

SUMMARY
Cachexia is a wasting syndrome characterized by pronounced skeletal muscle loss. In cancer, cachexia is associated with increased morbidity and mortality and decreased treatment tolerance. Although advances have been made in understanding the mechanisms of cachexia, translating these advances to the clinic has been challenging. One reason for this short-coming may be the current animal models, which fail to fully recapitulate the etiology of human cancer-induced tissue wasting. Because pancreatic ductal adenocarcinoma (PDA) presents with a high incidence of cachexia, we engineered a mouse model of PDA that we named KPP. KPP mice, similar to PDA patients, progressively lose skeletal and adipose mass as a consequence of their tumors. In addition, KPP muscles exhibit a similar gene ontology as cachectic patients. We envision that the KPP model will be a useful resource for advancing our mechanistic understanding and ability to treat cancer cachexia.
Graphical Abstract
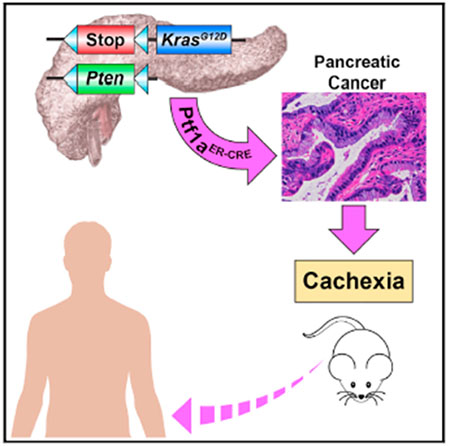
In Brief
Talbert et al. developed an inducible mouse model of cachexia caused by pancreatic cancer. This model exhibits features of the human condition, including the progressive depletion of muscle and adipose tissue associated with tumor progression.
INTRODUCTION
The defining symptom of cancer cachexia is unintentional weight loss because of depletion of skeletal muscle. This wasting cannot be prevented by supplemental nutrition and is often correlated with increased whole-body inflammation and anorexia (Argilés et al., 2014). Cachexia occurs in at least half of all cancer patients and is associated with advanced disease and palliative care. However, in gastrointestinal malignancies, especially pancreatic ductal adenocarcinoma (PDA), cachexia often presents much earlier, with approximately 70% of PDA patients meeting cachexia criteria at cancer diagnosis (Fearon et al., 2011; Nemer et al., 2017).
With a 9% 5-year survival rate and increasing incidence rates, PDA remains a devastating disease (American Cancer Society, 2018). Although resistance to current therapies and the difficultly of diagnosing early-stage malignancy are important contributors to the high mortality rate of PDA patients, the cachexia syndrome also contributes to poor survival in this disease by lowering patient tolerance of chemo- and radiotherapy (Andreyev et al., 1998; Bachmann et al., 2008; Kieler et al., 2017; Moningi et al., 2015; Pausch et al., 2012; von Haehling et al., 2016). Thus, as the search continues to more effectively diagnose and treat PDA, managing cachexia provides an additional strategy to decrease patient morbidity and mortality.
Despite the clear need to prevent and treat cachexia, therapies are currently lacking, as evidenced by two recent phase III clinical trials that failed to achieve their primary endpoints (Le-Rademacher et al., 2017; Temel et al., 2016). Although a number of factors likely contributed to the failure of these trials, one limitation may be the animal models currently used to study cancer cachexia. Two commonly used mouse models are xenograft colon-26 (C-26) and Lewis lung carcinoma (LLC) (Acharyya et al., 2004; Bonetto et al., 2016; He et al., 2013; Judge et al., 2014; Penna et al., 2010; Talbert et al., 2017; Zhang et al., 2017). Although these models have been useful for our laboratory and others in elucidating numerous mechanisms of tissue wasting, they often come under scrutiny for their inability to fully recapitulate the phenotype of human cancers, either by not forming spontaneous tumors or because of their inability to reconstitute a tumor microenvironment. These models are used in a tumor-agnostic manner and are also quite large, typically representing more than 10% of the entire body mass (Talbert et al., 2017; Zhang et al., 2017). They are also aggressive and induce cachexia on a timescale of only a few weeks (Roberts et al., 2013a; Talbert et al., 2014, 2017).
In this study, we tested for a disparity between mice and humans by comparing global gene expression changes in skeletal muscles from C-26 and LLC-bearing mice compared with biopsies obtained from PDA patients with cachexia. The results revealed little overlap in both the number and classification of genes expressed, emphasizing that, on the molecular level, these models may not be the most appropriate to study human cachexia. In addition to these xenograft models, the classic genetically engineered mouse model (GEMM) of PDA, called the Kras; p53; Cre (KPC) mouse, is gaining attraction in cachexia studies. However, as we reveal, this model also has limitations that question its usefulness to study cachexia. Such findings prompted us to design a mouse model of PDA to better reflect the features of human cancer-induced wasting.
RESULTS
The C-26 and LLC Models Do Not Resemble the Muscle Gene Expression Signature of PDA Patients
To assess the ability of the C-26 and LLC models to reflect PDA-induced cachexia, we compared differences between control and cachectic transcriptional profiles determined by RNA sequencing (RNA-seq) with differences between cachectic pancreatic cancer patients and control patients. In the C-26 model, using a false discovery rate (FDR) of < 0.05, we identified 3,474 differentially expressed genes (DEGs) between control and cachectic muscles (Figure 1A; Table S1), which is consistent with published microarray data (Bonetto et al., 2011; Cornwell et al., 2014; Judge et al., 2014). For the LLC model, we identified 788 DEGs (Figure 1A; Table S1), again within the range of published data (Blackwell et al., 2018). Although the LLC model demonstrated fewer DEGs than C-26 mice, gene ontology analysis showed that DEGs from both models were generally associated with processes of RNA translation (Figure S1A). Interestingly, when similar gene expression profiling was performed with cachectic PDA patients, only 141 DEGs were identified compared with weight-stable patients without cancer (Figure 1B; Table S1). Although these DEGs were too few to associate with a biological process by gene ontology, all five cellular components that associated with these DEGs involved the extracellular matrix (Figure S1B).
Figure 1. Comparing Xenograft Models with Cachectic Pancreatic Cancer Patients.
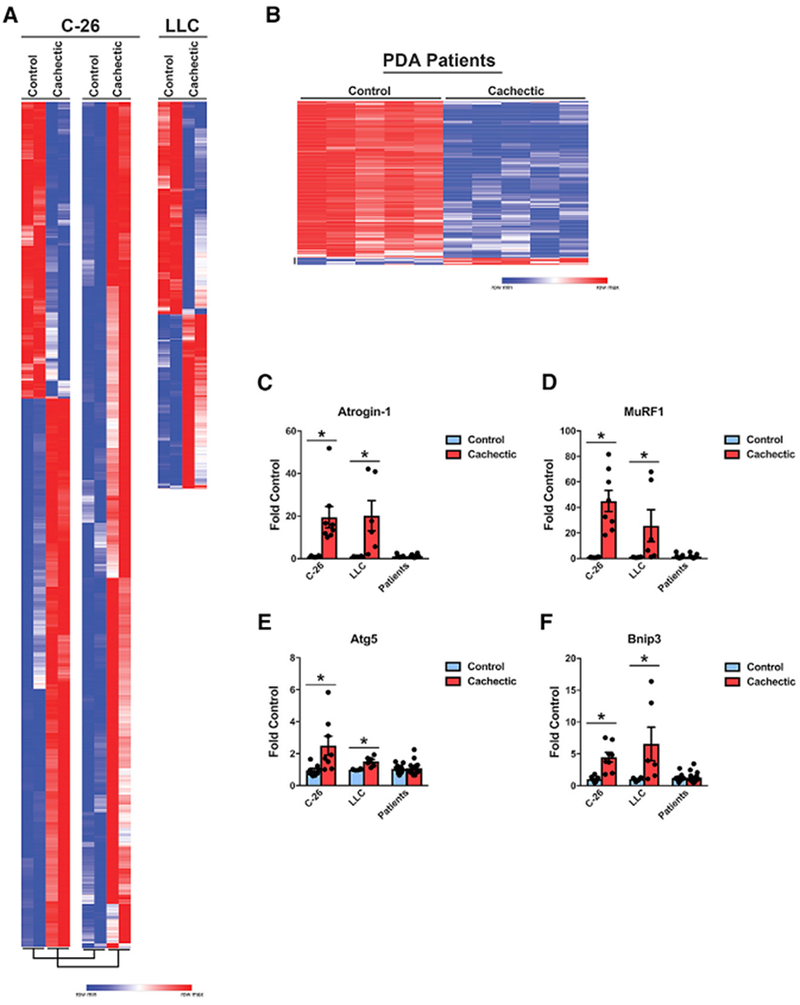
(A) RNA-seq of tibialis anterior (TA) muscle from C-26 and LLC tumor-bearing mice (n = 2 males per group).
(B) RNA-seq of n = 5 male non-cancer patients and 5 male cachectic PDA patients.
(C–F) Gene expression of (C) Atrogin-1, (D) MuRF1, (E) Atg5, and (F) Bnip3 (n = 8 males for C-26; n = 6 males for LLC; n = 11 controls, 17 cachectic patients, approximately equal male and female).
Patient characteristics for RNA-seq can be found in Table S2, and patient characteristics for RT-PCR can be found in Table S3. Bars represent mean ± SEM. Closed circles represent individual data points. *p < 0.05 in a t test.
See Figure S1 for additional information.
A striking feature of these results was that more than 95% of the DEGs from human muscle were decreased in cachectic patients, which we confirmed by qRT-PCR (Figure S1C); this is consistent with previous findings in cachectic patients with upper gastrointestinal (GI) malignancies (Gallagher et al., 2012). This gene expression pattern stands in stark contrast to the overwhelming upregulation of genes seen in the C-26 model (80% upregulated) or the approximately equal proportions of up-regulated and downregulated DEGs observed in LLC tumor-bearing mice.
These results suggested that the gene signatures in muscle from the standard C-26 and LLC models of cancer cachexia are distinct from that of muscle from cachectic pancreatic cancer patients. To test for a discordance specifically in relation to muscle atrophy, we measured the expression of genes coding for components of the ubiquitin proteasome and autophagy systems, which are considered to be causative in cancer-induced muscle wasting (Tisdale, 2009). The results showed the typical large increase in expression of the E3 ubiquitin ligases MuRF1 and Atrogin-1 in C-26 and LLC models, whereas no changes in the expression of these genes were seen in muscles from cachectic PDA patients (Figures 1C and 1D). Expression of the autophagy genes Atg5 and Bnip3 was also increased in muscle from C-26 and LLC mice but remained unchanged in cachectic PDA patients (Figures 1E and 1F). A similar lack of transcriptional changes in these genes in cachectic patients has been reported by other groups (Bonetto et al., 2013; D’Orlando et al., 2014). Collectively, we conclude from these data that xenograft C-26 and LLC models are not optimal to recapitulate the muscle wasting phenotype of human cancer-induced cachexia.
KPC Mice Exhibit Variability and Tissue Mass Changes that Do Not Correlate with Pancreatic Cancer Progression
Next we tested the established KPC GEMM of PDA (Hingorani et al., 2005), which has recently been described as a model of cachexia (Parajuli et al., 2018; Petruzzelli et al., 2014; Roberts et al., 2013b). KPC mice utilize the Pdx1 promoter to drive Cre recombinase to express mutant KrasG12D as well as either a floxed or a lox-stop-lox (LSL) mutant allele of Trp53, leading mice to a median survival of approximately 150 days (Figure S2A; Hingorani et al., 2005; Shakya et al., 2013). Consistent with previous reports, we too observed that KPC mice (Kras+/LSL-G12D, Trp53+/R270H, Pdx1+/Cre) that reached endpoint criteria weighed significantly less than their littermate controls (Figures 2A and S2B). This reduction in weight correlated with reduced hindlimb muscle (tibialis anterior [TA], quadriceps [QUAD], and gastrocnemius [GAST]) mass and lower white adipose tissue mass (Figures 2B, S2C, and 2C). However, when correlating body weight and muscle wasting with pancreas pathology, we were surprised to find that KPC mice tended to be smaller than their littermate control mice regardless of the severity of their pancreatic disease (Figure 2D, 2E, and S2D). In fact, some KPC mice with non-invasive pancreatic lesions (referred to as mPanIN) were among the smallest mice compared with their littermates, whereas other mice with advanced PDA were of similar weight as their littermate controls (Figure S2E). Similarly, KPC mice tended to have smaller muscle mass regardless of pancreas pathology (Figure 2F). Consistent with the heterogeneity of the model, an RNA-seq analysis of muscle from KPC mice similar to the one performed for the C-26 and LLC models returned only a single DEG (FDR < 0.05; Table S1). These results are not in keeping with PDA patients, where lower body and peripheral tissue masses are associated with cancer.
Figure 2. The KPC Mouse as a Model of Cancer Cachexia.
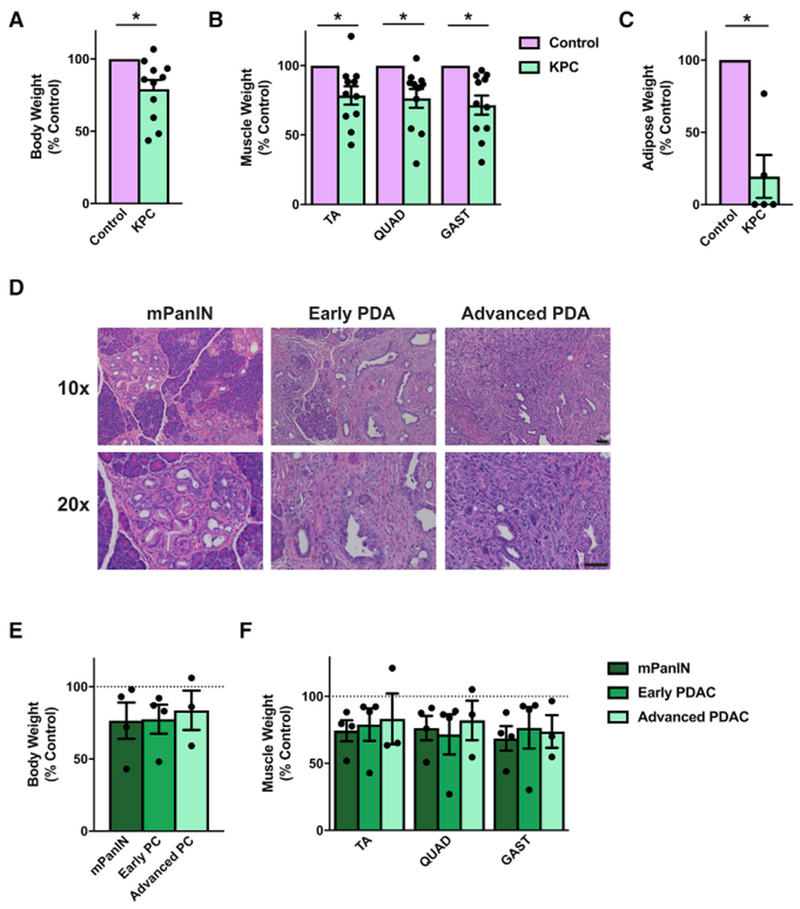
(A–C) KPC mice that have reached endpoint criteria have lower body weights (A; n = 11), muscle weights (B; n = 11), and gonadal white adipose tissue masses (C; n = 5) than littermate control mice.
(D–F) However, when pancreatic histopathology is considered (D), mice with mPanIN lesions (n = 4), early PDA (n = 4), and advanced PDA (n = 3) all tend to have lower body weights (E) and muscle weights (F).
Bars represent mean ± SEM. Closed circles represent individual data points. *p < 0.05 in a paired t test on absolute weights. Scale bars, 50 μm.
See Figure S2 for additional information.
Generation of the KPP Mouse
Given the limitations we identified in both xenograft models and KPC mice, we turned our efforts to generating another model of cancer-induced wasting. When developing this model, we considered several aspects reflective of human disease. First, we sought to model PDA because this disease has among the highest incidences and severities of cancer cachexia (Baracos et al., 2018). Next, because PDA develops in adults, we wanted to induce cancer in postnatal mice. Further, we sought to achieve a direct association between the development of PDA and an ensuing cachexia condition. Specifically, because patients with PDA-induced cachexia lose existing muscle and fat, we sought to model true tissue wasting as opposed to a condition of “perceived wasting,” where tissues do not gain mass because they fail to develop. Finally, we wanted a model that developed cachexia on a slower timescale than the rapid wasting that occurs in current xenograft models.
To meet these criteria, we started with the same LSL-KrasG12D mutant allele found in the KPC mouse but removed the stop cassette with a tamoxifen-inducible Cre recombinase driven by the Ptfla promoter, also known as p48, which remains active into adulthood in the pancreas (Pan et al., 2013). Our model also used two floxed alleles of the tumor suppressor Pten that were inactivated at the same time mutant Kras was induced (Stanger et al., 2005; Ying et al., 2011). Over 70% of human pancreatic tumors demonstrate reduced Pten protein expression, making Pten deletion relevant to pancreatic cancer patients (Jiang et al., 2014; Ying et al., 2011). Together, these mice exhibit the genotype Kras+/G12D,Ptf1a+/ER-Cre, Ptenf/f (Figure 3A), which we named KPP.
Figure 3. Generation of the KPP Mouse and Endpoint Analysis.
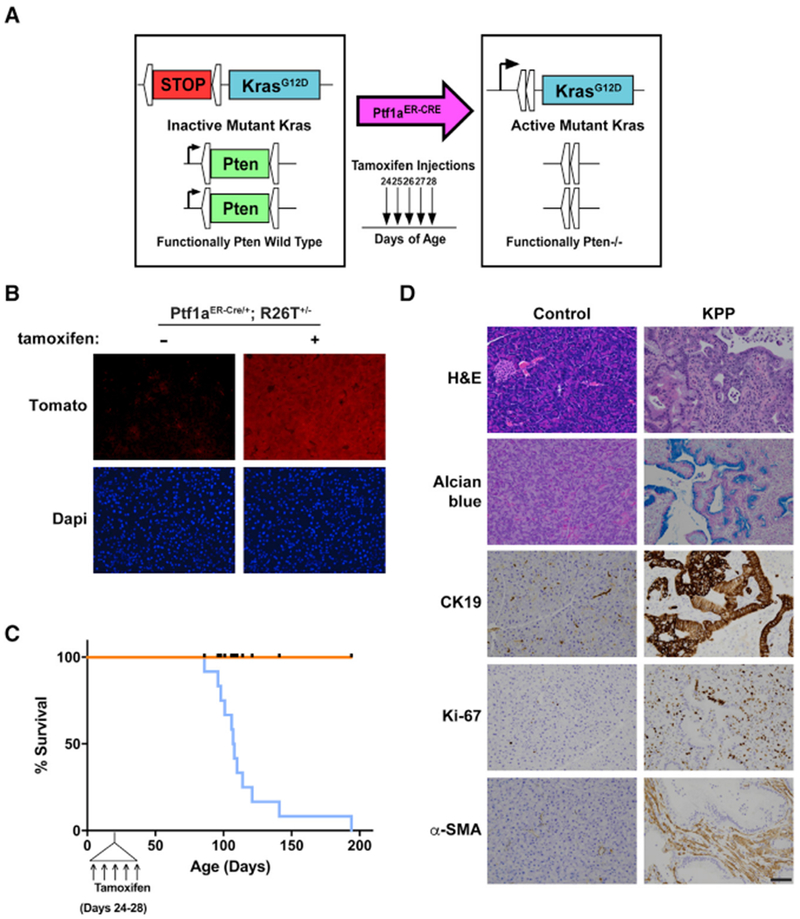
(A) The alleles of the KPP mouse.
(B) Naive Ptfa1ER-Cre/+;Rosa26Tomato+/− mice express background fluorescence in pancreas sections, whereas pancreata of tamoxifen-treated Ptfa1ER-Cre/+;Rosa26Tomato+/− mice exhibit a high fluorescence signal.
(C) KPP mice induced with tamoxifen between 24 and 28 days of age had an average survival of 107 days of age (n = 12). Ticks on the control line indicate points when a control mouse was euthanized for comparison with a littermate KPP mouse. Control genotypes include Kras+/+, Ptf1a+/ER-Cre, Ptenf/f (n = 6), Kras+/+, Ptf1a+/ER-Cre, Pten+/+ (n = 1), Kras+/+, Ptf1a+/ER-Cre, Pten+/+ (n = 1), Kras+/G12D, Ptf1a+/+, Ptenf/f (n = 1), and Kras+/+, Ptf1a+/+, Pten+/f (n = 3). Control genotypes are presented with their paired KPP mouse in Table S4.
(D) Endpoint KPP mice demonstrate moderately well-differentiated to well-differentiated PDA as evidenced by H&E, Alcian blue, CK19, Ki67, and α-smooth muscle actin (α-SMA) staining. Scale bar, 50 μm.
See Figure S3 for additional information.
We first confirmed that the Ptf1aER-Cre promoter is active only in the presence of tamoxifen by breeding Ptf1a+/ER-Cremice to Rosa26-tdTomato reporter mice (Madisen et al., 2010). Mice treated with five consecutive injections of tamoxifen expressed Tomato in acinar cells throughout the pancreas, whereas Tomato expression was not present in Ptf1a+/ER-Cre mice not treated with tamoxifen (Figure 3B). To initiate tumorigenesis, KPP mice were injected with tamoxifen between 24 and 28 days of age. At this age, there was no difference in body mass between control and KPP mice (Figure S3A). With 100% penetrance, KPP mice reached endpoint criteria (ascites, weight loss, and lethargy) at a median age of 107 days (Figure 3C). In comparison, all control mice showed no obvious phenotype at this age (genotypes for each control mouse can be found in Table S4). At the endpoint, the weights of KPP pancreata were significantly increased compared with littermate controls, consistent with tumor burden (Figure S3B). Histological analysis demonstrated well-differentiated to moderately well-differentiated ductal adenocarcinomas composed of anastomosing and branching glands lined by mucinous epithelium accompanied by abundant fibrous stromata. The pancreatic parenchyma was extensively replaced by the neoplasms (Figure 3D; Figure S3C). Cystic glands with intraluminal papillary projections were often noted. Expression of cytokeratin 19 (CK19) demonstrated the ductal phenotype of the glandular epithelium, and Alcian blue staining confirmed the presence of mucin. Additional staining for Ki-67 and α-smooth muscle actin revealed epithelial and stromal proliferative activity and the presence of large numbers of stromal myofibroblasts, respectively (Figure 3D; Figure S3C; Aichler et al., 2012).
KPP Mice Exhibit a Cachectic Phenotype
We next sought to evaluate whether KPP mice suffered from cachexia. KPP mice at their endpoint exhibited decreased body weight compared with their littermate controls (Figure 4A; Figure S4A) but, importantly, had similar tibia lengths (Figure 4B), suggesting that the lower body weights of KPP mice were not due to a failure to grow. KPP mice also exhibited decreased TA, QUAD, and GAST muscle masses (Figure 4C). In the GAST, this decrease in muscle weight was primarily due to decreased cross-sectional area (CSA) of type IIB or IIX muscle fibers (Figure 4D). KPP muscle also showed a trend toward reduced type IIA fiber size (p = 0.06), whereas type I fibers were spared, which is consistent with other mouse models of cancer cachexia (Acharyya et al., 2004; Roberts et al., 2013a). Functionally, muscles from KPP mice exhibited a decrease in total muscle force production at higher stimulation frequencies, but specific force was not altered (Figure 4E), suggesting that the sarcomere structure was intact. Consistent with this phenotype, the content of sarcomeric proteins actin and myosin per mass of muscle was unchanged in KPP mice (Figure S4B), which is unlike what we and others have reported in xenograft cachexia models (Acharyya et al., 2004; Bonetto et al., 2011) but in line with reports suggesting that sarcomeric protein content and specific force are maintained in muscles from cachectic cancer patients (Toth et al., 2013; Weber et al., 2009). Further, night cage activity was decreased, indicating that KPP mice exhibit reduced whole-body function (Figure 4F).
Figure 4. The KPP Mouse Develops Cachexia.
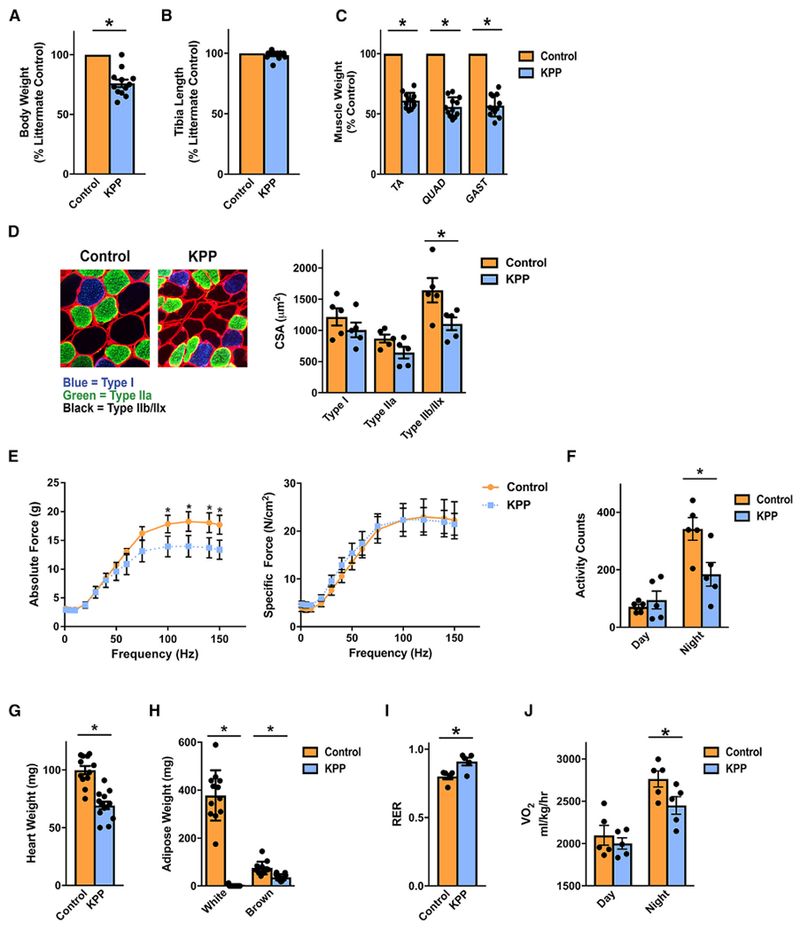
(A and B) KPP mice are smaller than their littermate controls when reaching endpoint criteria (A) but have similar tibia lengths (B).
(C) KPP mice exhibit reduced TA, quadriceps (QUAD), and gastrocnemius (GAST) muscle masses.
(D) Muscle wasting in KPP GAST is associated with lower type IIB or IIX fiber cross-sectional area, whereas type IIA fibers tended to be smaller (p = 0.06).
(E) Absolute ex vivo extensor digitorum longus (EDL) muscle force was significantly reduced at higher stimulation frequencies, but specific force was not altered.
(F) Endpoint KPP mice demonstrate decreased night (awake) activity counts, indicative of depressed whole-body function.
(G and H) KPP mice demonstrate decreased heart weight (G) and decreased masses of gonadal white and subscapular brown adipose (H).
(I) Consistent with decreased adipose tissue stores, KPP mice exhibit an increased daytime respiratory exchange ratio (RER).
(J) KPP mice exhibited decreased night oxygen consumption (VO2).
Bars represent mean ± SEM. Closed circles represent individual data points. *p < 0.05 in a paired t test. n = 12 per group; for cross-sectional area and metabolic data, n = 5 per group, and for muscle function data, n = 3 per group. Even when expressed as percent controls, statistical tests were conducted on absolute weights.
See Figure S4 for additional information.
In addition to reduced muscle mass and function, endpoint KPP mice had enlarged spleens (Figure S4C), which is a feature of both PDA and cachexia (Bonetto et al., 2016; Michaelis et al., 2017; Talbert et al., 2014). Furthermore, KPP mice had significantly lower heart masses (Figure 4G), which is also associated with many models of cancer cachexia (Bonetto et al., 2016; Michaelis et al., 2017; Talbert et al., 2014). KPP mice also had pronouncedly lower white and brown adipose tissue masses (Figure 4H). Consistent with depleted adipose tissue stores, KPP mice displayed an increased resting respiratory exchange ratio (RER), indicative of greater utilization of carbohydrate fuel stores (Figure 4I). Finally, KPP mice consumed less oxygen during dark hours (Figure 4J), which may suggest that carbohydrate utilization is increased or may simply reflect differences in activity (Figure 4F).
To ensure that these cachectic phenotypes were not limited to young mice, where tumor induction was initiated between 3 and 4 weeks of age, we attempted to mimic human PDA in older adults by injecting tamoxifen in 1-year-old KPP mice. Importantly, uninjected KPP mice showed no apparent pancreas pathology, confirming tight regulation of the Ptf1aER-Cre allele (Figure S4D). In contrast, 1-year-old KPP mice developed PDA with a median post-tumor initiation survival of 158 days (Figures S4D–S4F) and exhibited weight loss with associated lower masses of skeletal, cardiac, and adipose tissues (Figures S4G–S4J). Thus, induced cachexia occurs in adult KPP mice.
KPP Mice Undergo Progressive Wasting
Although patients can exhibit cachexia throughout the course of their disease, the incidence of cachexia tends to increase as the disease progresses (Dewys et al., 1980). To determine whether KPP mice exhibited progressive wasting, we induced tumorigenesis at 24-28 days of age and then euthanized cohorts of mice at 60, 75, and 90 days of age during disease progression, as evidenced by tumor burden (Figure S5A). The extent to which the pancreatic parenchyma of KPP mice was replaced by proliferating ductal profiles and accompanying stromal fibrosis increased remarkably with age (Figure 5A). Acinar-to-ductal metaplasia, pancreatic intraepithelial neoplasia (mPanIN), and cystic papillary neoplasms, all of which constitute non-invasive lesions, and occasional focally invasive (early carcinoma) lesions were predominant at 60 and 75 days. Notably, large areas of the exocrine and endocrine pancreas remained unaffected. In contrast, significantly larger portions of the pancreatic parenchyma were replaced by both non-invasive and invasive (advanced carcinoma) lesions at later time points, with severe parenchymal atrophy at 90 days and virtually no remaining histologically normal pancreas by the endpoint.
Figure 5. KPP Mice Exhibit Progressive Cachexia Associated with Advancing Pancreas Pathology.
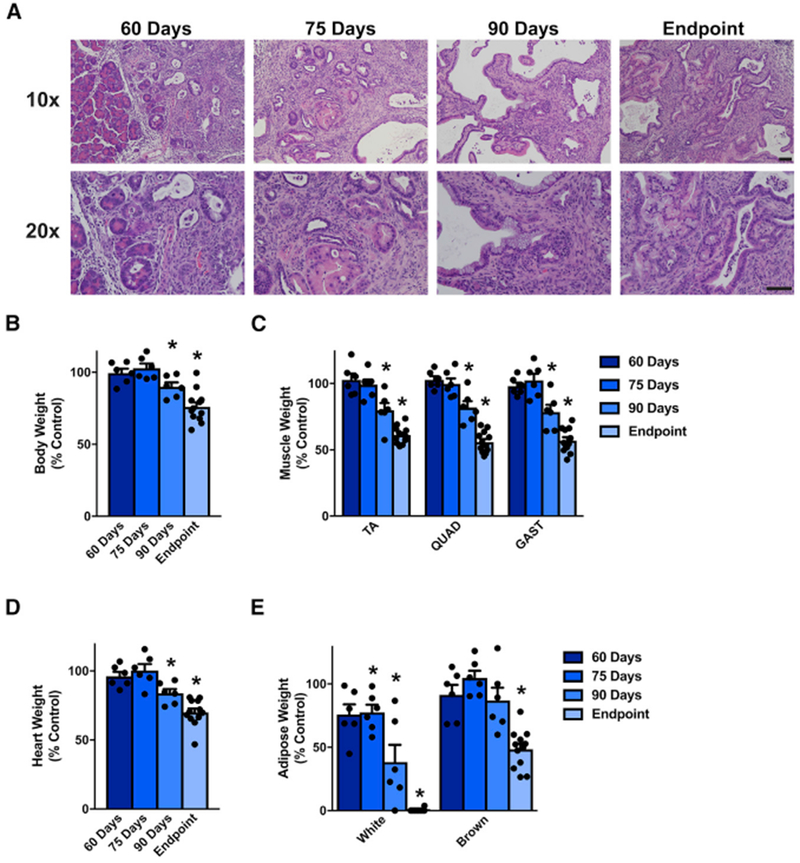
(A) Pancreatic disease in KPP mice progresses across their lifespan.
(B–E) Additionally, compared with their littermate controls, KPP mice progressively lose bodyweight (B) , skeletal muscle (C), heart mass (D), and white and brown adipose tissue (E).
Bars represent mean ± SEM. Closed circles represent individual data points. “Endpoint” KPP mice have reached Institutional Animal Care and Use Committee (IACUC) euthanasia criteria and are the same mice as shown in Figures 3 and 4 (n = 12). n = 5–6 mice per group for the 60-, 75-, and 90-day time points. Genotypes for each control mouse can be found in Table S6. *p < 0.05 in a paired t test on absolute weights. Scale bar, 50 μm.
See Figure S5 for additional information.
Although tibia length was not different between control and KPP mice across all time points (Figure S5B), KPP mice exhibited a progressive decline in body weight compared with their littermate controls (Figure 5B). Significantly, this decrease in weight matched a progressive decline in skeletal muscle (TA, QUAD, and GAST; Figure 5C), heart (Figure 5D), and adipose mass relative to controls (Figure 5E). In both male and female KPP mice, the decline in both absolute skeletal muscle began between days 75 and 90 (Figure S5C), showing that KPP mice exhibit progressive PDA-induced tissue loss, reflective of the human disease. Although KPP mice clearly lose existing adipose tissue, 60-day KPP mice tend to have less adipose tissue than their controls, suggesting that KPP mice may not put on adipose tissue as effectively as control mice during development (Figure S5D).
Cachectic Muscle from KPP Mice and PDA Patients Exhibits a Similar Gene Ontology
Finally, we compared transcriptional profiles between KPP mice and PDA patients with cachexia (Figures 6A and 6B). In sharp contrast to our findings with the C-26 and LLC xenograft models, only 172 DEGs were identified in KPP mice using an FDR of < 0.05. However, the five most significant biological process categories identified by gene ontology were related to the extracellular matrix, reminiscent of the cellular component gene ontology of cachectic PDA patients (Figure S6A). Furthermore, KPP mice exhibited smaller increases in the expression of E3 ubiquitin ligase and autophagy genes compared with the common xenograft models (Figures 6C–6F), and these modest increases only occurred during end-stage cachexia (Figure S6B). Together, these results imply that the cachexia phenotype of KPP mice may more closely resemble that of PDA patients.
Figure 6. The Gene Ontology of KPP Mice Resembles that of Cachectic PDA Patients.
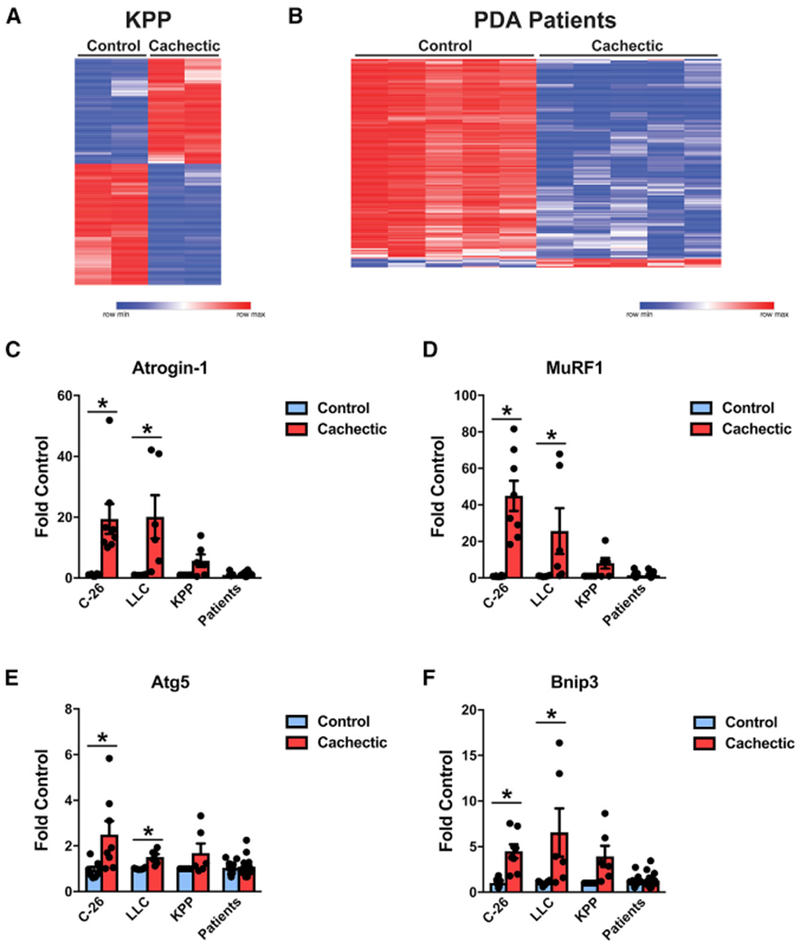
(A) RNA-seq of TA muscle from KPP mice (n = 2 males per group).
(B) RNA-seq of n = 5 male non-cancer patients and 5 male cachectic PDA patients.
(C–F) Gene expression of (C) Atrogin-1, (D) MuRF1, (E) Atg5, and (F) Bnip3 (n = 8 males for C-26; n = 6 males for LLC, n = 3 males and 3 females for KPP; n = 11 controls, 17 cachectic patients, approximately equal male and female).
Patient characteristics for RNA-seq can be found in Table S2, and patient characteristics for RT-PCR can be found in Table S3. Bars represent mean ± SEM. Closed circles represent individual data points. *p < 0.05 in a t test.
See Figure S6 for additional information.
DISCUSSION
Because mechanistic studies of cancer cachexia are difficult to complete in human patients, our current knowledge of mechanisms of cancer-induced wasting derives mainly from animal models of the disease. However, our work presented here suggests that, in many ways, two of the most commonly used xenograft mouse models of cachexia do not reflect gene expression changes seen in muscle of cachectic PDA patients. This is perhaps not surprising because both the C-26 and LLC models result in aggressive tumors and induce rapid wasting over an approximately 1- to 2-week period (Acharyya et al., 2004, 2005; He et al., 2013; Talbert et al., 2014, 2017; Zhang et al., 2017). In addition, the loss of skeletal muscle and adipose mass in these models is generally dependent on inflammatory factors whose concentration in the circulation far exceeds levels recorded in cachectic patients (Lerner et al., 2015, 2016a, 2016b; Llovera et al., 1998b; Strassmann et al., 1992; Talbert et al., 2018).
In an effort to better model the cachexia experienced by patients, a number of laboratories have recently published new mouse models of cancer cachexia, often utilizing more relevant cachexia-inducing cancers, with PDA being the most common. The three main approaches of these models have been (1) intraperitoneal injections of PDA cells, which localize to the pancreas; (2) orthotopic models of PDA in which cancer cells are injected directly into the pancreas; and (3) patient-derived xenograft (PDX) models in which a portion of a resected human pancreatic tumor is surgically attached to a mouse pancreas (Delitto et al., 2017; Go et al., 2017; Greco et al., 2015; Michaelis et al., 2017). Although these models have the advantage of establishing tumors within the pancreas, some are limited by their narrow window of wasting, similar to the C-26 and LLC models (Greco et al., 2015; Michaelis et al., 2017; Yang et al., 2019). Additionally, it is possible that the post-surgical inflammatory environment created by orthotopic injections and surgical tumor implantation contributes to the appearance of wasting rather than deriving from bona fide, tumor-induced tissue loss. Finally, use of human tumors or tumor cells necessitates the use of immunocompromised mice, which themselves may fall short in reconstituting the immune profile of cachexia. Thus, although these models may be an improvement over the C-26 or LLC models, they still face challenges in replicating human cancer cachexia.
Although a growing number of reports describe KPC mice as a model of cancer cachexia, in our view, this model also has significant limitations. Although human PDA is estimated to develop over the course of at least a decade (Yachida et al., 2010), no evidence suggests that mutant Kras is active prior to birth, as in the KPC mouse (Guz et al., 1995). Because neonatal skeletal muscle quadruples in size during the first 3 weeks of life (White et al., 2010), this raises the suspicion that prenatal activation of an oncogene might affect the development of skeletal muscle. This notion is supported by our findings that KPC mice tend to be smaller than their littermate controls regardless of pancreas pathology. An additional consideration with the use of KPC mice as a cachexia model is the propensity of these mice to develop other tumors outside of the pancreas (Gades et al., 2008; Lampson et al., 2012), which might influence how cachexia is interpreted. Admittedly, it may be possible to reduce the incidence of non-pancreas tumors by backcrossing mice into a pure strain, but this remains to be tested. Finally, we speculate that performing pre-clinical cachexia studies would be challenging in the KPC model because the slope of the survival curve of these mice is gradual (Hingorani et al., 2005; Shakya et al., 2013), meaning large cohorts would likely be required to achieve sufficient power to assess the effectiveness of an anti-cachexia compound.
For our purposes, we sought to use a GEMM of PDA that could overcome some of the shortcomings of the current models. Our findings show that the KPP mouse is suitable as a model of cancer cachexia for several reasons. The inducible nature of the Cre re-combinase utilized in the KPP mouse allows PDA and its associated cachexia to be induced after the rapid neonatal growth of mice has concluded. This allowed us to better model the actual loss of muscle and adipose tissue in patients with PDA, as opposed to potentially slowing development and giving the appearance of cachexia. We also showed that the inducible Cre is amenable to inducing tumor developments older mice, which allows manipulation of experimental conditions associated with PDA in older adults. Moreover, the slope of the survival curve of KPP mice is steeper than that of KPC mice (Figure S6C; Hingorani et al., 2005; Shakya et al., 2013), improving the feasibility of testing potential anti-cachexia compounds.
Although tumor-induced muscle wasting in KPP mice appears to more closely model the cachexia seen in PDA patients, this model is not without limitations. Because KPP mice lose normal pancreatic parenchyma, we cannot exclude the possibility, as reported recently (Danai et al., 2018), that pancreatic exocrine insufficiency contributes to tumor-induced wasting in this model. Further, it is possible that features of KPP mice are selective to PDA and, thus, less useful to study other tumor types that also promote cachexia. Despite these limitations, we view the KPP mouse as an improvement over existing animal models of cachexia and anticipate that KPP mice will prove to be a useful tool in improving our understanding of the mechanisms driving tissue wasting and in translating that understanding into new anti-cachexia therapies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Denis Guttridge (guttridg@musc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal experiments were approved by the Ohio State University Institutional Animal Care and Use Committee under protocol 2010A00000177 or the Medical University of South Carolina Institutional Animal Care and Use Committee under protocol 2018-00328. All animals were housed with littermates and given ad libitum access to standard chow and water. C-26 and LLC mice were allocated to tumor-bearing and control groups randomly, genetic models were allocated by genotype.
C-26 Mice
Experiments using the colon-26 (C-26) model were conducted as previously described (He et al., 2013; Talbert et al., 2014; Tanaka et al., 1990). Male CD2F1 mice from Charles River Laboratories were injected subcutaneously in the right flank with 1×106 C-26 cells under anesthesia. Control mice received an injection the same volume of phosphate buffered saline. Mice were euthanized by CO2 asphyxiation 21 days after injection, at which time tumor-bearing mice had reached IACUC euthanasia criteria of tumor volume or greater than 20% body weight loss.
LLC Mice
Experiments using the Lewis Lung Carcinoma (LLC) model were conducted as previously described (He et al., 2013; Llovera et al., 1998a; Talbert et al., 2017). Male C57BL/6J mice from Jackson Laboratories were injected intramuscularly in the right gluteus muscle with 5×105 LLC cells under anesthesia. Control mice received an injection the same volume of phosphate buffered saline. Mice were euthanized by CO2 asphyxiation 21 days after injection, at which time tumor-bearing mice had reached IACUC euthanasia criteria of tumor volume or > 20% weight loss.
KPC Mice
KPC mice (Kras+/LSL-G12D, Trp53+/R270H, Pdx1+/Cre) were provided by the Target Validation Shared Resource of The Ohio State Comprehensive Cancer Center. Mice were maintained on a mixed background (Shakya et al., 2013). Mice were euthanized by CO2 asphyxiation upon reaching IACUC-approved endpoint criteria, which included ascites, weight loss, lethargy, and severe skin papillomas. Both male and female KPC mice were utilized, with same-sex littermates were used as controls.
KPP Mice
Alleles used to generate KPP mice were obtained from Jackson Laboratory on a C57BL/6J background. The initial cohort of KPP mice was generated by breeding KrasLSL-G12D/+, Pten+/f mice to Ptf1aER-Cre/+, Pten+/f mice. Subsequent mice, including all mice euthanized prior to reaching endpoint criteria, were generated by breeding KrasLSL-G12D/+, Ptenf/f mice to Ptf1aER-Cre/+, Ptenf/f mice. Both male and female KPP mice were utilized, with same-sex littermates were used as controls. Genotypes of control mice appear in Tables S4, S5, and S6.
KPP mice and littermate controls were treated with tamoxifen by five consecutive daily intraperitoneal injections. Mice treated between 24-28 days of age received 1 mg tamoxifen/10 g of body weight, while year-old mice received 2 mg tamoxifen/dose.
Endpoint KPP mice euthanized upon reaching IACUC endpoint criteria, which included ascites, weight loss, lethargy, and a body condition score less than 1.5 (Ullman-Culleré and Foltz, 1999). Mice were euthanized at a surgical plane of isoflurane by cardiac puncture followed by cervical dislocation. Final body weights were collected after the draining of ascites fluid. Absence of pancreas pathology was confirmed in all mice utilized as controls by a veterinary pathologist.
Human Subjects
Patients undergoing attempted pancreatic resections at The Ohio State University Wexner Medical Center were asked to participate in a tissue banking protocol under which rectus abdominus muscle biopsies taken during surgery were deposited into a tissue bank run by The Ohio State University Comprehensive Cancer Center Pancreatic Cancer Program. Control muscle tissue was obtained from consenting volunteers without malignancy who were undergoing laparotomy for elective indications (e.g., hernia repair, ostomy reversal). All experiments were approved by The Ohio State University Cancer Institutional Review Board and all patients provided informed consent.
Patients with greater than 5% loss of their pre-illness weight were classified as cachectic (Fearon et al., 2011). When possible, self-reported weight loss was supported by a patient’s existing medical record. All tissue samples used in this analysis were from patients who had not received chemotherapy or radiation prior to surgery for this incidence of cancer. A previous history of a cancer was not an exclusionary criterion. Clinical characteristics of the patients from which muscle was used for RNA-Seq are included in Table S2, with characteristics of patients used for RT-PCR in Table S3. RNA-Seq was conducted on muscle from male subjects, while RT-PCR was performed on a roughly even mix of male and female subjects.
METHOD DETAILS
RNA-Seq and Gene Ontology
RNA was isolated from snap-frozen tibialis anterior (mouse) or rectus abdominus (human) muscle using Trizol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. RNA quality was assessed using an Agilent Bioanalyzer RNA 6000 Nano chip and RIN numbers for all samples were greater than 7. rRNA was removed from purified total RNA using Ribo-Zero Gold rRNA Removal kit (Illumina; MRZG12324). 200 ng of rRNA-depleted RNA was used for the construction of the library using the SureSelect Strand Specific RNA library prep kit (Agilent; G9691A) according to the manufacturer’s instruction, except for omitting the purification step before fragmentation. Barcoded libraries were pooled and sequenced using an Illumina HiSeq4000.
For muscle samples from mice, raw reads were aligned to Mus musculus GRCm38 from ensembl with tophat2v2.1.0 (Kim et al., 2013) which ran with gene model annotations provided by GRCm38 v83. Sequence for samples from multiple lanes were combined. Alignment QC was generated with RSeQCv2.6.4 (Wang et al., 2012) and picardv2.4.1 (http://broadinstitute.github.io/picard). Fragment counts were generated with featureCounts v1.4.5 (Liao et al., 2014) from the subread package for genes described by ensembl GRCm38 v83. For reads with multiple alignments, the primary alignment was counted by featureCounts. Strand-specific read counting was performed based on sequence that is reversely stranded. Differential expression was performed with limma (Ritchie et al., 2015) for genes with CPM > 2 in 4 or more samples.
For muscle samples from patients, raw reads for human samples were aligned to Homo sapiens GRCh38 with HISAT2 v2.0.5 (Kim et al., 2015). Alignment QC was generated with RSeQCv2.6.4 (Wang et al., 2012) and picardv2.4.1 (http://broadinstitute.github.io/ picard). Fragment counts were generated with featureCounts v1.4.5 (Liao et al., 2014) from the subread package for genes described by ensembl GRCh38 v86. For reads with multiple alignments, the primary alignment was counted by featureCounts. Strand-specific read counting was performed based on sequence that is reversely stranded. Differential expression was performed with limma (Ritchie et al., 2015) for genes with CPM > 2 in more than half the samples. For human samples, gene downregulation confirmed by real-time RT-PCR for 9 samples with RNA available. For both human and mouse samples, gene ontology was performed using the ToppFun application of the ToppGene Suite (Chen et al., 2009). Heatmaps were produced with Morpheus from the Broad Institute (https://software.broadinstitute.org/morpheus).
Real-Time RT-PCR
Messenger RNA was isolated from snap-frozen quadriceps (mouse) or rectus abdominus (human) muscle using Trizol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. M-MLV Reverse Transcriptase (Life Technologies) was used to reverse-transcribe total RNA to cDNA according to the manufacturer’s instructions. Real-time PCR was performed on an Applied Biosystems StepOnePlus instrument using SYBR Green mix (Bio-Rad, Hercules, CA). GAPDH was used as the housekeeping gene. Primer sequences appear in Table S7.
Pancreas Histopathology
Formalin-fixed, paraffin-embedded, hematoxylin-and-eosin-stained pancreata of KPC and KPP mice were assessed by a veterinary pathologist employing recommended nomenclature for pancreatic exocrine neoplasia (Hruban et al., 2006). To better characterize the lesions, pancreas sections from endpoint KPP mice were stained for Alcian blue to detect mucin and glycoproteins, Ki67 (1:200), α-smooth muscle actin (α-SMA, 1:5000), and cytokeratin 19 (CK19, 1:500). The sections were imaged using a Vectra Multispectral Imaging System (Perkin-Elmer).
CLAMS
Endpoint KPP mice (n = 5) were single-housed in a Comprehensive Lab Animal Monitoring System (CLAMS) unit (Columbus Instruments, Columbus, OH) for 24 hours prior to euthanasia. Environmental temperature was held constant at 25 °C. Mice were placed in the unit at 10 AM, and daytime data were collected from 12 PM until five minutes prior to lights out. Night data were collected from one hour after lights out until 5 minutes prior to lights on.
Muscle Fiber Size Measurements
Muscle fiber cross-sectional area (CSA) of individual fiber types was determined from 10 μM sections of gastrocnemius stained for laminin, myosin heavy chain Type IIA, and myosin heavy chain Type I by a modification of Minnaard (Minnaard et al., 2005). Muscle fiber CSA was determined using a semi-automated ImageJ plug-in designed in the Guttridge lab which outlines the internal laminin border of individual muscle fibers and then assigns fiber types based upon color of the fiber.
Muscle Function Measurements
Extensor digitorum longus (EDL) force production was measured across a force-frequency curve. Force was normalized to CSA as previously described (Segal et al., 1986).
Sarcomeric Protein Analysis
Muscle sarcomere components were analyzed as previously described (Blough et al., 1996).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was conducted with GraphPad Prism 7, and the details of these analyses appear in the figure legends with samples per group (n). Unless otherwise noted, data are presented as mean ± the standard error of the mean (SEM). For KPC and KPP mice, comparisons between groups were made by two-tailed paired t tests. For C-26 and LLC mice and comparisons between human subjects, unpaired two-tailed tests were used. Statistical analysis of tissue masses was conducted on absolute values, regardless of the presentation of the data. Statistical significance was defined a priori as p < 0.05.
DATA AND CODE AVAILABILITY
Raw RNA-Seq data files are available in the NCBI’s GEO: GSE133523 and GSE133524.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ki67 | Abcam | ab16667; RRID: AB_302459 |
| α-smooth muscle actin (α-SMA) | Abcam | ab5694; RRID: AB_2223021 |
| cytokeratin 19 (CK19) | Iowa Development Studies Hybridoma Bank | TROMA-III; RRID: AB_2133570 |
| laminin | ThermoScientific | MA1-06100; RRID: AB_559896 |
| myosin heavy chain Type I | Iowa Development Studies Hybridoma Bank | A4.840; RRID: AB_528384 |
| myosin heavy chain Type IIA | Iowa Development Studies Hybridoma Bank | sc-71; RRID: AB_2147165 |
| Biological Samples | ||
| rectus abdominus muscle biopsies | Pancreatic cancer patients and control patients undergoing abdominal surgery | N/A |
| Deposited Data | ||
| Raw RNA-Seq files (human) | This paper | GEO: GSE133523 |
| Raw RNA-Seq files (mouse) | This paper | GEO: GSE133524 |
| Experimental Models: Cell Lines | ||
| Colon-26 (C-26) | National Cancer Institute | N/A |
| Lewis Lung Carcinoma (LLC) | National Cancer Institute | N/A |
| Experimental Models: Organisms/Strains | ||
| KrasLSL-G12D (KPP) | The Jackson Laboratory | #008179 |
| Ptenf/f | The Jackson Laboratory | #006440 |
| Ptf1aER-Cre | The Jackson Laboratory | #019378 |
| Rosa26-tdTomato | The Jackson Laboratory | #007914 |
| KrasLSL-G12D (KPC) | NCI Frederick Mouse Repository | 01XJ6 |
| Pdx1Cre | NCI Frederick Mouse Repository | 01XL5 |
| Trp53+/R270H | NCI Frederick Mouse Repository | 01XM3 |
| Oligonucleotides | ||
| Primers for RT-PCR, see Table S7 | This paper, Talbert et al., 2014 | N/A |
| Software and Algorithms | ||
| tophat2 | Kim et al., 2013 | v2.1.0 |
| HISAT2 | Kim et al., 2015 | v2.0.5 |
| featureCounts | Liao et al., 2014 | v1.4.5 |
| RSeQC | Wang et al., 2012 | v2.6.4 |
Highlights.
Development of a mouse model of pancreatic adenocarcinoma (PDA)-induced cachexia
Model develops progressive wasting associated with advancing pancreas pathology
Induction of cachexia in adult KPP mice models tissue loss in PDA cancer patients
Gene ontology of cachectic muscles from KPP mice resembles that of PDA patients
ACKNOWLEDGMENTS
The authors would like to thank David Wang and Nivedita Ratman for thoughtful discussions of the manuscript, Lisa Baer for assistance with CLAMS; Tom Liu for assistance with the RNA-seq; Sabahattin Bicer for assistance with the myofilament gels; Jason Bice, Daphne Bryant, Raleigh Kladeny, and Melodie Parrish for histology assistance; Ericka Haverick, Karina Woodward, Angela Sarna, and Erica Williams for coordination of human subjects; and Jeffrey Chakedis, Heather Lewis, and Mitchell Ramsey for clinical data extraction. Funding was provided by NIH R21AR071021 (to D.C.G.), R01CA180057 (to D.C.G.), and K99AR071508 (to E.E.T.); and American Cancer Society postdoctoral fellowship PF-15-156-01-CSM (to E.E.T.) and a Weiss postdoctoral fellowship (to E.E.T.). Additional support was from The Ohio State Comprehensive Cancer Center P30CA016058, OSUCCC institutional funds and Hollings Cancer Center P30CA138313. A portion of this work was conducted in a facility constructed with support from NIH C06 RR015455.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.016.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, and Guttridge DC (2004). Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Invest 114, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, Ringel MD, Skipworth RJ, Fearon KC, Hollingsworth MA, et al. (2005). Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 8, 421–432. [DOI] [PubMed] [Google Scholar]
- Aichler M, Seiler C, Tost M, Siveke J, Mazur PK, Da Silva-Buttkus P, Bartsch DK, Langer P, Chiblak S, Dürr A, et al. (2012). Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J. Pathol 226, 723–734. [DOI] [PubMed] [Google Scholar]
- American Cancer Society (2018). Cancer Facts & Figures 2018 (American Cancer Society). [Google Scholar]
- Andreyev HJ, Norman AR, Oates J, and Cunningham D (1998). Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur. J. Cancer 34, 503–509. [DOI] [PubMed] [Google Scholar]
- Argilés JM, Busquets S, Stemmler B, and López-Soriano FJ (2014). Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer 14, 754–762. [DOI] [PubMed] [Google Scholar]
- Bachmann J, Heiligensetzer M, Krakowski-Roosen H, Buchler MW, Friess H, and Martignoni ME (2008). Cachexia worsens prognosis in patients with resectable pancreatic cancer. J. Gastrointest. Surg 12, 1193–1201. [DOI] [PubMed] [Google Scholar]
- Baracos VE, Martin L, Korc M, Guttridge DC, and Fearon KCH (2018). Cancer-associated cachexia. Nat. Rev. Dis. Primers 4, 17105. [DOI] [PubMed] [Google Scholar]
- Blackwell TA, Cervenka I, Khatri B, Brown JL, Rosa-Caldwell ME, Lee DE, Perry RA Jr., Brown LA, Haynie WS, Wiggs MP, et al. (2018). Transcriptomic analysis of the development of skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice. Physiol. Genomics 50, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blough ER, Rennie ER, Zhang F, and Reiser PJ (1996). Enhanced electrophoretic separation and resolution of myosin heavy chains in mammalian and avian skeletal muscles. Anal. Biochem 233, 31–35. [DOI] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, and Zimmers TA (2011). STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 6, e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Penna F, Aversa Z, Mercantini P, Baccino FM, Costelli P, Ziparo V, Lucia S, Rossi Fanelli F, and Muscaritoli M (2013). Early changes of muscle insulin-like growth factor-1 and myostatin gene expression in gastric cancer patients. Muscle Nerve 48, 387–392. [DOI] [PubMed] [Google Scholar]
- Bonetto A, Rupert JE, Barreto R, and Zimmers TA (2016). The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J. Vis. Exp Published online November 30, 2016 10.3791/54893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, and Jegga AG (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell EW, Mirbod A, Wu CL, Kandarian SC, and Jackman RW (2014). C26 cancer-induced muscle wasting is IKKb-dependent and NF-kap-paB-independent. PLoS ONE 9, e87776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orlando C, Marzetti E, Francois S, Lorenzi M, Conti V, di Stasio E, Rosa F, Brunelli S, Doglietto GB, Pacelli F, and Bossola M (2014). Gastric cancer does not affect the expression of atrophy-related genes in human skeletal muscle. Muscle Nerve 49, 528–533. [DOI] [PubMed] [Google Scholar]
- Danai LV, Babic A, Rosenthal MH, Dennstedt EA, Muir A, Lien EC, Mayers JR, Tai K, Lau AN, Jones-Sali P, et al. (2018). Altered exocrine function can drive adipose wasting in early pancreatic cancer. Nature 558, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, Gerber MH, George TJ Jr., Behrns KE, Hughes SJ, et al. (2017). Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 8, 1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr., Engstrom PF, Ezdinli EZ, et al. ; Eastern Cooperative Oncology Group (1980). Prognostic effect of weight loss prior to chemotherapy in cancer patients. Am. J. Med 69, 491–497. [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. (2011). Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495. [DOI] [PubMed] [Google Scholar]
- Gades NM, Ohash A, Mills LD, Rowley MA, Predmore KS, Marler RJ, and Couch FJ (2008). Spontaneous vulvar papillomas in a colony of mice used for pancreatic cancer research. Comp. Med 58, 271–275. [PMC free article] [PubMed] [Google Scholar]
- Gallagher IJ, Stephens NA, MacDonald AJ, Skipworth RJ, Husi H, Greig CA, Ross JA, Timmons JA, and Fearon KC (2012). Suppression of skeletal muscle turnover in cancer cachexia: evidence from the transcrip-tome in sequential human muscle biopsies. Clin. Cancer Res 18, 2817–2827. [DOI] [PubMed] [Google Scholar]
- Go KL, Delitto D, Judge SM, Gerber MH, George TJ Jr., Behrns KE, Hughes SJ, Judge AR, and Trevino JG (2017). Orthotopic Patient-Derived Pancreatic Cancer Xenografts Engraft Into the Pancreatic Parenchyma, Metastasize, and Induce Muscle Wasting to Recapitulate the Human Disease. Pancreas 46, 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco SH, Tomkötter L, Vahle AK, Rokosh R, Avanzi A, Mahmood SK, Deutsch M, Alothman S, Alqunaibit D, Ochi A, et al. (2015). TGF-β Blockade Reduces Mortality and Metabolic Changes in a Validated Murine Model of Pancreatic Cancer Cachexia. PLoS ONE 10, e0132786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, and Teitelman G (1995). Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 121, 11–18. [DOI] [PubMed] [Google Scholar]
- He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J,Wang J, Bloomston M, Muscarella P, Nau P, et al. (2013). NF-kB-medi-ated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest 123, 4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein A, Klimstra DS, et al. (2006). Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 66, 95–106. [DOI] [PubMed] [Google Scholar]
- Jiang K, Lawson D, Cohen C, and Siddiqui MT (2014). Galectin-3 and PTEN expression in pancreatic ductal adenocarcinoma, pancreatic neuroendocrine neoplasms and gastrointestinal tumors on fine-needle aspiration cytology. Acta Cytol. 58, 281–287. [DOI] [PubMed] [Google Scholar]
- Judge SM, Wu CL, Beharry AW, Roberts BM, Ferreira LF, Kandarian SC, and Judge AR (2014). Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 14, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieler M, Unseld M, Bianconi D, and Prager GW (2017). Cross-over comparison and new chemotherapy regimens in metastatic pancreatic cancer. Memo. 10, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson BL, Kendall SD, Ancrile BB, Morrison MM, Shealy MJ, Barrientos KS, Crowe MS, Kashatus DF, White RR, Gurley SB, et al. (2012). Targeting eNOS in pancreatic cancer. Cancer Res. 72, 4472–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le-Rademacher JG, Crawford J, Evans WJ, and Jatoi A (2017). Overcoming obstacles in the design of cancer anorexia/weight loss trials. Crit. Rev. Oncol. Hematol 117, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L, Hayes TG, Tao N, Krieger B, Feng B, Wu Z, Nicoletti R, Chiu MI, Gyuris J, and Garcia JM (2015). Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachexia Sarcopenia Muscle 6, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L, Gyuris J, Nicoletti R, Gifford J, Krieger B, and Jatoi A (2016a). Growth differentiating factor-15 (GDF-15): A potential biomarker and therapeutic target for cancer-associated weight loss. Oncol. Lett 12, 4219–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, Mazsa E, Siddiquee Z, Wang R, Huang L, et al. (2016b). MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 7, 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Llovera M, García-Martínez C, López-Soriano J, Agell N, López-Soriano FJ, Garcia I, and Argilés JM (1998a). Protein turnover in skeletal muscle of tumour-bearing transgenic mice overexpressing the soluble TNF receptor-1. Cancer Lett. 130, 19–27. [DOI] [PubMed] [Google Scholar]
- Llovera M, García-Martínez C, López-Soriano J, Carbó N, Agell N, Ló-pez-Soriano FJ, and Argiles JM (1998b). Role of TNF receptor 1 in protein turnover during cancer cachexia using gene knockout mice. Mol. Cell. Endocrinol 142, 183–189. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, and Marks DL (2017). Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 8, 824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnaard R, Drost MR, Wagenmakers AJ, van Kranenburg GP, Kuipers H, and Hesselink MK (2005). Skeletal Muscle wasting and contractile performance in septic rats. Muscle Nerve 31, 339–348. [DOI] [PubMed] [Google Scholar]
- Moningi S, Walker AJ, Hsu CC, Reese JB, Wang JY, Fan KY, Rosati M, Laheru DA, Weiss MJ, Wolfgang CL, et al. (2015). Correlation of clinical stage and performance status with quality of life in patients seen in a pancreas multidisciplinary clinic. J. Oncol. Pract 11, e216–e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemer L, Krishna SG, Shah ZK, Conwell DL, Cruz-Monserrate Z, Dillhoff M, Guttridge DC, Hinton A, Manilchuk A, Pawlik TM, et al. (2017). Predictors of Pancreatic Cancer-Associated Weight Loss and Nutritional Interventions. Pancreas 46, 1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, and Wright CV (2013). Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli P, Kumar S, Loumaye A, Singh P, Eragamreddy S, Nguyen TL, Ozkan S, Razzaque MS, Prunier C, Thissen JP, and Atfi A (2018). Twist1 Activation in Muscle Progenitor Cells Causes Muscle Loss Akin to Cancer Cachexia. Dev. Cell 45, 712–725. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pausch T, Hartwig W, Hinz U, Swolana T, Bundy BD, Hackert T, Grenacher L, Büchler MW, and Werner J (2012). Cachexia but not obesity worsens the postoperative outcome after pancreatoduodenectomy in pancreatic cancer. Surgery 152 (3, Suppl 1), S81–S88. [DOI] [PubMed] [Google Scholar]
- Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, and Costelli P (2010). Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 5, e13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, Swarbrick M, Rose-John S, Rincon M, Robertson G, et al. (2014). A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 20, 433–447. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BM, Frye GS, Ahn B, Ferreira LF, and Judge AR (2013a). Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem. Biophys. Res. Commun 435, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, et al. (2013b). Depletion of stromal cells expressing fibroblast activation protein-a from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med 210, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS, White TP, and Faulkner JA (1986). Architecture, composition, and contractile properties of rat soleus muscle grafts. Am. J. Physiol 250, C474–C479. [DOI] [PubMed] [Google Scholar]
- Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, et al. (2013). Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 73, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, Greenwood A, Cheng KH, McLaughlin M, Brown D, et al. (2005). Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 8, 185–195. [DOI] [PubMed] [Google Scholar]
- Strassmann G, Fong M, Kenney JS, and Jacob CO (1992). Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Invest 89, 1681–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert EE, Metzger GA, He WA, and Guttridge DC (2014). Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J. Cachexia Sarcopenia Muscle 5, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert EE, Yang J, Mace TA, Farren MR, Farris AB, Young GS, Elnaggar O, Che Z, Timmers CD, Rajasekera P, et al. (2017). Dual Inhibition of MEK and PI3K/Akt Rescues Cancer Cachexia through both Tumor-Extrinsic and -Intrinsic Activities. Mol. Cancer Ther 16, 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert EE, Lewis HL, Farren MR, Ramsey ML, Chakedis JM, Rajasekera P, Haverick E, Sarna A, Bloomston M, Pawlik TM, et al. (2018). Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachexia Sarcopenia Muscle 9, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Eda H, Tanaka T, Udagawa T, Ishikawa T, Horii I, Ishitsuka H, Kataoka T, and Taguchi T (1990). Experimental cancer cachexia induced by transplantable colon 26 adenocarcinoma in mice. Cancer Res. 50, 2290–2295. [PubMed] [Google Scholar]
- Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, and Fearon KC (2016). Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 17, 519–531. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ (2009). Mechanisms of cancer cachexia. Physiol. Rev 89, 381–410. [DOI] [PubMed] [Google Scholar]
- Toth MJ, Miller MS, Callahan DM, Sweeny AP, Nunez I, Grunberg SM, Der-Torossian H, Couch ME, and Dittus K (2013). Molecular mechanisms underlying skeletal muscle weakness in human cancer: reduced myosin-actin cross-bridge formation and kinetics. J. Appl. Physiol 114, 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman-Culleré MH, and Foltz CJ (1999). Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab. Anim. Sci 49, 319–323. [PubMed] [Google Scholar]
- von Haehling S, Anker MS, and Anker SD (2016). Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016. J. Cachexia Sarcopenia Muscle 7, 507–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Weber MA, Krakowski-Roosen H, Schröder L, Kinscherf R, Krix M, Kopp-Schneider A, Essig M, Bachert P, Kauczor HU, and Hildebrandt W (2009). Morphology, metabolism, microcirculation, and strength of skeletal muscles in cancer-related cachexia. Acta Oncol. 48, 116–124. [DOI] [PubMed] [Google Scholar]
- White RB, Bidrinx AS, Gnocchi VF, and Zammit PS (2010). Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol 10,21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang Z, Zhang Y, Ni X, Zhang G, Cui X, Liu M, Xu C, Zhang Q, Zhu H, et al. (2019). ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 156, 722–734.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, Fletcher-Sananikone E, Zhang H, Liu Y, Wang W, et al. (2011). PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kB-cytokine network. Cancer Discov. 1, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Liu Z, Ding H, Miao H, Garcia JM, and Li YP (2017). Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci. Rep 7, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RNA-Seq data files are available in the NCBI’s GEO: GSE133523 and GSE133524.