have revealed potential biomarkers associated with SCLC (13,14). To further investigate the crucial biomarkers associated with SCLC and to overcome the limitations regarding the results that are due to different technology platforms or small sample sizes in various studies, integrated bioinformatics methods were adopted in the present study in order to conduct investigations across multiple platforms with larger sample sizes.
Materials and methods
Gene expression profile data
The GEO database was used to obtain 4 microarray expression profiles: GSE60052 (15), GSE43346 (16), GSE15240 (17) and GSE6044 (18).
The inclusion criteria for the gene expression profiles were as follows: i) The tumor tissue samples were from patients with SCLC, and the control group was normal tissue; ii) the number of samples was >10; iii) all included studies were published in the English language; iv) the included studies provided enough data or raw data to enable further bioinformatics analysis; and v) the research subjects were human. The characteristics of all the gene expression profiles are presented in Table I.
Table I.
Basic characteristics of the gene expression profile data.
| Record | Tissue | Platform | Normal | Tumor | Country | Race | (Refs.) |
|---|---|---|---|---|---|---|---|
| GSE60052 | SCLC | GPL11154 Illumina HiSeq 2000 (Homo sapiens) | 7 | 79 | USA | Asian | (15) |
| GSE43346 | SCLC | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 42 | 23 | Japan | Asian | (16) |
| GSE15240 | SCLC | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 3 | 42 | USA | Caucasian | (17) |
| GSE6044 | SCLC | GPL201 [HG-Focus] Affymetrix Human HG-Focus Target Array | 5 | 9 | Germany | Caucasian | (18) |
SCLC, small cell lung cancer.
Integrated analysis of microarray datasets
R software (version 3.5.1; 64-bit; http://www.r-project.org/) (19) was used to identify relevant DEGs. The expression data for each gene expression profile were downloaded from the GEO database. For values not reported in logarithmic form, log2 conversion was performed. If standardized data were not available, the raw data were downloaded. For CEL files, the affy package (19) was used to read the expression data. If a gene had multiple probes in the same chip, the average value of all probes was taken as the expression value of the gene. The limma package (20) was used to standardize the data for each experiment. Finally, the RobustRankAggreg package (21) was used to integrate the DEGs identified in the four gene expression profiles. The included DEGs conformed to the following criteria: |log2FC| >1.5 (where FC means fold change) and adjusted P<0.05.
Functional and pathway enrichment analysis
FunRich software (version 3.1.3; 64-bit) (22) was used to perform the functional and pathway enrichment analyses. The FunRich tool was designed to process a variety of gene and protein datasets and to perform powerful functional enrichment analyses (22). The GO database (geneontology.org) was chosen for the functional pathway enrichment analysis of downregulated and upregulated DEGs in the present study. This database contains information associated with biological processes (BPs), cellular components (CCs) and molecular functions. The DOSE package (23) for R software was selected for the KEGG pathway enrichment analysis. P<0.05 was considered to indicate a statistically significant result.
PPI network construction and analysis of modules
The STRING database (version 10.5; string-db.org/) is an online database that facilitates searches for interactions between proteins (24). The DEGs with interaction scores >0.9 were mapped into PPIs. Then, Cytoscape (version 3.6.1; 64-bit; www.cytoscape.org/) (25) was used to identify hub genes. Cytoscape is an open-source software platform for visualizing complex networks and integrating those networks with any type of attributed data (25). The modules with degree cut-off=2, node score cut-off=0.2 and K-core value =2 were screened by Molecular Complex Detection (MCODE) (26). The modules with MCODE scores >10 and >15 nodes were selected as notable modules. FunRich software and R software were used to perform functional and KEGG pathway enrichment analyses, respectively, of the DEGs in these different modules. The genes with degrees >20 were considered hub genes.
Expression level analysis of hub genes
Oncomine (oncomine.org) is currently the largest oncogene chip database and integrated data mining platform in the world; it is intended to be used to mine cancer gene information. To date, the database has collected data from 729 gene expression datasets and >90,000 cancer and normal tissue samples. It can be used to analyze the differential expression of individual genes in samples of major cancers and their associated normal tissues and to visualize the results (27). The expression value data for normal and SCLC tissues were downloaded from Oncomine (pre-processed expression levels were Log2 normalized and median centered), and GraphPad Prism software (version 7.0; 64-bit; GraphPad Software, Inc.) was used to draw box diagrams and identify significant differences using unpaired Student's t-test. P<0.05 was considered to indicate a statistically significant result.
Results
DEG identification
In all four microarray expression profiles (GSE60052, GSE43346, GSE15240 and GSE6044), 412 DEGs were identified, which contained 57 normal tissue samples and 153 SCLC tissue samples. Compared with normal tissues, SCLC tissues had 146 upregulated genes and 266 downregulated genes. To visualize the differential expression levels in each microarray expression profile following standardization, volcano maps were used for the 4 datasets (Fig. 1). Red points represent upregulated genes (log2FC >1.5 and adjusted P<0.05), whereas green points represent downregulation genes (log2FC <-1.5 and adjusted P<0.05). The black points represent genes with no significant difference (adjusted P≥0.05). Then, a cluster heat map was used to present the differences in the expression levels of the top 20 DEGs according to adjusted P-values integrated from the four different microarray expression profiles (Fig. 2). The top 20 integrated DEGs presented the smallest adjusted P-values, and were S1PR1, MAD2L1, CDKN2A, STIL, NDC80, NCAPG, PAD51AP1, TTK, PRM2, EZH2, PRC1, UBE2C, RFC4, CENPF, TCP2A, HMGB3, TYMS, SOX4, MCM2 and SMC4.
Figure 1.

Differential expression data between two sets of samples. (A) GSE60052 data. (B) GSE43346 data. (C) GSE15240 data. (D) GSE6044 data. The red points represent upregulated genes screened on the basis of | log2FC|>1.5 and an adjusted P<0.05. The green points represent downregulation of the expression of genes screened on the basis of |log2FC|>1.5 and an adjusted P<0.05. The black points represent genes with no significant difference. FC, fold change.
Figure 2.

Heatmap of the top 20 integrated differentially expressed genes screened on the basis of |log2FC|>1.5 and adjusted P<0.05. Red indicates the genes that were upregulated, green indicates the genes that were downregulated, and black indicates no significant change in gene expression. GEO, Gene Expression Omnibus; SCLC, small cell lung cancer.
Enrichment analyses
For the upregulated and downregulated DEGs, GO and KEGG pathway enrichment analyses were performed. The top 5 enriched results are presented in Figs. 3–5. For upregulated DEGs, the enriched BPs were cell division, DNA replication, G1/S transition of the mitotic cell cycle, mitotic cell cycle and mitotic sister chromatid segregation. The enriched BPs among the downregulated genes included the cytokine-mediated signaling pathway, positive regulation of angiogenesis, the inflammatory response, cell adhesion and vasculogenesis. Regarding the enriched CCs, the upregulated DEGs were enriched in the nucleoplasm, nucleus, spindle, midbody and kinetochore, while the downregulated DEGs were enriched in extracellular vesicular exosomes, the extracellular space, the cell surface, focal adhesion and the extracellular region. In addition, with regard to molecular function, the upregulated DEGs were associated with microtubule binding, ATP binding, microtubule motor activity and protein kinase binding, while the downregulated DEGs were associated with protein binding, protein homodimerization activity, integrin binding, β-amyloid binding and tumor necrosis factor binding (all adjusted P-values <0.05).
Figure 3.

Gene Ontology analysis of the upregulated differentially expressed genes. (A) Biological process. (B) Cellular component. (C) Molecular function.
Figure 5.

Kyoto Encyclopedia of Genes and Genomes pathway analysis of DEGs. (A) Upregulated DEGs. (B) Downregulated DEGs. The x-axes represent the number of target genes belonging to a pathway; the bigger the number, the higher level of enrichment, and the color of the bar reflects the different P-value range. DEGs, differentially expressed genes.
There were 5 KEGG pathways that were significantly associated with the upregulated genes (all adjusted P-values <0.05; Fig. 5), including the cell cycle, DNA replication, mismatch repair, homologous recombination and oocyte meiosis (Fig. 5A). Complement and coagulation cascades, fluid shear stress and atherosclerosis, Staphylococcus aureus infection, cytokine-cytokine receptor interaction and viral myocarditis were primarily associated with downregulated DEGs (Fig. 5B).
Construction of PPI network and module identification
To predict the interactions between recognized DEGs at the protein level, a PPI network was constructed using the STRING database. In total, 410 nodes and 852 edges were presented in this PPI network (Fig. 6). According to the inclusion criteria used within the present study, one significant module was identified (Fig. 7). In this module, 8 hub genes with degrees >20 were identified: BIRC5, NDC80, BUB1B, CENPE, KIF2C, PLK1, CDC20 and MAD2L1 (Table II). The GO pathway enrichment analysis revealed that microtubule binding, tubulin binding, microtubule motor activity, motor activity, protein C-terminus binding and ATPase activity were the primarily enriched pathways. Via the KEGG analysis, the following four crucial pathways were identified: The cell cycle, oocyte meiosis, human T-cell leukemia virus 1 infection and progesterone-mediated oocyte maturation (Fig. 8).
Figure 6.

Protein-protein interaction network. Circles represent genes, lines represent the protein-protein associations of genes, and the results within the circle represent the structures of the proteins. Red nodes represent upregulated genes and green nodes represent downregulated genes.
Figure 7.
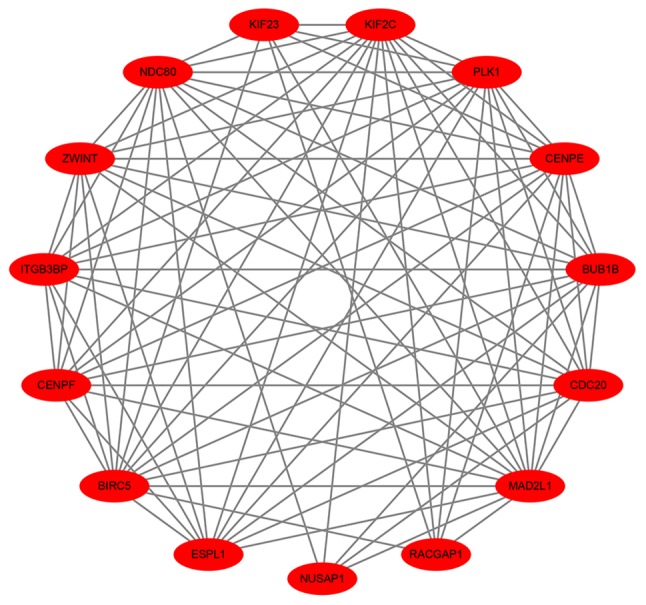
Protein-protein interaction network of differentially expressed genes in the significant module. Circles represent genes, lines represent the protein-protein associations of genes, and the results within the circle represent the structures of the proteins. Red nodes represent upregulated genes.
Table II.
Hub genes with a high degree of connectivity.
| Gene | Degree | Type | MCODE Cluster |
|---|---|---|---|
| MAD2L1 | 36 | Upregulated | Cluster 1 |
| CDC20 | 33 | Upregulated | Cluster 1 |
| PLK1 | 29 | Upregulated | Cluster 1 |
| CENPE | 25 | Upregulated | Cluster 1 |
| KIF2C | 25 | Upregulated | Cluster 1 |
| BUB1B | 23 | Upregulated | Cluster 1 |
| NDC80 | 22 | Upregulated | Cluster 1 |
| BIRC5 | 20 | Upregulated | Cluster 1 |
Figure 8.
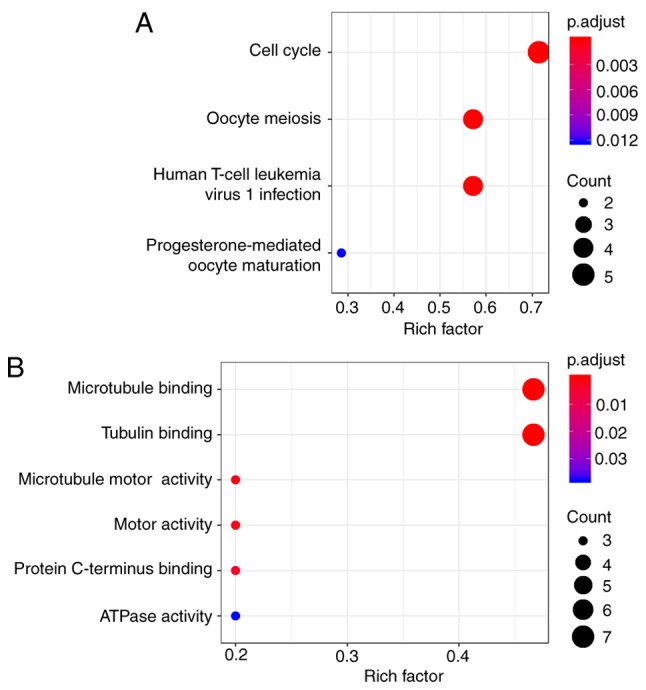
Enrichment analyses of the significant module. (A) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. (B) Gene Ontology enrichment analysis. The x-axes represent the rich factor, which is the ratio of the number of target genes belonging to a pathway to the number of all the annotated genes located in the pathway. The higher rich factor represents the higher level of enrichment. The size of the dot indicates the number of target genes in the pathway, and the color of the dot reflects the different P-value range.
Expression level analysis of hub genes
Using the Ocomine database, two important results were obtained from the Garber et al (28) and Bhattacharjee et al (29) lung cancer gene expression data. From the analysis of their data, it can be concluded that when compared with normal tissues, SCLC tissues had five highly expressed hub genes. These genes were the same as those identified in the bioinformatics investigation of the present study, except CENPE, PLK1 and BIRC5. Three of the identified genes were associated with cell cycle pathways and were highly expressed (CDC20, BUB1B and MAD2L1) (all P<0.05; Fig. 9).
Figure 9.

(A-H) Analysis of the expression of hub genes in the Oncomine database. The preprocessed expression levels were log2-normalized and median-centered. The black dots represent the sample size. BIRC5, baculoviral inhibitor of apoptosis repeat-containing 5; NDC80, kinetochore protein NDC80 homolog; BUB1B, BUB1 mitotic checkpoint serine/threonine kinase B; CENPE, centromere-associated protein E; KIF2C, kinesin family member 2C; PLK1, polo-like kinase 1; CDC20, cell division cycle protein 20; MAD2L1, mitotic arrest deficient 2 like 1; SCLC, small cell lung cancer.
Discussion
SCLC has been termed a recalcitrant cancer. Despite being sensitive to radiotherapy and chemotherapy, the recurrence rate is very high. Although there have been breakthroughs in the treatment of SCLC, the pathogenesis remains unclear (30–32). Therefore, the discovery of potential molecular mechanisms underlying SCLC may be very important for the treatment and diagnosis of SCLC. In particular, the development of high-throughput sequencing facilitates the search for potentially involved genes and tumor mechanisms. Cancer is a polygenic genetic disease; with the development of tumors, cancer cells are still constantly dividing, multiplying and accumulating mutations, and their genomes are unstable. Bioinformatics, on the other hand, uses genetic information for molecular diagnoses and drug therapies. To the best of our knowledge, the present study is currently the first to use four gene chips for cross-platform analysis. The sample size was increased, the effect of batch difference was eliminated using the RobustRankAggreg package, and the results were more accurate. The more accurate evaluation of the previously discovered genes and pathways will provide a theoretical basis for subsequent studies, and also guide future studies.
In the present study, 412 DEGs were identified, including 146 upregulated genes and 266 downregulated genes. The upregulated genes were primarily enriched in the cell cycle, DNA replication, mismatch repair, homologous recombination and oocyte meiosis. The downregulated genes were primarily enriched in cytokine-cytokine receptor interactions. Among these DEGs, a crucial module containing 8 hub genes was selected from the PPI network. Furthermore, the important enriched module pathways containing the 8 hub genes revealed that the cell cycle was the most significant pathway. A previous study (32) that performed whole genome sequencing on a large sample of 152 freshly frozen samples from patients diagnosed with stage I–IV SCLC revealed that a loss of the tumor suppressors TP53 and retinoblastoma protein 1 promoted the development of SCLC. The present study obtained 5 new promising hub genes, which were highly expressed in SCLC tissues compared with normal tissue, and three of those genes were strongly associated with the cell cycle pathway (TP53, TP73 and RB1; all P-values <0.05).
An abnormal cell cycle may lead to the occurrence of a variety of different types of tumor, such as hepatocellular carcinoma (33,34), breast cancer (35), pancreatic cancer (36), ovarian cancer (37) and NSCLC in non-smoking females (38). Wang et al (39) used different microarray expression profiles in their study of primitive neuro-ectodermal tumors, and revealed the same four pathways that were identified in the present study, and several of the same genes in the modules that they identified as being important. These similarities may be the result of both tumors being neuroendocrine tumors. These results support the view that disordered cell cycle regulation may contribute to the development of SCLC. Previous studies (14,40) have also indicated that the cell cycle pathway is an important pathway involved in the development and progression of SCLC. It has also been demonstrated that certain associated drugs can restore the abnormal cell cycle in SCLC, thus playing anti-tumor roles (41).
Following the discovery of cell division cycle protein 20 (CDC20) ~40 years ago, the original researchers demonstrated that CDC20 mutations can cause abnormal cessation of mitosis, resulting in abnormal chromosome separation in anaphase (42). Wan et al (43) revealed that the anaphase-promoting complex (APC) CDC20 plays an important role in the regulation of the cell cycle and apoptosis, and it can also inhibit the apoptosis-induced resistance of cancer cells to chemotherapy and radiotherapy. Furthermore, CDC20 is highly expressed in a number of different types of tumor, and is associated with the poor prognosis of patients, such as those with colorectal cancer (44), hepatocellular carcinoma (45), gastric cancer (46), bladder cancer (47) and cervical cancer (48). Taniguchi et al (49) discovered that downregulation of CDC20 can increase the sensitivity of pancreatic cancer cells to radiotherapy and paclitaxel. In addition, Wan et al (43) demonstrated that hyperactive CDC20 plays an important role in chemoresistance, and is associated with the human T-cell leukemia virus 1 infection pathway. The downregulation of CDC20 expression slows the growth of both NSCLC and SCLC and the formation rate of lung cancer cell colonies (50). In conclusion, CDC20 is expected to be a new target for research on SCLC.
The BUB1B gene encodes a kinase involved in the spindle assembly checkpoint and chromosome separation (51). During the G2 phase of mitosis, BUB1B binds to CDC20 and inhibits APC/cyclostome (APC/C) activity, allowing cyclin B to accumulate before mitosis begins and slows the cell cycle (52). This gene also contributes to the progression of certain types of tumor and is associated with the prognosis of patients with hepatocellular carcinoma (53) and glioblastoma (54). Ma et al (55) revealed that high expression levels of BUB1B may promote the tolerance of glioblastoma to radiotherapy. Chen et al (56) used gene expression profiling of the GSE40791 (57) database to demonstrate that BUB1B is a candidate gene associated with the pathogenesis of lung adenocarcinoma. Therefore, this gene is a promising candidate, and should be the focus of future research on SCLC.
Another gene enriched in cell cycle pathways is mitotic arrest deficient 2 like 1 (MAD2L1), which is a component of the mitotic spindle assembly checkpoint that ensures that all chromosomes are properly aligned at the metaphase plate (58). The destruction of the function of MAD2L1 in mammalian cells can affect the function of the spindle, leading to cell aneuploidy or the occurrence of tumors. Partial deletion of MAD2L1 (MAD2L1+/− cells) accompanied by chromosomal instability can cause lung cancer in mice (59). MAD2L1 is highly expressed in patients with breast cancer (60) and gastric cancer (61), and its expression is associated with the tumor stage. A recent study (62) also demonstrated that patients with lung adenocarcinoma with high expression levels of MAD2L1 have worse prognoses than those with low expression levels of MAD2L1.
The kinesin family member 2C (KIF2C) gene encodes a protein that is part of the kinase-like protein family. This protein plays a role in regulating microtubule dynamics in cells, and is important for chromosome separation during anaphase (63). The GO analysis in the present study suggests that it has functional enrichment in two aspects: Microtubule binding and tubulin binding. Therefore, its abnormal expression affects mitosis and can lead to cancer. In addition, KIF2C has been highly expressed not only in patients with esophageal squamous cell carcinoma (64) and glioma (65) with poor prognoses, but also in those with adenocarcinoma of the lung, as indicated by two different bioinformatics analyses (66,67).
Nuclear division cycle 80 (Ndc80) is an isotetrameric protein complex that plays an important role in cell mitosis (68). A liver cancer-associated study (69) revealed that liver cancer tumor tissue samples from 47 patients had higher Ndc80 mRNA expression levels than the adjacent normal tissue samples. The virus carrying Ndc80-siRNA silences the Ndc80 gene, inhibits the in vitro proliferation of the cultured liver cancer SMMC-7721 cell line and induces cell apoptosis, demonstrating that Ndc80 primarily acts through its effect on overcoming cell cycle arrest and antiapoptosis mechanism-induced liver cancer. In a study that focused on osteosarcoma, Ndc80 mRNA expression was higher in 84.6% of tumor tissues compared with in adjacent normal tissues, and the expression level was associated with the Tumor-Node-Metastasis stage (70) and distant metastasis of the tumor. In addition, the level of Ndc80 was indicated to have value as an independent prognostic evaluation index (71). Yuan et al (72) screened 13 GSE datasets and revealed that NDC80 and 7 other hub genes can interact with ZW10-interacting protein and participate in the formation of both NSCLC and SCLC.
There were limitations to the present study. First, the conclusions were drawn based on data from public databases rather than actual experiments, meaning that the quality of the data cannot be guaranteed, and that the results may be inaccurate. Secondly, the inability to include samples from all ethnic groups, such as African-Americans, may have affected the levels of gene expression observed. Finally, due to the lack of public data on the prognosis of patients with SCLC, the impact of the identified genes on survival was not further investigated, and factors such as the sex, age and tumor stage of patients were not included. All of these factors could affect the conclusions of the present study.
In conclusion, the present study has revealed five genes that may be associated with the pathogenesis of SCLC: NDC80, BUB1B, KIF2C, CDC20 and MAD2L1. The cell cycle may play an important role in the development of SCLC; however, more well-designed experiments with larger sample sizes need to be performed in order to confirm these conclusions. The results from the present study will provide new ideas for the treatment of SCLC.
Figure 4.

Gene Ontology analysis of the downregulated differentially expressed genes. (A) Biological process. (B) Cellular component. (C) Molecular function.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
YL and GY conceived and designed the study, acquired and analyzed data and wrote the manuscript. XW and PZ contributed to data analysis and manuscript drafting. XF and CH designed the study and revised the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Landwehr MS, Watson SE, Macpherson CF, Novak KA, Johnson RH. The cost of cancer: A retrospective analysis of the financial impact of cancer on young adults. Cancer Med. 2016;5:863–870. doi: 10.1002/cam4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao H, Niu Y, Li M, Fang S, Guo L. Identification of DJ-1 as a contributor to multidrug resistance in human small-cell lung cancer using proteomic analysis. Int J Exp Pathol. 2017;98:67–74. doi: 10.1111/iep.12221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang C, Huang M, Chen W, Zhu W, Meng H, Guo L, Wei T, Zhang J. N-acetylglucosaminyltransferase V modulates radiosensitivity and migration of small cell lung cancer through epithelial-mesenchymal transition. FEBS J. 2015;282:4295–4306. doi: 10.1111/febs.13419. [DOI] [PubMed] [Google Scholar]
- 5.Demedts IK, Vermaelen KY, van Meerbeeck JP. Treatment of extensive-stage small cell lung carcinoma: Current status and future prospects. Eur Respir J. 2010;35:202–215. doi: 10.1183/09031936.00105009. [DOI] [PubMed] [Google Scholar]
- 6.Lam WK. Lung cancer in Asian women-the environment and genes. Respirology. 2005;10:408–417. doi: 10.1111/j.1440-1843.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- 7.Hosgood HD, III, Boffetta P, Greenland S, Lee YC, McLaughlin J, Seow A, Duell EJ, Andrew AS, Zaridze D, Szeszenia-Dabrowska N, et al. In-home coal and wood use and lung cancer risk: A pooled analysis of the international lung cancer consortium. Environ Health Perspect. 2010;118:1743–1747. doi: 10.1289/ehp.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu PF, Lee CH, Wang MJ, Goggins WB, Chiang TA, Huang MS, Ko YC. Cancer aggregation and complex segregation analysis of families with female non-smoking lung cancer probands in Taiwan. Eur J Cancer. 2004;40:260–266. doi: 10.1016/j.ejca.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5:588–599. doi: 10.1038/ncponc1187. [DOI] [PubMed] [Google Scholar]
- 10.Matamala N, Vargas MT, González-Cámpora R, Miñambres R, Arias JI, Menéndez P, Andrés-León E, Gómez-López G, Yanowsky K, Calvete-Candenas J, et al. Tumor microRNA expression profiling identifies circulating microRNAs for early breast cancer detection. Clin Chem. 2015;61:1098–1106. doi: 10.1373/clinchem.2015.238691. [DOI] [PubMed] [Google Scholar]
- 11.Lusito E, Felice B, D'Ario G, Ogier A, Montani F, Di Fiore PP, Bianchi F. Unraveling the role of low-frequency mutated genes in breast cancer. Bioinformatics. 2019;35:36–46. doi: 10.1093/bioinformatics/bty520. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Yang Y, Cheng L, Cheng Y, Zhou HH, Tan ZR. Identification of common genes refers to colorectal carcinogenesis with paired cancer and noncancer samples. Dis Markers. 2018;2018:3452739. doi: 10.1155/2018/3452739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H, Wei S, Zhang L, Yuan C, Duan Y, Wang Q. Secreted phosphoprotein 1 promotes the development of small cell lung cancer cells by inhibiting autophagy and apoptosis. Pathol Oncol Res. 2018 doi: 10.1007/s12253-018-0504-7. [DOI] [PubMed] [Google Scholar]
- 14.Ni Z, Wang X, Zhang T, Li L, Li J. Comprehensive analysis of differential expression profiles reveals potential biomarkers associated with the cell cycle and regulated by p53 in human small cell lung cancer. Exp Ther Med. 2018;15:3273–3282. doi: 10.3892/etm.2018.5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, Conley S, Zhong H, Liu Z, Brohawn P, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12:e1005895. doi: 10.1371/journal.pgen.1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T, Yamanaka R, Tanaka Y, Nukiwa T, Marquez VE, et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep. 2013;3:1911. doi: 10.1038/srep01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, Watkins DN. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009;69:3364–3373. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohrbeck A, Neukirchen J, Rosskopf M, Pardillos GG, Geddert H, Schwalen A, Gabbert HE, von Haeseler A, Pitschke G, Schott M, et al. Gene expression profiling for molecular distinction and characterization of laser captured primary lung cancers. J Transl Med. 2008;6:69. doi: 10.1186/1479-5876-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 20.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pathan M, Keerthikumar S, Chisanga D, Alessandro R, Ang CS, Askenase P, Batagov AO, Benito-Martin A, Camussi G, Clayton A, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6:1321455. doi: 10.1080/20013078.2017.1321455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu G, Wang LG, Yan GR, He QY. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S, Pico AR, Bader GD, Ideker T. A travel guide to cytoscape plugins. Nat Methods. 2012;9:1069–1076. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pietanza MC, Byers LA, Minna JD, Rudin CM. Small cell lung cancer: Will recent progress lead to improved outcomes? Clin Cancer Res. 2015;21:2244–2255. doi: 10.1158/1078-0432.CCR-14-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karachaliou N, Pilotto S, Lazzari C, Bria E, de Marinis F, Rosell R. Cellular and molecular biology of small cell lung cancer: An overview. Transl Lung Cancer Res. 2016;5:2–15. doi: 10.3978/j.issn.2218-6751.2016.01.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Zhong Y, Chen YJ, Chen H. SOX11 regulates apoptosis and cell cycle in hepatocellular carcinoma via Wnt/β-catenin signaling pathway. Biotechnol Appl Biochem. 2019;66:240–246. doi: 10.1002/bab.1718. [DOI] [PubMed] [Google Scholar]
- 34.An MJ, Kim DH, Kim CH, Kim M, Rhee S, Seo SB, Kim JW. Histone demethylase KDM3B regulates the transcriptional network of cell-cycle genes in hepatocarcinoma HepG2 cells. Biochem Biophys Res Commun. 2019;508:576–582. doi: 10.1016/j.bbrc.2018.11.179. [DOI] [PubMed] [Google Scholar]
- 35.Sun X, Hu Y, Wu J, Shi L, Zhu L, Xi PW, Wei JF, Ding Q. RBMS2 inhibits the proliferation by stabilizing P21 mRNA in breast cancer. J Exp Clin Cancer Res. 2018;37:298. doi: 10.1186/s13046-018-0968-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsushita Y, Furutani Y, Matsuoka R, Furukawa T. Hot water extract of Agaricus blazei Murrill specifically inhibits growth and induces apoptosis in human pancreatic cancer cells. BMC Complement Altern Med. 2018;18:319. doi: 10.1186/s12906-018-2385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee KS, Kim SW, Lee HS. Orostachys japonicus induce p53-dependent cell cycle arrest through the MAPK signaling pathway in OVCAR-3 human ovarian cancer cells. Food Sci Nutr. 2018;6:2395–2401. doi: 10.1002/fsn3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang G, Chen Q, Xiao J, Zhang H, Wang Z, Lin X. Identification of genes and analysis of prognostic values in nonsmoking females with non-small cell lung carcinoma by bioinformatics analyses. Cancer Manag Res. 2018;10:4287–4295. doi: 10.2147/CMAR.S174409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GY, Li L, Liu B, Han X, Wang CH, Wang JW. Integrated bioinformatic analysis unveils significant genes and pathways in the pathogenesis of supratentorial primitive neuroectodermal tumor. Onco Targets Ther. 2018;11:1849–1859. doi: 10.2147/OTT.S148776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hubaux R, Thu KL, Coe BP, MacAulay C, Lam S, Lam WL. EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J Thorac Oncol. 2013;8:1102–1106. doi: 10.1097/JTO.0b013e318298762f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin YC, Su JH, Lin SC, Chang CC, Hsia TC, Tung YT, Lin CC. A soft coral-derived compound, 11-dehydrosinulariolide, induces G2/M cell cycle arrest and apoptosis in small cell lung cancer. Mar Drugs. 2018;16 doi: 10.3390/md16120479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hartwell LH, Culotti J, Reid B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc Natl Acad Sci USA. 1970;66:352–359. doi: 10.1073/pnas.66.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J, et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev Cell. 2014;29:377–391. doi: 10.1016/j.devcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu WJ, Hu KS, Wang DS, Zeng ZL, Zhang DS, Chen DL, Bai L, Xu RH. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J Transl Med. 2013;11:142. doi: 10.1186/1479-5876-11-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Gao JZ, Du JL, Huang ZX, Wei LX. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. Int J Oncol. 2014;45:1547–1555. doi: 10.3892/ijo.2014.2559. [DOI] [PubMed] [Google Scholar]
- 46.Ding ZY, Wu HR, Zhang JM, Huang GR, Ji DD. Expression characteristics of CDC20 in gastric cancer and its correlation with poor prognosis. Int J Clin Exp Pathol. 2014;7:722–727. [PMC free article] [PubMed] [Google Scholar]
- 47.Choi JW, Kim Y, Lee JH, Kim YS. High expression of spindle assembly checkpoint proteins CDC20 and MAD2 is associated with poor prognosis in urothelial bladder cancer. Virchows Arch. 2013;463:681–687. doi: 10.1007/s00428-013-1473-6. [DOI] [PubMed] [Google Scholar]
- 48.Kim Y, Choi JW, Lee JH, Kim YS. MAD2 and CDC20 are upregulated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the uterine cervix. Int J Gynecol Pathol. 2014;33:517–523. doi: 10.1097/PGP.0000000000000082. [DOI] [PubMed] [Google Scholar]
- 49.Taniguchi K, Momiyama N, Ueda M, Matsuyama R, Mori R, Fujii Y, Ichikawa Y, Endo I, Togo S, Shimada H. Targeting of CDC20 via small interfering RNA causes enhancement of the cytotoxicity of chemoradiation. Anticancer Res. 2008;28:1559–1563. [PubMed] [Google Scholar]
- 50.Kidokoro T, Tanikawa C, Furukawa Y, Katagiri T, Nakamura Y, Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008;27:1562–1571. doi: 10.1038/sj.onc.1210799. [DOI] [PubMed] [Google Scholar]
- 51.Guo Y, Kim C, Ahmad S, Zhang J, Mao Y. CENP-E-dependent BubR1 autophosphorylation enhances chromosome alignment and the mitotic checkpoint. J Cell Biol. 2012;198:205–217. doi: 10.1083/jcb.201202152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malureanu LA, Jeganathan KB, Hamada M, Wasilewski L, Davenport J, van Deursen JM. BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell. 2009;16:118–131. doi: 10.1016/j.devcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhuang L, Yang Z, Meng Z. Upregulation of BUB1B, CCNB1, CDC7, CDC20, and MCM3 in tumor tissues predicted worse overall survival and disease-free survival in hepatocellular carcinoma patients. Biomed Res Int. 2018;2018:7897346. doi: 10.1155/2018/7897346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee E, Pain M, Wang H, Herman JA, Toledo CM, DeLuca JG, Yong RL, Paddison P, Zhu J. Sensitivity to BUB1B inhibition defines an alternative classification of glioblastoma. Cancer Res. 2017;77:5518–5529. doi: 10.1158/0008-5472.CAN-17-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Q, Liu Y, Shang L, Yu J, Qu Q. The FOXM1/BUB1B signaling pathway is essential for the tumorigenicity and radioresistance of glioblastoma. Oncol Rep. 2017;38:3367–3375. doi: 10.3892/or.2017.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L, Zhuo D, Chen J, Yuan H. Screening feature genes of lung carcinoma with DNA microarray analysis. Int J Clin Exp Med. 2015;8:12161–12171. [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, Van Deursen J, Galardy PJ. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest. 2012;122:4362–4374. doi: 10.1172/JCI63084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng Y, Li K, Diao D, Zhu K, Shi L, Zhang H, Yuan D, Guo Q, Wu X, Liu D, Dang C. Expression of KIAA0101 protein is associated with poor survival of esophageal cancer patients and resistance to cisplatin treatment in vitro. Lab Invest. 2013;93:1276–1287. doi: 10.1038/labinvest.2013.124. [DOI] [PubMed] [Google Scholar]
- 59.Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci USA. 2004;101:4459–4464. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan B, Xu Y, Woo JH, Wang Y, Bae YK, Yoon DS, Wersto RP, Tully E, Wilsbach K, Gabrielson E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006;12:405–410. doi: 10.1158/1078-0432.CCR-05-0903. [DOI] [PubMed] [Google Scholar]
- 61.Kim HS, Park KH, Kim SA, Wen J, Park SW, Park B, Gham CW, Hyung WJ, Noh SH, Kim HK, Song SY. Frequent mutations of human Mad2, but not Bub1, in gastric cancers cause defective mitotic spindle checkpoint. Mutat Res. 2005;578:187–201. doi: 10.1016/j.mrfmmm.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 62.Shi YX, Zhu T, Zou T, Zhuo W, Chen YX, Huang MS, Zheng W, Wang CJ, Li X, Mao XY, et al. Prognostic and predictive values of CDK1 and MAD2L1 in lung adenocarcinoma. Oncotarget. 2016;7:85235–85243. doi: 10.18632/oncotarget.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wordeman L, Wagenbach M, von Dassow G. MCAK facilitates chromosome movement by promoting kinetochore microtubule turnover. J Cell Biol. 2007;179:869–879. doi: 10.1083/jcb.200707120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duan H, Zhang X, Wang FX, Cai MY, Ma GW, Yang H, Fu JH, Tan ZH, Fu XY, Ma QL, et al. KIF-2C expression is correlated with poor prognosis of operable esophageal squamous cell carcinoma male patients. Oncotarget. 2016;7:80493–80507. doi: 10.18632/oncotarget.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bie L, Zhao G, Wang YP, Zhang B. Kinesin family member 2C (KIF2C/MCAK) is a novel marker for prognosis in human gliomas. Clin Neurol Neurosurg. 2012;114:356–360. doi: 10.1016/j.clineuro.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 66.Bidkhori G, Narimani Z, Hosseini Ashtiani S, Moeini A, Nowzari-Dalini A, Masoudi-Nejad A. Reconstruction of an integrated genome-scale co-expression network reveals key modules involved in lung adenocarcinoma. PLoS One. 2013;8:e67552. doi: 10.1371/journal.pone.0067552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song YJ, Tan J, Gao XH, Wang LX. Integrated analysis reveals key genes with prognostic value in lung adenocarcinoma. Cancer Manag Res. 2018;10:6097–6108. doi: 10.2147/CMAR.S168636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.D'Archivio S, Wickstead B. Trypanosome outer kinetochore proteins suggest conservation of chromosome segregation machinery across eukaryotes. J Cell Biol. 2017;216:379–391. doi: 10.1083/jcb.201608043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ju LL, Chen L, Li JH, Wang YF, Lu RJ, Bian ZL, Shao JG. Effect of NDC80 in human hepatocellular carcinoma. World J Gastroenterol. 2017;23:3675–3683. doi: 10.3748/wjg.v23.i20.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang L, Dou X, Liu T, Lu W, Ma Y, Yang Y. Tumor size and lymph node metastasis are prognostic markers of small cell lung cancer in a Chinese population. Medicine (Baltimore) 2018;97:e11712. doi: 10.1097/MD.0000000000011712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu B, Wu DP, Xie RT, Liu LG, Yan XB. Elevated NDC80 expression is associated with poor prognosis in osteosarcoma patients. Eur Rev Med Pharmacol Sci. 2017;21:2045–2053. [PubMed] [Google Scholar]
- 72.Yuan W, Xie S, Wang M, Pan S, Huang X, Xiong M, Xiao RJ, Xiong J, Zhang QP, Shao L. Bioinformatic analysis of prognostic value of ZW10 interacting protein in lung cancer. Onco Targets Ther. 2018;11:1683–1695. doi: 10.2147/OTT.S149012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.