

SUMMARY
Inflammation coordinates tissue regeneration via damaged cell removal and stem cell activation. Hematopoietic stem cells (HSC) survive inflammatory stress that kills other blood cells, but the mechanisms underlying this effect remains poorly understood. Here, we find that tumor necrosis factor α (TNFα) acts differently on HSCs and progenitors, thus facilitating hematopoietic clearance and promoting regeneration. We show that while inducing myeloid progenitor apoptosis, TNFα promotes HSC survival and myeloid differentiation by activating a strong and specific p65/nuclear factor-κB (NF-κB)-dependent gene program that primarily prevents necroptosis rather than apoptosis, induces immunomodulatory functions and poises HSCs for myeloid cell production. These TNFα-driven mechanisms are critical for HSC response to inflammatory stress, but are also hijacked in aged and malignant HSCs. Our results reveal several TNFα-mediated pro-survival mechanisms unique to HSCs, highlight an important role for necroptosis in HSC killing, and establish TNFα as a major pro-survival and pro-regeneration factor for HSCs.
Keywords: Hematopoietic stem cells (HSCs), inflammation, myeloid regeneration, TNFα, necroptosis, NF-κB, stem cell activation, leukemia, cIAP2, PD-L1
eTOC BLURB
Passegué and colleagues reveal a complex role for TNFα in hematopoietic regeneration. Inflammation-induced TNFα eliminates myeloid progenitors, but prevents HSC necroptosis and initiates emergency myelopoiesis through NF-κB-dependent mechanisms, hence promoting HSC survival and hematopoietic regeneration. Persistent TNFα-driven hematopoietic regeneration underlies chronic inflammation and can contribute to aging and malignant hematopoiesis.
Graphical Abstract
INTRODUCTION
Tissue regeneration is achieved through coordinated regulation of tissue destruction via removal of damaged cells, and tissue repair via the release of pro-regenerative signals, often pro-inflammatory cytokines, and production of replacement cells by activated tissue-specific stem cells (Naik et al., 2018). Hematopoietic stem cells (HSC) produce all mature blood cells and regenerate the blood system throughout life (Orkin and Zon, 2008). The vast majority of adult HSCs are kept quiescent in the bone marrow (BM) and survive environmental stresses that kill most progenitor and mature cells (Mohrin et al., 2010; Warr et al., 2013), thereby remaining able to initiate blood regeneration. HSCs respond to a wide range of insults by activating their metabolism and cell cycle machinery, mobilizing into the circulation, and changing their differentiation output to increase the production of specific blood lineages (Baldridge et al., 2010; Essers et al., 2009; Pietras et al., 2016). While all these mechanisms allow for HSC maintenance and rapid recovery of blood homeostasis after injury, how they are triggered and integrated to mediate an effective regenerative response remains elusive.
The tumor necrosis factor-α (TNFα) is one of the most intensively studied pro-inflammatory cytokines with central roles in mammalian immunity and cellular homeostasis (Silke and Hartland, 2013). TNFα is a prototypical death ligand induced upon infection that can elicit two types of programmed cell death, apoptosis and necroptosis (Brenner et al., 2015). The fate of a TNFα-exposed cell depends on the cellular context and, particularly, on the ubiquitylation status of the receptor interacting serine/threonine kinase 1 (RIPK1) enzyme, which is in large part regulated by the inhibitor of apoptosis protein (cIAP) E3 ubiquitin ligases. Upon TNFα binding to its receptors (TNF-R), poly-ubiquitylated RIPK1 acts as a scaffold protein necessary for activating pro-survival signaling, essentially the canonical nuclear factor-κB (NF-κB) pathway. In situations where RIPK1 remains non-ubiquitylated, such as upon cIAP depletion, RIPK1 assembles with initiator caspases (CASP) including CASP-8, whose activation leads to the execution of classical CASP-3/7-mediated apoptosis. In contrast, in cells where caspases are inactivated, RIPK1 phosphorylates RIPK3, which in turn phosphorylates the mixed lineage kinase domain-like (MLKL) protein resulting in the execution of necroptosis (Brenner et al., 2015).
Despite such detailed molecular understanding of TNFα mechanisms of action, its role in HSC regulation remains controversial. Earlier studies report conflicting results for the effect of TNFα on growth and proliferation of human hematopoietic progenitor in vitro, ranging from potent inhibition (Broxmeyer et al., 1986) to actual stimulation (Caux et al., 1990). Similarly, studies using TNF-R knockout mice show opposite effects as to whether TNFα acts in a positive (Pearl-Yafe et al., 2010) or negative (Pronk et al., 2011) manner on the survival of HSC-enriched populations. The impact of TNFα on HSC engraftment potential is also highly debated since both promoting and inhibitory effects are observed depending on the experimental setting (Ishida et al., 2017; Pronk et al., 2011). In addition, TNFα, like other classical inflammatory signals, has emerged as an essential regulator of HSC ontogeny during embryonic development (Espin-Palazon et al., 2018). RIPK1 has also been suggested to be critical for protection of hematopoietic cells from TNFα-driven necroptosis (Roderick et al., 2014), yet the ability of TNFα to induce necroptosis in the most primitive HSCs, and the importance of TNFα-driven necroptosis in regulating HSC viability and function remain largely un-addressed. Here, we set out to clarify the effect of TNFα on adult HSC function, and to understand how TNFα-dependent mechanisms are integrated to coordinate hematopoietic regeneration during inflammation.
RESULTS
Differential TNFα cytotoxicity on HSCs and committed progenitors
To assess the effect of TNFα on hematopoiesis, wild-type (WT) mice were injected with various concentrations of TNFα (0.5 to μ4 g per injection) using a previously published 48h delivery protocol with 3 injections, 12h apart, followed by a 24h wait period (Figure 1A), which at the dose of 2 μg TNFα reduced BM cellularity and compromised HSC engraftment potential (Pronk et al., 2011). In fact, TNFα treatment induced a dose-dependent reduction in BM cellularity, which was already evident at 0.5 μg TNFα (Figure 1B). Flow cytometry analyses (Figure S1A) also indicated a dose-dependent elimination of BM mature B cells and granulocytes (Gr) (Figures 1C and S1B), and their committed progenitors including common lymphoid progenitors (CLP), common myeloid progenitors (CMP), granulocyte/macrophage progenitors (GMP) and megakaryocyte/erythrocyte progenitors (MEP) (Figures 1D and S1C–S1E). In contrast, the changes in the HSC-containing LSK (Lin−/c-Kit+/Sca-1+) compartment, which consists mainly of distinct populations of lineage-biased multipotent progenitors (MPP) (Pietras et al., 2015), were more contrasted. While lymphoid-primed MPP4 also showed a dose-dependent elimination, despite an initial increase at 0.5 μg TNFα, myeloid-primed MPP2/3 displayed a sustained expansion and HSCs maintained rather constant numbers up to the highest dose of 4 μg TNFα (Figures 1E, 1F, S1F and S1G). These results highlight the potent cytotoxic effect on TNFα on nearly all BM hematopoietic populations, except for the most immatures HSCs and MPP2/3.
Figure 1. Differential cytotoxicity of TNFα on HSCs and hematopoietic progenitors.

(A) Experimental design for repeated in vivo TNFα injections in wild type (WT) mice; h, hours.
(B) BM cellularity ± TNFα (n = 4–8 mice/group from 5 independent experiments).
(C–F) Absolute numbers of the indicated BM populations ± TNFα: (C) mature cells, (D) myeloid progenitors, (E) MPPs and (F) HSCs (n = 4–8 mice/group from 5 independent experiments).
(G) Experimental design for in vitro TNFα treatment; d, days.
(H) Expansion of BM HSCs and GMPs after 72h culture ± TNFα (n = 9 pools of 300 cells/group from 3 independent experiments). Results are expressed as fold expansion compared to the number of plated cells/condition.
(I) CASP-3/7 activity in BM HSCs and GMPs after 24h culture ± TNFα (n = 14 pools of 200 cells/group from 4 independent experiments). Results are expressed as fold changes compared to untreated HSCs (set to 1).
(J) Plating efficiency in methylcellulose of BM HSCs and GMPs after 24h culture in cytokine-rich or -poor (media ± TNFα (n = 9 pools of 100 cells/group from 3 independent experiments). Colonies are scored after 7 days.
(K) Autophagy flux in BM GFP-LC3 HSCs after 8h culture in cytokine-rich or -poor media ± TNFα and bafilomycin A (BafA) (n = 3 biological replicates from 3 independent experiments). Results are calculated as percent changes of GFP-LC3 MFI between +BafA vs. −BafA conditions per treatment; −cyto, no cytokines.
Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
See also Figure S1.
To further investigate the differential effect of TNFα on HSCs and their progeny, we isolated HSCs and GMPs from WT mouse BM and performed in vitro cultures with or without (±) TNFα (Figure 1G). When expanded for 72h (3 days) in cytokine-rich media, HSCs were not inhibited by TNFα regardless of its concentration (1 ng to 10 μg/ml), whereas GMPs exhibited a consistent ~50% reduction in cell number with as low as 10 ng/ml TNFα (Figure 1H). Consistently, HSCs did not activate CASP-3/7 upon 24h exposure with 1 μg/ml TNFα, while GMPs strongly did (Figure 1I). Cultured MPP2/3 were also resistant to TNFα cytotoxicity, whilst MPP4, CMPs and MEPs were all susceptible (Figure S1H). Of note, the growth of HSC-containing LSK cells was reportedly suppressed by TNFα when cultured with only SCF and G-CSF (Pronk et al., 2011). However, the expansion rate and colony-forming ability of both HSCs and GMPs was already compromised in these “cytokine-poor” conditions (Figure 1J, S1I and S1J), suggesting a possible confounding effect of other stress-response mechanisms activated by cytokine deprivation. In fact, we directly demonstrated induction of autophagy in cytokine-poor conditions using HSCs isolated from autophagy reporter GFP-LC3 mice (Mizushima et al., 2004; Warr et al., 2013), and showed a strong inhibition of this protective response upon TNFα treatment (Figure 1K). Autophagy inhibition by TNFα in these cytokine-poor culture conditions may therefore sensitize HSCs to cell death. Collectively, these results demonstrate that HSCs are highly resistant to TNFα cytotoxicity, although their survival can be affected by other environmental stresses, whereas GMPs are eliminated by TNFα in a dose-dependent manner by apoptosis. They also show that the resistance to TNFα extends to myeloid-biased MPP2/3, but disappears shortly afterwards, resulting in a broad hematopoietic clearance initiated at the MPP4 level in the LSK compartment.
TNFα drives myeloid regeneration from HSCs
We next investigated the kinetics of TNFα response and associated hematopoietic regeneration using the same delivery protocol (3 injections, 12h apart) and 2 μg TNFα (Figure 2A). We confirmed significantly increased TNFα levels in the BM fluid of 24h injected mice, which rapidly became undetectable at 48h (Figure S2A). Interestingly, BM HSC numbers transiently increased at 24h, then returned to basal levels at 48h and remained stable thereafter. In contrast, BM GMPs and Grs were depleted as early as 3h after the first TNFα injection, with GMP numbers fully recovering by 96h. For Grs, we observed a transient overshoot by 96–144h, indicative of ongoing myeloid regeneration and GMP cluster formation (Herault et al., 2017), before normalizing by 192h. To address the possibility of an egress of GMPs and Grs from the BM in response to TNFα injections, we tracked their numbers in the spleen (SP) and peripheral blood (PB) (Figure 2B). These kinetics analyses confirmed that HSCs expanded mainly in the BM at 24h, with a small number of HSCs migrating and accumulating in the SP by 96h. Interestingly, the rapid depletion of BM GMPs was not accompanied by a complementary increase in the periphery until the appearance of splenic GMPs by 96h, likely as a consequence of in situ differentiation of homed HSCs (Figure 2B). Indeed, BM HSCs did not engage apoptosis at any time points, whereas BM GMPs showed massively increased CASP-3/7 activity at 3h and 24h (Figure 2C), thus confirming their direct elimination by apoptosis. In contrast, we observed a transient increase in PB Grs at 3h that could be explained by the known egress of TNFα-exposed Grs into the circulation (Sadik et al., 2011), although we cannot formally exclude a quick differentiation of some TNFα-exposed GMPs producing Grs that were immediately released in the circulation. However, this increase in PB Grs disappeared by 24h and two additional injections of TNFα, indicating a mostly depleting effect of TNFα on Grs. Collectively, these results demonstrate that TNFα transiently expands HSCs while rapidly eliminating GMPs and Grs, which is followed by hematopoietic regeneration upon return to normal TNFα levels.
Figure 2. TNFα drives HSCs to mount a regenerative response.

(A) Experimental design and absolute numbers of the indicated BM populations ± TNFα (n = 4–14 mice/group from 9 independent experiments).
(B) Absolute numbers of the indicated BM, PB and SP populations ± TNFα (n = 3–6 mice/group from 3 independent experiments; * vs. 0h in BM, ° vs. 0h in PB, # vs. 0h in SP).
(C) CASP-3/7 activity in BM HSCs and GMPs ± TNFα (n = 3 mice/group from 3 independent experiments). Results are expressed as fold changes compared to 0h HSCs (set to 1).
(D) BM HSC cell cycle distribution ± TNFα (n = 3–6 mice/group from 4 independent experiments; * vs. 0h).
(E) BM long-term HSCs (HSCLT) and MPP1 frequency ± TNFα (n = 3–9 mice/group from 8 independent experiments; * vs. 0h).
(F) BM CD41+ HSCs frequency ± TNFα (n = 3–9 mice per group from 8 independent experiments).
(G) PU.1 level in BM PU.1-eYFP HSCs ± TNFα (n = 4–5 mice/group from 2 independent experiments); MFI, mean fluorescent intensity.
(H) In vitro myeloid differentiation of BM HSCs ± TNFα (n = 3 pools of 500–1000 cells/group from 3 independent experiments). Results are shown as percentage of Mac-1+/FcγR+ mature myeloid cells upon 6-day culture.
Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
To understand how TNFα triggers regeneration, we first assessed the cell cycle of HSCs isolated from TNFα-treated mice (Figure 2D). As expected, HSCs quickly exited quiescence and started cycling by 24h, an effect that persisted for at least 96h but was fully restored by 192h. Concomitantly, HSCs transiently shifted to a metabolically activated CD34+ MPP1 fate (Cabezas-Wallscheid et al., 2014) and a CD41+ myeloid-primed state (Yamamoto et al., 2013), which was quickly reverted by 96h (Figures 2E and 2F). Fluidigm qRT-PCR analyses of 3h and 48h HSCs revealed a sustained inhibition of quiescence-enforcing mechanisms and activation of the cell cycle and metabolic machineries (Figure S2B and Table S1). They also confirmed the strong induction of the master myeloid regulator Spi1 (PU.1) in 3h HSCs, which was subsequently attenuated by 48h. Using PU.1-eYFP knock-in mice, we confirmed elevated levels of PU.1 protein in TNFα-treated HSCs, which quickly peaked by 24h before slowly declining (Figure 2G and S2C). We also observed accelerated differentiation into myeloid cells from cultured 24h HSCs (Figures 2H and S2D), which directly demonstrates their myeloid-priming. In vitro, the exposure of HSCs to TNFα also accelerated cell division (Figure S2E), increased PU.1 levels (Figure S2F), and promoted myeloid differentiation (Figure S2G), although the produced myeloid cells were directly killed by apoptosis due to the constant presence of TNFα in the culture media, leading to blunted expansion over time (Figures S2H and S2I). Together, these results demonstrate that TNFα activates a strong regenerative program in HSCs, leading to cell cycle activation and myeloid priming likely through precocious PU.1 activation. However, such TNFα-driven myeloid regeneration is futile when TNFα is still present, demonstrating that a tapering of TNFα release is critical for effective restoration of blood homeostasis.
TNFα-mediated NF-κB activity prevents HSC necroptosis
To understand the mechanisms by which TNFα preserves HSCs while killing GMPs, we focused on the canonical NF-κB pro-survival signaling pathway (Figure S3A). We confirmed that both HSCs and GMPs express the two TNFα receptors, with GMPs having higher levels TNF-R1 than HSCs (Figure S3B). Using NF-κB-eGFP reporter mice, we showed the immediate activation of NF-κB in TNFα-exposed HSCs and GMPs, with stronger NF-κB activity observed in HSCs (Figure 3A). We also found higher expression and more robust nuclear translocation of p65/RelA and p50/Nfkb1 in TNFα-treated HSCs compared to GMPs (Figures 3B). Importantly, treatment with the IKK inhibitor (IKKi) BMS-345541 partially inhibited p65 nuclear translocation (Figure S3C), and rendered HSCs partly, and GMPs completely, susceptible to TNFα cytotoxicity (Figure 3C). However, p50 deficiency in p50−/− mice had no effect on HSC survival, while further promoting GMP death by apoptosis (Figure 3C). In contrast, p65 loss in Mx1-Cre:p65f/f conditional knockout (p65cKO) mice completely abrogated HSC resistance to TNFα cytotoxicity, with p65cKO HSCs and GMPs being both killed by apoptosis (Figures 3C and 3D). Interestingly, the capacity of TNFα to activate p65 was maintained in p50−/− HSCs (Figure S3D), which suggest that the transcriptional program responsible for HSC survival might be activated solely by p65, perhaps through p65 homodimers or other atypical heterodimers without p50. Collectively, these results indicate that HSCs entirely rely on strong p65-dependent NF-κB activity to survive TNFα exposure.
Figure 3. TNFα-mediated p65/NF-κB activation protects HSCs from necroptosis.

(A) NF-κB activity in BM NF-κB-eGFP HSCs and GMPs 3h post-TNFα injection (n = 3 mice/group from 2 independent experiments). Results are shown as eGFP MFI levels in the corresponding gates.
(B) p65 and p50 localization in BM HSCs and GMPs after 3h culture ± TNFα; scale bars, 5 μm.
(C) NF-κB inhibition due to IKK inhibitor (IKKi, 2 μM BMS-345541; left), p50 deficiency (p50−/− mice; middle) or p65 deficiency (p65cKO mice; right) on 72h expansion (n =6–15 pools of 300 cells/group from 3–5 independent experiments, top) and 24h CASP-3/7 activity (n = 9–15 pools of 200 cells/group from 3–4 independent experiments, bottom) in BM HSCs and GMPs cultured ± TNFα. Results are expressed as fold expansion compared to the number of plated cells/condition (top), or fold changes compared to untreated WT or Ctrl HSCs (set to 1, bottom).
(D) Absolute numbers of BM HSCs and GMPs in Ctrl and p65cKO mice ± TNFα (n = 3–5 mice/group from 5 independent experiments; * vs. Ctrl).
(E) Expansion of the indicated IKKi-treated BM HSCs after 72h culture ± TNFα (n = 9–63 pools of 300–500 cells/group from 21 independent experiments). Results are expressed as fold expansion compared to the number of plated cells/condition.
(F) Expansion of the indicated BM HSCs after 72h culture ± TNFα and pan-caspase inhibitor (CASPi, 20 μM zVAD-fmk) (n = 8–9 pools of 300 cells/group from 3 independent experiments). Results are expressed as fold expansion compared to the number of plated cells/condition.
Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
See also Figure S3.
To further characterize the cell death pathways that are blocked by the strong NF-κB induction in TNFα-treated HSCs, we next isolated HSCs from various apoptosis (i.e., Mx1-Cre:Casp8f/f, Bid−/−, and Mx1-Cre:Bak−/−:Baxf/f) and necroptosis (i.e., Ripk1 kinase dead (Ripk1KD), Ripk3−/−, and Mlkl−/−) deficient mice (Figure S3A), and submitted them to 72h in vitro expansion assay ± TNFα and IKKi to partially block TNFα-induced NF-κB activity. Surprisingly, while apoptosis inhibition did not rescue TNFα cytotoxicity caused by partial NF-κB blockade, necroptosis inhibition fully restored HSC survival (Figure 3E). The importance of necroptosis in TNFα-mediated HSC elimination was reinforced in p65cKO HSCs with complete NF-κB blockade, since only concurrent inhibition of both apoptosis via treatment with the pan-caspase inhibitor (CASPi) zVAD-fmk, and necroptosis via Ripk3 deficiency in Mx1-Cre:p65f/f:Ripk3−/− mice could restore HSC survival (Figures 3F). These results were recapitulated by treating p65cKO HSCs with both zVAD-fmk and the RIPK1 kinase inhibitor (RIPK1i) GSK’963 (Berger et al., 2015) (Figure S3E). In contrast, the cytotoxic effects of TNFα on GMPs could not be rescued by either apoptosis or necroptosis inhibition, or a combination of both (Figures S3E–S3G), suggesting the involvement of an additional form of programmed cell death when apoptosis is inhibited. Collectively, these results demonstrate that TNFα-induced p65/NF-κB activity protects HSCs primarily from necroptosis, which is the first form of cell death unleashed upon decreased NF-κB activity (Figure S3H).
TNFα-mediated pro-survival gene signature in HSCs
To gain a broader understanding of the mechanisms dictating differential survival outcomes following TNFα exposure, we performed RNA sequencing (RNA-seq) analyses on both HSCs and GMPs exposed to TNFα in culture for 3h and 12h, or isolated from TNFα-treated mice at 3h (Figure 4A). We selected these conditions to extract common gene expression signatures, which are independent of the treatment conditions and reflect an early response when p65/NF-κB is differentially activated between the two population and necroptosis or apoptosis not yet engaged. Principal component analysis (PCA) confirmed that TNFα-treated cells are transcriptionally distinct from untreated cells, with a greater number of significantly affected genes in HSCs compared to GMPs (Figures 4B, 4C and Table S2). We also observed a larger effect upon in vivo treatment compared to in vitro exposure, likely reflecting the contribution of indirect environmental effects not observed upon direct TNFα stimulation By using only genes that were upregulated in all 3 TNFα treatment conditions, which had a false discovery ratio (FDR) < 0.1 and fold changes (FC) > 3, we extracted a stringent TNFα signature of 62 genes in HSCs and 51 genes in GMPs (Figures 4C, 4D and Tables S3–S5). Eighteen of those genes were commonly upregulated in both cell types and represented core regulators of the NF-κB pathway (Zhang et al., 2017) (Table S6). Strikingly, the remaining 33 GMP-specific genes highlighted a clear pro-death signature, with enrichment of gene ontology (GO) terms including apoptosis and inflammation, and upregulation of genes such as Tnf (TNFα) itself, Lta (Lymphotoxin-α) and Il6 (IL-6). In contrast, the remaining 44 HSC-specific genes defined a strong pro-survival signature, characterized by anti-inflammatory GO terms and anti-apoptotic genes including Birc3 (cIAP2), which directly blocks the cytotoxic effect of TNFα by ubiquitylating RIPK1 (Brenner et al., 2015; Wallach et al., 2016), as well as Cd274 (PD-L1) and Pdcd1lg2 (PD-L2), which are ligands for the PD-1 receptor that induce immune tolerance (Okazaki et al., 2013). Taken together, these molecular data provide two novel TNFα gene signatures that reflect the actual pro-survival and pro-death effect of TNFα on HSCs and GMPs, respectively.
Figure 4. Identification of HSC-specific pro-survival TNFα signature genes.

(A-D) Identification of TNFα signature genes in BM HSCs and GMPs by RNA-seq (n = 3 biological replicates/group): (A) experimental design with both in vitro and in vivo ± TNFα treatment conditions, (B) 3D principal component analysis (PCA) plots, (C) Venn diagrams showing the number of significantly upregulated genes/condition, and (D) composite heatmap showing the log2 fold changes and Venn diagrams the number of TNFα signature genes with representative examples; FC, fold change; FDR, false discovery ratio.
(E) Quantitative RT-PCR analyses for IAP family genes in BM Ctrl and p65cKO HSCs cultured for 12h ± TNFα (n = 3 biological replicates/group from 3 independent experiments). Results are expressed as log2 fold change compared to −TNFα Ctrl HSCs.
(F) p65 localization in BM HSCs cultured for 3h ± TNFα and cIAP inhibitor (cIAPi, 5 μM LCL-161); scale bar, 5 μm.
(G) Expansion of BM HSCs after 72h culture ± TNFα and cIAPi (5 μM LCL-161 or 4.5 μM AEG40730) (n = 6–9 pools of 300 cells/group from 3 independent experiments). Results are expressed as fold expansion compared to the number of plated cells/condition.
(H) Quantitative RT-PCR analyses for PD-1 ligands in Ctrl and p65cKO HSCs cultured for 12h ± TNFα (n = 3 biological replicates/group from 3 independent experiments). Results are expressed as log2 fold change compared to −TNFα Ctrl HSCs.
(I) PD-L1/PD-L2 levels on HSCs cultured for 24h ± TNFα (n = 4 biological replicates from 4 independent experiments). Results are expressed as log2 fold changes in MFI in +TNFα vs. −TNFα conditions (* vs. −TNFα).
(J) T cell suppression assay showing the division rates of CFSE-labelled BM CD8+ T cells co-cultured for 72h ± BM HSCs, TNFα and PD-1 blocking antibody (αPD-1, 20 μg/ml). Results are representative of 2 independent experiments.
Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we investigated the role of the uncovered signature genes in mediating HSC survival upon TNFα exposure. We first confirmed that Birc3 was the only cIAP family members induced in a p65-dependent manner in TNFα-treated HSCs (Figure 4E). We then used two synthetic second mitochondrial-derived activator of caspases (SMAC) mimetics, the dimeric AEG40730 and monomeric LCL-161 compounds (Fulda and Vucic, 2012), to rapidly trigger cIAP degradation in TNFα-treated HSCs. Remarkably, despite preserved p65 nuclear translocation (Figure 4F), cIAP inhibition (cIAPi) rendered HSCs highly sensitive to TNFα cytotoxicity (Figures 4G). We also confirmed PD-L1 and PD-L2 up-regulation in TNFα-treated HSCs both at the transcriptional level, mainly via p65 activation, and surface protein expression (Figures 4H and 4I). Moreover, we showed in co-culture experiments that TNFα-treated HSCs could block the increased proliferation of BM CD8+ T cells induced by TNFα exposure, which was reverted by treatment with an anti-PD-1 blocking antibody (Figure 4J). Together, these results demonstrate the functional role of the TNFα signature genes in protecting HSCs from necroptosis via cIAP2 induction, and in providing immunomodulatory capacity to suppress T cell proliferation through upregulation of PD-L1 and PD-L2.
Rapid NF-κB deactivation renders active HSCs susceptible to necroptosis
To directly assess the effect of TNFα on HSC regenerative function in vivo, we transplanted HSCs isolated from TNFα-treated WT mice at various time post-TNFα injections (Figure 5A). As expected, the engraftment potential of TNFα-treated HSCs matched their activation state, with 3h HSCs, which had just started entering the cell cycle, having minimally decreased engraftment compared to PBS-treated (0h) HSCs, 48h HSCs, which had exited quiescence, having severely compromised reconstitution ability, and 192h HSCs, which had restored quiescence, having normal engraftment and BM reconstitution potential (Figures 5B and 5C). However, no major changes in lineage distributions was observed from TNFα-treated HSCs at any time points (Figures S4A and S4B). Remarkably, 48h HSCs showed no nuclear p65 signal in contrast to 3h and 24h HSCs (Figures 5D and 5E), which reflected a deactivation of the NF-κB pathway that was also observed at the molecular level with rapid downregulation of canonical NF-κB target genes and the pro-survival gene Birc3 in 48h HSCs (Figures S4C and S4D). This transient NF-κB signaling response is likely a direct consequence of TNFα levels having returned to baseline by 48h (Figure S2A). Interestingly, 48h HSCs also showed impaired growth in 72h expansion assay, which could be recovered upon in vitro TNFα supplementation and re-activation of the NF-κB pathway (Figures S4E–S4G). Moreover, injection of TNFα to lethally-irradiated WT recipients (3 injections, 12h apart, starting 12h post-transplantation) significantly improved the engraftment ability of 48h HSCs, while having no effects on the reconstitution capability of 0h HSCs (Figure S4H and S4I). Collectively, these results demonstrate a transient impairment of the engraftment potential of TNFα-treated HSCs, which correlates with both cell cycle activation and status of the NF-κB pathway. They indicate that fully activated HSCs with deactivated NF-κB response have the lowest regeneration potential, which stress the importance of the strength and duration of TNFα signaling in dictating HSC engraftment capability. They also show no sustained myeloid skewing for the output of TNFα-exposed HSCs, presumably because of the transient nature of the myeloid priming that reverts within a few days upon loss of TNFα signaling.
Figure 5. Necroptosis-mediated elimination of NF-κB-deactivated HSCs.

(A–C) Engraftment potential of BM HSCs ± TNFα (n=5–16 mice/group from 3 independent experiments): (A) experimental design, (B) donor-derived chimerism in PB and (C) absolute numbers of donor-derived BM HSCs at 4 months post-transplantation; mo, months.
(D-E) NF-κB-deactivation following TNFα treatment: (D) representative images and (E) quantification of nuclear p65 in BM HSCs ± TNFα (n = 3–9 biological replicates/group from 8 independent experiments); scale bar, 5 μm.
(F) Experimental design and changes in absolute numbers of the indicated BM populations ± TNFα (n = 4–11 mice/group from 5 independent experiments; * vs. WT).
(G–J) Necroptosis inhibition due to Ripk3 deficiency (Ripk3−/− mice, top) and Mlkl deficiency (Mlkl−/− mice, bottom) on the functionality of 48h TNFα-exposed BM HSCs: (G) experimental design, (H) expansion after 72h culture (n = 7–9 pools of 300 cells/group from 3 independent experiments; results are expressed as fold expansion compared to the number of plated cells/condition), and (I) donor chimerism in PB and (J) absolute number of donor-derived BM HSCs at 4 months post-transplantation (n = 6–25 mice/group from 4–5 independent experiments).
Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
See also Figures S4 and S5.
To further dissect the involvement of necroptosis, we performed kinetics analyses of TNFα-treated necroptosis-deficient mice (Figures 5F and S5A–S5C). Interestingly, while HSC numbers consistently increased at 24h and then contracted at 48h in WT mice, they were either maintained or further increased at 48h in Ripk3−/− and Mlkl−/− mice. In contrast, GMP and Gr numbers were similarly depleted in Ripk3−/− and Mlkl−/− mice at 24h, but were regenerated faster than in WT mice at 48h. HSCs from necroptosis-deficient mice also showed similar cell cycle activation compared to WT HSCs upon TNFα exposure (Figures S5D and S5E). These results indicate that HSCs undergo necroptosis during the 24–48h time window post-TNFα injection, when NF-κB is deactivated, and that inhibition of necroptosis in Ripk3−/− and Mlkl−/− mice results in increased numbers of activated HSCs that participate to a faster regeneration of myeloid lineage cells and recovery of BM cellularity (Figure S5F). Moreover, the defective expansion potential of 48h HSCs was fully rescued by inhibition of necroptosis in 48h Ripk3−/− and Mlkl−/− HSCs (Figures 5G and 5H). The impaired engraftment potential of 48h HSCs was also significantly improved by necroptosis inhibition (Figures 5I and 5J), although this rescue was only partial likely because of the persisting cell cycle activation of 48h Ripk3−/− and Mlkl−/− HSCs at the time of transplantation (Figures S5D and S5E). Taken together, these results demonstrate that HSCs become susceptible to necroptosis-mediated cell killing during the particular phase following TNFα exposure when they are actively proliferating but devoid of p65/NF-κB-mediated pro-survival signaling. Moreover, they identify two instances when necroptosis plays an active role in controlling the size of the HSC compartment in vivo.
TNFα protects HSCs from necroptosis and promotes myeloid priming during inflammation
To address the importance of TNFα during inflammation, we used a well-established model of chronic inflammation with repeated injections of polyinosinic:polycytidylic acid (pIC) mimicking an anti-viral immune response (Essers et al., 2009; Pietras et al., 2014) (Figure 6A). Multiplex cytokine measurement confirmed TNFα as one of the most increased cytokines in the BM fluid of WT mice treated with 10 mg/kg pIC for 13 days (7 injections, every other day, followed by a 24h wait period) (Figures 6B and S6A). We previously reported that ESAM-selected HSCs (HSCESAM) were free of contaminating progenitors and essentially preserved during pIC-induced inflammation, while other progenitors including GMPs were depleted (Figures 6C and S6B) (Pietras et al., 2014). In contrast, pIC-induced inflammation strongly depleted HSCESAM in Tnf−/− mice, while the reduction in GMPs remained very similar between pIC-treated WT and Tnf−/− mice (Figure S6C). The lack of TNFα signaling did not affect the cell cycle activation of pIC-treated HSCESAM (Figure S6D), but it partially attenuated their myeloid priming as demonstrated by the delayed myeloid differentiation from pIC-treated Tnf−/− HSCESAM in vitro (Figure 6D and S6E). Furthermore, we observed severely compromised NF-κB activation in response to pIC treatment in the absence of TNFα, as shown by the lack of reporter activity in NF-κB-eGFP:Tnf−/−HSCESAM (Figure 6E) and p65 nuclear localization in isolated Tnf−/− HSCESAM (Figures 6F and 6G). In fact, TNFα appeared as the major source of NF-κB activation during inflammation, as removal of IL-1 signaling, another known NF-κB inducer secreted in response to pIC treatment (Figure S6A), did not compromise p65 nuclear translocation in pIC-treated Il1r1−/− HSCESAM (Figures 6F and 6G). Furthermore, blockade of necroptosis in Tnf−/−:Ripk3−/− or Tnf−/−:Mlkl−/− mice completely rescued the loss of HSCESAM induced by pIC treatment in the absence of TNFα (Figures 6H and 6I), demonstrating that HSCs are directly eliminated by necroptosis during inflammation when TNFα-driven NF-κB signaling is abrogated. The lack of TNFα signaling also attenuated the elimination by apoptosis of pIC-treated GMPs (Figure S6F), but the overall reduction of HSC number and their impaired myeloid priming likely masked this rescue effect on GMP survival due to a globally impaired regenerative response in pIC-treated Tnf−/− mice. Finally, we confirmed the importance of TNFα for HSC maintenance in other inflammatory contexts, including 48h LPS challenge that elicits an anti-bacterial immune response (Figures S7A) and repeated 5-fluorouracil (5-FU) injections that trigger damage-associated BM ablation (Figure S7B). In both cases, TNFα deficiency in Tnf−/− mice resulted in a significant loss of HSC numbers, although it still remains to be determined whether TNFα itself or other ligands induced by TNFα act directly on HSCs to prevent their depletion. Taken together, these results highlight the importance of TNFα in maintaining NF-κB activity during inflammation, which protects HSCs from necroptosis-mediated elimination and drives myeloid regeneration.
Figure 6. TNFα-driven NF-κB activity protects HSCs from necroptosis during inflammation.

(A) Experimental design for repeated pIC injections.
(B) Luminex measurement of TNFα concentration in BM fluid of WT mice ± pIC (n = 3 biological replicates/group from 3 mice in 1 experiment)
(C) Absolute number of BM HSCESAM in WT and Tnf−/− mice ± pIC (n = 7–8 mice/group from 5 independent experiments).
(D) In vitro myeloid differentiation of BM HSCESAM from WT and Tnf−/− mice ± pIC (n = 3 pools of 500–1000 cells/group from 3 independent experiments). Results are shown as percentage of Mac-1+/FcγR+ mature myeloid cells upon 6-day culture.
(E) NF-κB activity in BM NF-κB-eGFP HSCESAM isolated from the indicated mice ± pIC (n = 3–5 mice/group from 5 independent experiments). Results are shown as eGFP MFI levels in the corresponding gates.
(F-G) TNFα-driven NF-κB activity: (F) representative images and (G) quantification of nuclear p65 in BM HSCESAM in the indicated mice ± pIC (n = 3–4 biological replicates/group from 4 independent experiments); scale bar, 5 μm.
(H) Absolute number of BM HSCESAM in the indicated mice ± pIC (n = 5–7 mice/group from 4 independent experiments).(I) Absolute number of BM HSCESAM in the indicated mice ± pIC (n = 7–15 mice/group from 8 independent experiments).
Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
See also Figures S6 and S7.
Hijacking of TNFα-mediated mechanisms in dysregulated hematopoiesis
TNFα levels are constitutively elevated during aging and in hematological malignancies such as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) (Kovtonyuk et al., 2016). To understand how TNFα could contribute to HSC dysregulation in these conditions, we used our pro-survival HSC-specific TNFα signature genes (HTSG) to perform gene set enrichment analyses (GSEA) on both mouse and human RNA-seq datasets. Analyses of young (8–12 week old) and aged (22 month old) mouse HSCs revealed a specific enrichment for HTSG in old HSCs (Figure 7A), consistent with the increased NF-κB activity reported in aged HSCs (Chambers et al., 2007). Moreover, analyses of human AML (Gentles et al., 2010) and MDS (Woll et al., 2014) patient samples also showed a specific enrichment for HSTG in AML leukemic stem cells (LSC) and MDS HSCs (M-HSC), which was not observed in normal HSCs or with the GMP-specific TNFα signature genes (GTSG) (Figures 7A and 7B). This is in line with the constitutive NF-κB activation reported in human AML patients (Guzman et al., 2001), and confirmed here in HSC-derived LSCs from a BCR/ABL-driven leukemia mouse model (Figure S7C). In contrast, we found a significant enrichment for GTSG in AML blasts (Figure 7B), illustrating the fact that a high proportion of cells respond to TNFα in AML patients (Stirewalt et al., 2008). Taken together, these results demonstrate that HTSG successfully detects TNFα response in aged and malignant stem cells.
Figure 7. HSC-specific TNFα signature genes in normal and abnormal hematopoiesis.

(A) GSEA for HSC-specific TNFα signature genes (HTSG) in the indicated mouse aging and human MDS and AML patient RNA-Seq datasets; oHSC, old HSCs; yHSC, young HSCs; M-HSC, MDS stem cells; M-GMP, MDS progenitors; LSC, leukemic stem cells; LPC, leukemic progenitor cells; NES, normalized enrichment score.
(B) GSEA for GMP-specific TNFα signature genes (GTSG) in the indicated human MDS and AML patient RNA-Seq datasets.
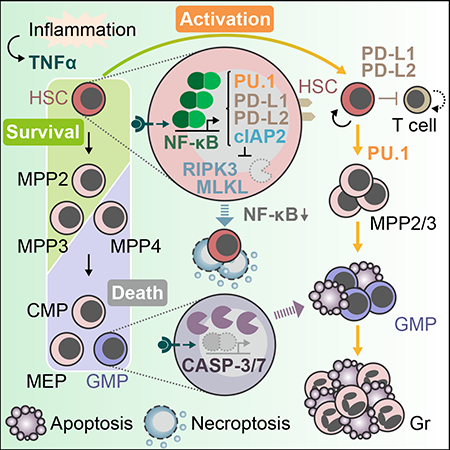
(C) Model for the role of TNFα in HSC and progenitor regulation.
See also Figure S7.
DISCUSSION
Here, we provide a novel integrated picture of the role of TNFα as a key pro-regenerative inflammatory cytokine in the local BM niche microenvironment (Figure 7C). We show that regardless of its concentration, TNFα primarily provides pro-survival cues to HSCs, while inducing apoptosis in a dose-dependent manner in committed progenitors and mature cells. These opposing effects on survival outcome are, in large part, attributable to differences in the strength of canonical NF-κB activation, with a dominant role for p65-mediated regulations in HSCs. In fact, we demonstrate that a strong TNFα-driven p65/NF-κB signaling leading to robust cIAP2 expression is essential for protecting HSCs from engaging cell death mechanisms, especially necroptosis. TNFα also rapidly induces emergency myelopoiesis leading to HSC activation and PU.1-mediated myeloid priming, which together facilitate the rapid recovery of myeloid lineage cells and restoration of blood homeostasis upon normalization of TNFα levels. However, during this period of reduced TNFα signaling HSCs become susceptible to necroptosis-mediated killing due to p65/NF-κB-cIAP2 pathway deactivation, which together with their cycling status contribute to a contraction of the HSC pool and a significant, but transient, loss of their engraftment potential. In addition, TNFα endows HSCs with the capability of suppressing T cell response via the PD-L1/PD-L2:PD-1 pathway, which may protect HSCs from excessive immune activation as demonstrated in other contexts (Zheng et al., 2011). However, it remains to be established whether and how PD-L1/PD-L2 upregulation protects HSCs from T-cell mediated cytotoxicity following in vivo activation by TNFa. Together, our results decipher the molecular mechanisms by which TNFα regulates survival, cellular activation, myeloid commitment, and immune tolerance in HSCs, while at the same time promoting cell death in most of their downstream progeny.
Our findings establish several guiding principles that help explain much of the controversies surrounding the effect of TNFα on HSC survival and function. First, the effect of TNFα is highly cell type dependent, with only HSCs and MPP2/3 molecularly geared to survive TNFα exposure. One issue with many previous studies, including the work we used as reference for our treatment conditions, is the utilization of poorly purified populations for in vitro expansion assay, usually LSK cells, which contain a large fraction of MPP4 that are highly susceptible to TNFα cytotoxicity. Hence, the reported pro-death effect of TNFα on HSC-containing LSK cells (Pronk et al., 2011) likely reflects the elimination of these susceptible and abundant cells. A similar explanation applies to methylcellulose cultures containing TNFα (Ishida et al., 2017), since the produced myeloid cells are likely killed by TNFα as we showed here for TNFα-containing liquid cultures. Second, the effect of TNFα is influenced by the cytokine milieu. In particular, HSCs can become susceptible to TNFα cytotoxicity when cultured in limited cytokine conditions (Pronk et al., 2011), which we reveal to be the indirect consequence of TNFα-mediated blockade of a protective autophagy response induced by cytokine deprivation (Warr et al., 2013). This demonstrate that careful attention must be paid to the cytokine milieu when testing for TNFα responses in vitro, and that rich cytokine conditions are better at mimicking in vivo conditions including inflammation. Third, the effect of TNFα is very transient, with the pro-survival p65/NF-κB-cIAP2 pathway being quickly deactivated in the absence of persistent TNFα signaling. When this happens while HSCs are cycling, it creates a window of susceptibility to necroptosis killing that only disappears when HSCs re-enter quiescence. We identify three different instances where necroptosis-mediated killing of HSC occurs in vivo: 1) following acute TNFα induction to cull the number of activated HSCs, 2) upon transplantation of TNFα-exposed HSCs to limit their engraftment potential, and 3) during chronic inflammation without TNFα signaling to deplete the HSC pool. However, it is likely that elimination of HSCs via necroptosis is an under-appreciated feature of many other conditions including bone marrow failure syndrome as previously suggested in RIPK1-deficient inflammatory context (Rickard et al., 2014; Roderick et al., 2014). This susceptibility to necroptosis coupled with differences in cell cycle status is also likely at the heart of the diversity of engraftment behavior reported in the literature for TNFα-exposed HSCs.
Our results highlight the importance of cIAP2 in preventing the engagement of cell death mechanisms in TNFα-exposed HSCs. The half-life of cIAP2 can be as short as 3h (Lee et al., 2015), and we show quick decreases of Birc3 expression upon NF-κB deactivation in HSCs. Rapid cIAP depletion has been reported to trigger the formation of a pro-death complex, which can initiate necroptosis when CASP-8 activity is inhibited (Brenner et al., 2015). Since only necroptosis, and not apoptosis, is induced upon NF-κB deactivation, it suggests the involvement of some endogenous caspase inhibitors that could still be activated by low level p65 signaling. One such possibility is a short isoform of the cellular FLICE-like inhibitory protein (cFLIP), which is a known p65 target directly inhibiting CASP-8 activity (Brenner et al., 2015), and is induced in TNFα-treated HSCs according to our RNA-seq analyses. This would be consistent with the engagement of apoptotic killing solely when NF-κB signaling is completely abrogated in p65-deficient TNFα-treated HSCs. The observation that HSCs can also be killed by necroptosis in pIC-treated Tnf−/− mice indicates that necroptosis can be triggered independently of TNFα, likely via other exogenous factors such as interferons (IFN), Toll-like receptor ligands or other death ligands. Given that IFNα is induced in the serum of pIC-treated mice (Pietras et al., 2014) and is one of the known necroptosis inducers (Wallach et al., 2016), it suggests that TNFα could protect HSCs from IFNα-induced necroptosis during inflammation.
Our results demonstrate that TNFα coordinates hematopoietic regeneration and stimulates emergency myelopoiesis from HSCs via precocious activation of PU.1, as recently confirmed in an independently study (Etzrodt et al., 2018) and already observed for IL-1 (Pietras et al., 2016). However, in contrast to the long-lasting effect of IL-1, which has a prominent reprograming effect recently confirmed at the epigenetic level and termed trained immunity (Naik et al., 2018), TNFα only shows a transient ability to poise HSCs for myeloid differentiation, which rapidly disappears upon interruption of TNFα signaling. Nevertheless, TNFα signaling, and not IL-1 signaling, appears the key driver of p65/NF-κB activation and enhanced myeloid differentiation during pIC-induced inflammation. In addition, TNFα-mediated PU.1 upregulation could contribute to the re-establishment of HSC quiescence by inhibiting cell cycle activators (Staber et al., 2013), thus helping protect HSCs from necroptosis and terminating the regenerative response. The observation that TNFα eliminates myeloid progenitors by apoptosis may also appear counterintuitive, as they are an important source of mature myeloid cells that act as a first line of defense (Sadik et al., 2011). In fact, while TNFα always stimulates myelopoiesis, it only kills GMPs in a dose-dependent manner. Moreover, while HSCs are known to be highly resistant to pathogen infection (Baldridge et al., 2010), virally infected hematopoietic progenitors including GMPs can serve as a reservoir for viral latency in the BM and a mediator of viral dissemination to other organs (Kondo et al., 1994). Since TNFα signaling is designed to detect cells that are infected (Silke and Hartland, 2013), and TNFα levels closely correlate with viral activity (Docke et al., 1994), it is conceivable that such differential and dose-dependent TNFα killing mechanism has been developed to eliminate infected progenitors, while preserving HSCs to provide new replacement progenitors, hence preventing the establishment of latent infection. Our results are also compatible with the fact that some specialized immune cells such as macrophage or T cells, which are required for pathogen or damaged cell clearance, are activated and not killed by TNFα. Therefore, the mechanism we describe here may act as a fail-safe mechanism to determine the level of cell killing according to the severity of the ongoing infection. It will now be interesting to test this possibility using an established infection model. In addition, to strictly dissect the direct vs. indirect effect of TNFα on HSCs during inflammation, one would need to develop a sophisticated system where only HSCs, but not the other stromal and hematopoietic cell types, are unable to respond to TNFα.
Finally, while TNFα and NF-κB gene signatures are the most ubiquitous inflammatory signatures in gene expression analysis software, they are also the vaguest in terms of functional significance as they do not pinpoint to a particular cellular outcome. In this regard, our work identifies two novel TNFα gene signatures representative of a TNFα-mediated pro-survival response in HSCs and pro-death effect in GMPs, which could potentially prove critical for deciphering the pathogenesis of human leukemia and identifying new treatment strategies. Although the precise role of elevated TNFα in aged aged organisms or patients with hematological disorder remains to be investigated, it is intriguing to speculate that by keeping MDS-HSCs alive, TNFα might favor the progression of MDS to AML, and conversely that blockade of TNFα-mediated pro-survival effects through additional mutations might contribute to the development of BM failure syndromes (Ogawa, 2016). Moreover, our results raise the possibility of un-anticipated broad hematopoietic toxicity associated with the clinical use of TNFα inhibitors for treating inflammatory diseases, as already suggested by anecdotal case reports showing the development of pancytopenia in patients receiving anti-TNFα therapy for rheumatoid arthritis (Day, 2002). Understanding how dysregulation of these TNFα-dependent mechanisms contribute to the development of hematological disorders would help further develop therapeutic interventions targeting dysregulated HSCs while sparing normal HSCs.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Emmanuelle Passegué, ep2828@cumc.columbia.edu. Mouse TNFα was obtained from Genentech under an MTA. GSK’963 was obtained from GlaxoSmithKline under an MTA.
Experimental Model and Subject Details
Mice
CD45.2 C57BL/6J, CD45.1 C57BL/6-BoyJ (B6.SJL-Ptprca Pepcb/BoyJ), p50−/− (B6.Cg-Nfkb1tm1Bal/J) and Il1r1−/− (B6.129S7-Il1r1tm1Imx/J) mice were purchased from the Jackson Laboratory and bred in our animal facility. Mlkl−/− mice (Murphy et al., 2013) were obtained from Dr. A. Warren, Walter and Eliza Hall Institute of Medical Research, Australia. Ripk1D138N/D138N (Ripk1KD) mice (Newton et al., 2014) were obtained from Dr. K. Newton, Genentech. Tnf−/− (Pasparakis et al., 1996) and Ripk3−/− mice (He et al., 2009) were obtained from Dr. A. Ma, UCSF. Casp8f/f mice (Beisner et al., 2005) were obtained from Dr. S. M. Hedrick, UCSD. Bid−/− mice (Yin et al., 1999) were obtained from Dr. S. Zinkel, Vanderbilt University. Bak−/−Baxf/f, GFP-LC3, PU.1-eYFP and Scl-tTA:TRE-BCR/ABL mice were described previously (Warr et al., 2013). p65f/f mice (Algul et al., 2007) were obtained from Dr. R. Schmid, Technical University of Munich, Germany. NF-κB-eGFP mice (Magness et al., 2004) were obtained from Dr. C. Jobin, University of North Carolina, and maintained as hemizygotes. Six- to 12-week-old mice were used as donor for cell isolation and in vivo experiments, and eight- to 12-week-old congenic mice were used as recipients for HSC transplantation experiments. Respective littermates or age-matched WT animals were used as controls. No specific randomization or blinding protocol was used, and both male and female animals were used indifferently in the study. For Mx1-Cre-mediated deletion, four- to 6-week-old Mx1-Cre-positive or -negative mice were injected intraperitoneally once (p65 flox allele) or three times every other day (Casp8 and Bax flox alleles) with 250 μg of poly I:C (pIC, GE Healthcare, 27473201) in 200 μl PBS. pIC injection was completed at least 4 weeks prior to each experiment, and pIC-injected Mx1-Cre-negative mice were used as controls. All mice were maintained in mouse facilities at UCSF or Columbia University in accordance with IACUC approved protocols approved at each institution.
Method Details
In vivo assays
For in vivo TNFα treatment, mice were injected retro-orbitally with 0.5 to 4 μg mouse TNFα (Genentech) in 100 μl PBS or 100 μl PBS alone (untreated animals) up to 3 times every 12 hours. For pIC-induced inflammation experiments, mice were injected intraperitoneally with 10 mg/kg pIC in PBS or PBS alone every other day for 13 days (7 times). For LPS-induced inflammation experiments, mice were injected intraperitoneally with 2 mg/kg LPS (Sigma-Aldrich, L2880) in PBS (1 time). For repeated 5-fluorouracil (5-FU) treatment, mice were injected intraperitoneally with 150 mg/kg 5-FU (Sigma-Aldrich, F6627) in PBS every month for 3 months (3 times). For transplantation experiments, recipient mice were lethally irradiated (11 Gy, delivered in split doses 3 hours apart) using a 137Cs source (J.L. Shepherd), and injected retro-orbitally with 500 donor HSCs together with 3×105 Sca-1-depleted BM cells within the next 36 hours following irradiation. For in vivo TNFα supplementation after transplantation, recipient mice were injected retro-orbitally with 2 μg mouse TNFα in 100 μl PBS or 100 μl PBS alone for 3 times 12 hours apart starting 12 hours after transplantation. Transplanted mice were maintained on antibiotic-containing water for 4 weeks, and analyzed for donor hematopoietic chimerism by monthly bleeding. PB was obtained from retro-orbital plexus, and collected in tubes containing 4 ml of ACK (150 mM NH4Cl and 10 mM KHCO3) and 10 mM EDTA for flow cytometry analyses, or in EDTA-coated tubes (Becton Dickinson) for complete blood counts using a Genesis (Oxford Science) hematology system.
Flow Cytometry
Harvest and staining of hematopoietic stem and progenitor cells were performed as described previously (Pietras et al., 2016) using Hank’s buffered saline solution (HBSS) containing 2% heat-inactivated FBS (Sigma-Aldrich). Briefly, single-cell suspensions of BM cells were obtained by either crushing leg, arm and pelvic bones together with sternum or by flushing only leg bones, and splenocytes by mechanical dissociation between two slides of whole spleens. The entire circulating PB cell compartment was collected by transcardiac perfusion with 20 ml of 10 mM EDTA/PBS. Erythrocytes were lysed by ACK lysis buffer containing 0.02% NaN3, and contaminating bone fragments were further removed by centrifugation on a Ficoll gradient (Histopaque 1119, Sigma Aldrich). BM cellularity was then determined using a ViCELL-XR automated cell counter (Beckman-Coulter), and the absolute cell number for each population was calculated by multiplying the frequency of the population by the BM cellularity. For cell sorting, BM cells were enriched for c-Kit+ cells using c-Kit microbeads (Miltenyi Biotec) or T cells using the mouse Pan T Cell Isolation Kit II (Miltenyi Biotec). Bead-labelled cells were separated with either an automated AutoMACS, or manual AutoMACS Pro or LS Columns according to cell number and the manufacturer’s instruction (Miltenyi Biotec). For immature cell analyses and sorting, unfractionated and c-Kit-enriched BM cells were incubated with a cocktail of unconjugated rat anti-lineage (Lin) antibodies (CD3, CD5, CD8, B220 from BioLegend, and CD4, Ter-119, Gr-1 and Mac-1 from eBioscience) followed by goat anti-rat-PE-Cy5 (Invitrogen, A10691), and subsequent blocking with purified rat IgG (Sigma-Aldrich). Cells were then stained with Sca-1-PB (BioLegend, 108120), c-Kit-APC-Cy7 (BioLegend, 105826), CD48-A647 (BioLegend, 103416), CD150-PE (BioLegend, 115904), and Flk2-Bio (eBioscience, 13-1351-85) followed by SA-PE-Cy7 (eBioscience, 25-4317-82), -Qdot605 (Invitrogen, Q10101MP) or –BV605 (BioLegend, 405229) for HSCs/MPPs, together with CD34-FITC (eBioscience, 11-0341-85) and FcγR-PerCP-eFluor710 (eBioscience, 46-0161-82) or FcγR-PE-Cy7 (BioLegend, 101317) for myeloid progenitors. For identification of HSCLT, MPP1, CD41+ HSCs and CLPs, extended antibody panel including Lin/PE-Cy5, Sca-1-BV421 (BioLegend, 108128), c-Kit-APC-Cy7, Flk2-Bio/SA-BV605, CD48-A700 (BioLegend, 103426), CD150-BV650 (BioLegend, 115931), CD34-FITC, CD41-BV510 (BioLegend, 133923) and IL-7Rα-PE (eBioscience, 12-1271-82) was used. For TNFα receptor expression, TNF-R1-PE (BioLegend, 113003) or TNF-R2-PE (BioLegend, 113405) was combined with the Lin/PE-Cy5, Sca-1-PB, c-Kit-APC-Cy7, Flk2-Bio/SA-BV605, CD48-A647, CD150-BV605, CD34-FITC and FcγR-PE-Cy7. For sorting T cells, T cell-enriched BM cells were stained with CD4-FITC (BD Pharmingen, 553729), CD8-PE (BioLegend, 100708) and SA-BV605, and BV605+ non-T cell fraction was excluded during sorting. For mature cell analyses, unfractionated BM cells were stained with Gr-1-eFluor450 (eBioscience, 48-5931-82), Mac-1-PE-Cy7 (eBioscience, 25-0112-82), B220-APC-eFluor780 (eBioscience, 47-0452-82), and CD19-PerCP-Cy5.5 (BD Pharmingen, 551001). For NF-κB-eGFP mice, staining was performed either with Lin-PE-Cy5, Sca-1-PB, c-Kit-APC-Cy7, Flk2-Bio/SA-Qdot605, CD48-A647, CD150-PE-Cy7 (BioLegend, 115913) to assess HSCs/MPPs, or Lin-PE-Cy5, Sca-1-PB, c-Kit-APC-Cy7, FcγR-PerCP-eFluor710 and CD34-Bio (BioLegend, 119304)/SA-Qdot605 to assess myeloid progenitors. For PU.1-eYFP mice, BM cells were stained with Lin-PE-Cy5, Sca-1-PB, c-Kit-APC-Cy7, Flk2-PE (eBioscience, 12-1351-82), CD34-Bio/SA-BV605, CD48-A647 and CD150-BV650. For transplantation experiments, PB chimerism and lineage distribution of donor-derived cells were assessed by cell staining with Gr-1-eFluor450, Mac-1-PE-Cy7, B220-APC-eFluor780, CD3-eFluor660 (eBioscience, 50-0032-82), Ter-119-PE-Cy5 (eBioscience, 15-5921-83), CD45.1-PE (eBioscience, 12-0453-83) and CD45.2-FITC (eBioscience, 11-0454-85). For enumeration of donor-derived HSCs, unfractionated BM cells were stained with Lin/PE-Cy5, Sca-1-PB, c-Kit-APC-Cy7, Flk2-Bio/SA-Qdot605, CD48-A647, CD150-PE-Cy7, CD45.1-PE and CD45.2-FITC. For BM analyses in pIC-treated mice, ESAM-FITC (BioLegend, 136205) was used together with Lin/PE-Cy5, Sca-1-PB, c-Kit-APC-Cy7, Flk2-Bio/SA-Qdot605, CD48-A647 and CD150-PE-Cy7 to exclude contaminating myeloid progenitors due to Sca-1 upregulation (Pietras et al., 2014). For pIC-treated NF-κB-eGFP mice, ESAM-FITC and CD48-A647 were replaced by ESAM-APC (BioLegend, 136208) and CD48-PerCP-Cy5.5 (BioLegend, 103421), respectively. For myeloid differentiation and surface marker tracking, expanded HSCs were stained using c-Kit-APC-Cy7, Sca-1-PB, FcγR-PerCP-eFluor710 or -PE-Cy7 and Mac-1-PE-Cy7 or -FITC (eBioscience, 11-0112-82). For PD-L1 and PD-L2 expression, HSCs were cultured for 24 hours and stained with PD-L1-PE (BioLegend, 124307) or PD-L2-PE (BioLegend, 107205). Live cells were finally re-suspended in 2% FBS/HBSS containing 1 μg/ml propidium iodide (PI) to exclude dead cells. For intracellular Ki67/DAPI staining, unfractionated BM cells were first stained with Lin/PE-Cy5, Sca-1-PE-Cy7 (BioLegend, 108113), c-Kit-APC-Cy7, CD48-A647 and CD150-PE, and then fixed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences) for 20 min on ice. After washing with Perm/Wash (BD Biosciences), cells were stained with anti-Ki67-FITC (eBioscience, 11-5698-80) in Perm/Wash for 30 min on ice, washed with Perm/Wash and then re-suspended in Perm/Wash containing 1 μg/ml DAPI (Molecular Probes, D1306) or Hoechst 33342 (Molecular Probes, H3570) before analysis. Cell sorting was performed on FACS Aria II (Becton Dickenson) and each population was double sorted to maximize cell purity. All data were collected on FACS Aria II, LSR II or Celesta (Becton Dickenson), and analyzed with FlowJo (Treestar).
In vitro assays
All cultures were performed at 37°C in a 5% CO2 water jacket incubator (Thermo Scientific). For liquid culture assays, cells were grown in Iscove’s modified Dulbecco’s media (IMDM) containing 5% FBS (StemCell Technology), 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate and 50 μM 2-mercaptoethanol, supplemented with the following cytokines unless otherwise indicated (all from PeproTech): SCF (25 ng/ml), TPO (25 ng/ml), Flt3-L (25 ng/ml), IL-11 (25 ng/ml), IL-3 (10 ng/ml), GM-CSF (10 ng/ml) and EPO (4 U/ml). For liquid culture with cytokine-poor media, only SCF (25 ng/ml) and G-CSF (25 ng/ml, PeproTech) were used. TNFα (Genentech) was added at 1 μg/ml unless otherwise indicated. For 3-day expansion assays, 300–500 cells were directly sorted per well of a 96-well plates in 200 μl of culture media, then TNFα and the other drugs and/or cytokines were added, and cells were manually counted after 3 days using a hemocytometer and trypan blue exclusion of dead cells. BMS-345541 (2 μM, Sigma-Aldrich, B9935), LCL-161 (5 μM, ChemiTek, CT-LCL161) and AEG40730 (4.5 μM, Tocris, 5330) were added at the same time than TNFα, while GSK’963 (10 μM, GSK) and zVAD-fmk (20 μM, Bachem, N-1510) were added 1 hour before the addition of TNFα. Bafilomycin A (Sigma-Aldrich, B1793) was used at 5 nM. For clonogenic methylcellulose colony assays, 100 HSCs or 250 GMPs were cultured for 24 hours with or without TNFα in liquid media, and then transferred into a 35-mm dish containing 1 ml methylcellulose (StemCell Technologies, M3231) supplemented with L-glutamine, penicillin/streptomycin and cytokines described above. Colonies were manually counted under a microscope after 7 days of culture. For apoptosis assays, 200 cells were seeded per well of a 384-well white luminescence plate in 40 μl of liquid media and 40 μl of Caspase-Glo 3/7 reagent (Promega, G8091) were added to each well. Plates were then shaken at 300 rpm for 1 min, incubated for 60 min at room temperature and read on a luminometer (Synergy2, BioTek or Victor 3V, Perkin Elmer). Background luminescence was determined with 40 μl of culture media without cells and subtracted before calculation of fold changes. For myeloid differentiation and surface marker tracking, 1,000–2,000 HSCs were directly sorted per well of a 96-well plates and cultured for up to 8 days. Cytokines were refreshed every other day by replacing ~30% of the total volume with fresh media, and cells were analyzed by flow cytometry after different culture periods. For CFSE dilution assay, sorted BM CD8+ T cells were labelled with 2.5 μM CFSE (Molecular Probes, C1157) as described previously (Mohrin et al., 2010) and used in T cell suppression assays. CFSE-labelled T cells were either plated alone (10,000 T cells) or together with HSCs (5,000 T cells and 5,000 HSCs) per well of a 384-well plates in 20 μl of IMDM supplemented with 5% FBS (Corning), 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate and 50 μM 2-mercaptoethanol, SCF (25 ng/ml), TPO (25 ng/ml), Flt3-L (25 ng/ml), IL-11 (25 ng/ml), IL-3 (10 ng/ml), GM-CSF (10 ng/ml), EPO (4 U/ml) and IL-2 (100 U/ml, PeproTech). TNFα was then added together with PD-1 blocking antibody (BioLegend, 114107) or isotype IgG control (BioLegend, 400621), both used at 20 μg/ml. Antibodies were replenished every day and co-cultures were performed for 3 days prior to CFSE analyses.
Immunofluorescence staining
Cells (500–2,000 cells per slide) were pipetted onto poly-L-lysine coated sides (Sigma-Aldrich, P0425–72EA), incubated at 4°C for 30 min, fixed with 4% PFA for 10 min at room temperature, permeabilized in 0.3% Triton X-100/PBS for 2 min at room temperature and blocked in 1% BSA/PBS for 1 hour at room temperature. Slides were then incubated overnight at 4°C with either rabbit anti-p65 (Cell Signaling, 8242) or rabbit anti-p50 (Cell Signaling, 13586). Slides were washed 3 times in PBS and incubated for 1 hour at room temperature in 1% BSA/PBS with A594-conjugated goat anti-rabbit IgG (Invitrogen, A11037). Slides were then washed 3 times in PBS, incubated with 1 μg/ml DAPI/PBS for 5 min and washed twice in PBS. Slides were finally mounted using VectaShield (Vector Laboratories, H-1000) or ProLong Diamond (Invitrogen, P36961). Cells were imaged on a SP5 Upright Confocal Microscope (Leica, with 63 × objective) or an Eclipse Ti inverted microscope (Nikon, with 60 × objective) and images were processed using Fiji or Photoshop CC (Adobe). For quantification, fifty to 100 cells per condition were randomly captured and nuclear p65 was scored by eye or quantified with Fiji using integrated density in the nucleus.
Gene expression analyses
For RNA-sequencing analyses, RNA was purified from 10,000 HSCs and GMPs isolated from ± TNFα-injected mice or cultured ± TNFα using RNeasy Plus Micro Kit (Qiagen, 74034). RNA integrity number (RIN) was determined by Bioanalyzer (Agilent Technologies) and RNA samples with RIN > 8.0 were subjected to further processing. Double-stranded cDNA was generated using Ovation RNA-Seq System V2 (Nugen, 7102), and sequencing libraries were prepared using LTP Library Preparation Kit (Kapa Biosystems, KR0453). Different adaptors were used for multiplexing samples in one lane, and pooled libraries were sequenced on Illumina HiSeq 3000 with 50 bp single end read length. Data quality was verified on Sequencing Analysis Viewer (Illumina) and demultiplexing was performed with CASAVA 1.8.2 (Illumina). Adapter trimming, alignment, and gene-level expression quantification was performed using STAR. Principal component analysis was performed using standard packages in R, and plots were generated using the first 3 principal components. Normalization and pairwise differential expression analyses were performed in R using the DESeq2 package. For each type of TNFα treatment, genes were regarded as highly upregulated if their expression levels were more than 3-fold elevated compared to control with false discovery ratio less than 0.1. These highly upregulated genes were visualized by area-proportional Venn diagrams, and TNFα signature genes were defined in each cell type as overlapping genes across all three TNFα treatment. A heat map showing fold changes for the entire TNFα signature genes were generated with hierarchical clustering of samples with Euclidean distance. Gene Ontology (GO) analyses were performed using DAVID 6.8 on common, HSC- or GMP-specific TNFα signature genes. Gene set enrichment analyses (GSEA) were performed using the weighted enrichment score for microarray data (GSE9476 and GSE24006) and the classic enrichment score with pre-ranked gene list according to log2 fold change for RNA-seq data (GSE55689 and ours). Fluidigm gene expression analyses were done on the 96.96 Dynamic Array IFC, and analyses were performed as previously described (Herault et al., 2017). In brief, 100 HSCs were directly sorted per well of a 96-well plates containing 5 μl CellsDirect lysis buffer (Invitrogen, 11753–100), reverse-transcribed and pre-amplified for 18 cycles using SuperScript III Platinum Taq Mix (Invitrogen, 12574–026) with a custom-made set of 96 proprietary target-specific primers (Fluidigm). The resulting cDNA was analyzed on a Biomark system (Fluidigm) using EvaGreen SYBR dye (Bio-Rad, 172–5211). Data were collected with Biomark Data Collection Software (Fluidigm) and analyzed using Biomark qPCR software with a quality threshold of 0.65 and linear baseline correction. Melt curves and melting temperature values for each assay reaction were checked individually, and reactions with melt curves showing multiple peaks or poor quality were discarded, leaving 87 genes excluding housekeeping genes (Actb, Gapdh, Gusb and Hprt) for further analyses. For gene expression quantification, data were exported as a Microsoft Excel .csv file and analyzed by the Ct method using Gusb for normalization. Violin plots were generated using BoxPlotR. For quantitative RT-PCR analyses, RNA was isolated using RNeasy Plus Micro Kit, and reverse-transcribed using SuperScript III kit and random hexamers (Invitrogen, 18080–051). Runs were performed on a 7900HT Fast Real-Time PCR system or QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems) using SYBR Green reagents (Kapa Biosystems, KK4603 or KK4620) and cDNA equivalent of 200 cells per reaction. Values were normalized to Actb expression levels.
Cytokine analyses
For collecting BM fluids, the four long bones (two femurs and two tibiae) of each mouse were flushed with the same 200 μl of 2% FBS/HBSS using a 0.3 ml insulin syringe with a 28G needle and spun at 500 × g for 5 min to remove BM cells. Supernatants were further clarified by spinning down at 12,000 × g for 10 min, and samples were subsequently stored at −80°C until use. For TNFα measurement after TNFα injection, 100 μl of 2×-diluted samples were analyzed with a mouse TNFα ELISA Kit (eBioscience, 88-7324-22) according to the manufacturer’s protocol. For cytokine measurement after pIC injection, 50 μl of 2×-diluted samples were analyzed with a Luminex Cytokine Mouse 20-plex panel (Thermo Scientific, LMC0006M) using a BioPlex instrument (Bio-Rad) according to the manufacturer’s instructions.
Quantitation and Statistical Analysis
All experiments were repeated as indicated; n indicates the numbers of biological replicates. Data are expressed as mean ± standard error (SEM). Mice for treatment and transplantation were randomized, samples were alternated whenever possible, and no blinding protocol was used. No statistical method was used to predetermine sample size. Pairwise statistical significance was evaluated by two-tailed Student’s t-test. P values < 0.05 were considered statistically significant.
Data and Software Availability
RNA-seq data used to identify TNFα signature genes have been deposited online with the Gene Expression Omnibus (GEO) under accession number GSE115403. RNA-seq data used for GSEA analysis between mouse old vs. young HSCs have been deposited to GEO under accession number GSE127522. Source Data for all the figures are provided with the paper. All other data are available from the corresponding author upon reasonable request.
Supplementary Material
Table S2. Genes upregulated in HSCs and GMPs after TNFα treatment, related to Figure 4
List of upregulated genes (FDR < 0.1, fold change > 3) in HSCs or GMPs treated with TNFα under each condition (in vitro for 3h and 12h and in vivo for 3h) relative to −TNFα control. Genes are ranked based on fold change, and P value, FDR and normalized read count of each biological replicate are also displayed. Related Venn diagrams are shown in Figure 4.
Table S6. GO analysis of TNFα signature genes, related to Figure 4
List of GO terms enriched in each TNFα signature, enrichment P value and contributing genes are displayed. GO terms are ranked based on P value.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Armenian hamster anti-mouse CD48-A647 (HM48-1) | BioLegend | 103416 |
| Armenian hamster anti-mouse CD48-A700 (HM48-1) | BioLegend | 103426 |
| Armenian hamster anti-mouse CD48-PerCP-Cy5.5 (HM48-1) | BioLegend | 103421 |
| Armenian hamster anti-mouse TNF-RI (55R-286) | BioLegend | 113003 |
| Armenian hamster anti-mouse TNF-RII (TR75-89) | BioLegend | 113405 |
| Goat anti-rabbit IgG-A594 | Invitrogen | A11037 |
| Goat anti-rat IgG-PE-Cy5 | Invitrogen | A10691 |
| Mouse anti-mouse CD45.1-PE (A20) | eBioscience | 12-0453-83 |
| Mouse anti-mouse CD45.2-FITC (104) | eBioscience | 11-0454-85 |
| Rabbit anti-mouse p50 (D4P4D) | Cell Signaling | 13586 |
| Rabbit anti-mouse p65 (D14E12) | Cell Signaling | 8242 |
| Rat anti-mouse B220 (RA3-6B2) | BioLegend | 103202 |
| Rat anti-mouse B220-APC-eFluor780 (RA3-6B2) | eBioscience | 47-0452-82 |
| Rat anti-mouse CD3 (17A2) | BioLegend | 100202 |
| Rat anti-mouse CD3-eFluor660 (17A2) | eBioscience | 50-0032-82 |
| Rat anti-mouse CD4 (GK1.5) | eBioscience | 16-0041-85 |
| Rat anti-mouse CD4-FITC (GK1.5) | BD Pharmingen | 553729 |
| Rat anti-mouse CD5 (53-7.3) | BioLegend | 100602 |
| Rat anti mouse CD8 (53-6.7) | BioLegend | 100702 |
| Rat anti-mouse CD8-PE (53-6.7) | BioLegend | 100708 |
| Rat anti-mouse CD19-PerCP-Cy5.5 (1D3) | BD Pharmingen | 551001 |
| Rat anti-mouse CD34-Bio (MEC14.7) | BioLegend | 119304 |
| Rat anti-mouse CD34-FITC (RAM34) | eBioscience | 11-0341-85 |
| Rat anti-mouse CD41-BV510 (MWReg30) | BioLegend | 133923 |
| Rat anti-mouse CD150-BV650 (TC15-12F12.2) | BioLegend | 115931 |
| Rat anti-mouse CD150-PE (TC15-12F12.2) | BioLegend | 115904 |
| Rat anti-mouse CD150-PE-Cy7 (TC15-12F12.2) | BioLegend | 115913 |
| Rat anti-mouse c-Kit-APC-Cy7 (2B8) | BioLegend | 105826 |
| Rat anti-mouse ESAM-APC (1G8) | BioLegend | 136208 |
| Rat anti-mouse ESAM-FITC (1G8) | BioLegend | 136205 |
| Rat anti-mouse FcγR-PECy7 (93) | BioLegend | 101317 |
| Rat anti-mouse FcγR-PerCP-eFluor710 (93) | eBioscience | 46-0161-82 |
| Rat anti-mouse Flk2-Bio (A2F10) | eBioscience | 13-1351-85 |
| Rat anti-mouse Flk2-PE (A2F10) | eBioscience | 12-1351-82 |
| Rat anti-mouse Gr-1 (RB6-8C5) | eBioscience | 14-5931-85 |
| Rat anti-mouse Gr-1-eFluor450 (RB6-8C5) | eBioscience | 48-5931-82 |
| Rat anti-mouse IL-7Rα-PE (A7R34) | eBioscience | 12-1271-82 |
| Rat anti-mouse Ki67-FITC (SolA15) | eBioscience | 11-5698-80 |
| Rat anti-mouse Mac-1 (M1/70) | eBioscience | 16-0112-85 |
| Rat anti-mouse Mac-1-FITC (M1/70) | eBioscience | 11-0112-82 |
| Rat anti-mouse Mac-1-PE-Cy7 (M1/70) | eBioscience | 25-0112-82 |
| Rat anti-mouse PD-L1-PE (10F.9G2) | BioLegend | 124307 |
| Rat anti-mouse PD-L2-PE (TY25) | BioLegend | 107205 |
| Rat anti-mouse PD-1 (RMP1-14) | BioLegend | 114107 |
| Rat anti-mouse Sca-1-BV421 (D7) | BioLegend | 108128 |
| Rat anti-mouse Sca-1-PB (D7) | BioLegend | 108120 |
| Rat anti-mouse Sca-1-PE-Cy7 (D7) | BioLegend | 108113 |
| Rat anti-mouse Ter-119 (TER-119) | eBioscience | 16-5921-85 |
| Rat anti-mouse Ter-119-PE-Cy5 (TER-119) | eBioscience | 15-5921-83 |
| Streptavidin-BV605 | BioLegend | 405229 |
| Streptavidin-PE-Cy7 | eBioscience | 25-4317-82 |
| Streptavidin-Qdot605 | Invitrogen | Q10101MP |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-FU | Sigma-Aldrich | F6627 |
| AEG 40730 | Tocris | 5330 |
| Bafilomycin A | Sigma-Aldrich | B1793 |
| BMS-345541 | Sigma-Aldrich | B9935 |
| CFSE | Molecular probes | C1157 |
| GSK’963 | Berger et al., 2015 | N/A |
| LCL-161 | ChemiTek | CT-LCL161 |
| LPS | Sigma-Aldrich | L2880 |
| Poly I:C | GE Healthcare | 27473201 |
| zVAD-fmk | Bachem | N-1510 |
| Recombinant human EPO | PeproTech | 100–64 |
| Recombinant mouse Flt3-L | PeproTech | 250-31L |
| Recombinant mouse G-CSF | PeproTech | 250-05 |
| Recombinant mouse GM-CSF | PeproTech | 315–03 |
| Recombinant mouse IL-11 | PeproTech | 220–11 |
| Recombinant human IL-2 | PeproTech | 200–02 |
| Recombinant mouse IL-3 | PeproTech | 213–13 |
| Recombinant mouse SCF | PeproTech | 250–03 |
| Recombinant mouse TNFα | Genentech | N/A |
| Recombinant mouse TPO | PeproTech | 315–14 |
| Critical Commercial Assays | ||
| Caspase-Glo 3/7 | Promega | G8091 |
| CellsDirect One-Step qRT-PCR Kit | Invitrogen | 11753–100 |
| Cytofix/Cytoperm buffer | BD Biosciences | 554722 |
| Cytokine 20-Plex Mouse Panel | Thermo Scientific | LMC0006M |
| KAPA SYBR® FAST ABI Prism | Kapa Biosystems | KK4603 |
| KAPA SYBR® FAST ROX Low | Kapa Biosystems | KK4620 |
| LTP Library Preparation Kit | Kapa Biosystems | KR0453 |
| Methylcellulose Methocult M3231 | StemCell Technologies | 03231 |
| Mouse c-Kit microbeads | Miltenyi Biotec | 130-091-224 |
| Mouse Pan T Cell Isolation Kit II | Miltenyi Biotec | 130-095-130 |
| Mouse TNFα ELISA Kit | eBioscience | 88-7324-22 |
| Ovation RNA-Seq System V2 | Nugen | 7102 |
| Perm/Wash buffer | BD Biosciences | 554723 |
| RNeasy Plus Micro Kit | Qiagen | 74034 |
| SsoFast EvaGreen Supermix With Low ROX | Bio-Rad | 172–5211 |
| SuperScript III First-Strand Synthesis System | Invitrogen | 18080–044 |
| Deposited Data | ||
| RNA-seq: Raw and analyzed data for TNFα-treated mouse HSCs and GMPs | This paper | GSE115403 |
| RNA-seq: Raw and analyzed data for young and old mouse HSCs | This paper | GSE127522 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | 000664 |
| Mouse: C57BL/6N | Charles River | 027 |
| Mouse: Bak−/−Baxf/f | Jackson Laboratories | 006329 |
| Mouse: Bid−/− | Jackson Laboratories | 008887 |
| Mouse: Casp8f/f | Jackson Laboratories | 027002 |
| Mouse: Il1r1−/− | Jackson Laboratories | 003245 |
| Mouse: Mlkl−/− | Murphy et al., 2013 | N/A |
| Mouse: Mx1-Cre | Jackson Laboratories | 003556 |
| Mouse: NF-κB-eGFP | Magness et al., 2004 | N/A |
| Mouse: p50−/− | Jackson Laboratories | 006097 |
| Mouse: p65f/f | Algul et al., 2007 | N/A |
| Mouse: Ptprca Pepcb/BoyJ | Jackson Laboratories | 002014 |
| Mouse: PU.1-eYFP | Kirstetter et al., 2006 | N/A |
| Mouse: Ripk1D138N/D138N | Newton et al., 2014 | N/A |
| Mouse: Ripk3−/− | He et al., 2009 | N/A |
| Mouse: Scl-tTA:TRE-BCR/ABL | Reynaud et al., 2011 | N/A |
| Mouse: Tnf−/− | Jackson Laboratories | 005540 |
| Oligonucleotides | ||
| qRT-PCR primer: mouse Actb Forward: GACGGCCAGGTCATCACTATTG | This paper | N/A |
| qRT-PCR primer: mouse Actb Reverse: AGGAAGGCTGGAAAAGAGCC | This paper | N/A |
| qRT-PCR primer: mouse Birc2 Forward: TGTGGCCTGATGTTGGATAAC | Primer Bank | 6680698a1 |
| qRT-PCR primer: mouse Birc2 Reverse: GGTGACGAATGTGCAAATCTACT | Primer Bank | 6680698a1 |
| qRT-PCR primer: mouse Birc3 Forward: ACGCAGCAATCGTGCATTTTG | Primer Bank | 6680696a1 |
| qRT-PCR primer: mouse Birc3 Reverse: CCTATAACGAGGTCACTGACGG | Primer Bank | 6680696a1 |
| qRT-PCR primer: mouse Cd274 Forward: GCTCCAAAGGACTTGTACGTG | Primer Bank | 11230798a1 |
| qRT-PCR primer: mouse Cd274 Reverse: TGATCTGAAGGGCAGCATTTC | Primer Bank | 11230798a1 |
| qRT-PCR primer: mouse Pgcd1lg2 Forward: CTGCCGATACTGAACCTGAGC | Primer Bank | 10946740a1 |
| qRT-PCR primer: mouse Pgcd1lg2 Reverse: GCGGTCAAAATCGCACTCC | Primer Bank | 10946740a1 |
| qRT-PCR primer: mouse Xiap Forward: CGAGCTGGGTTTCTTTATACCG | Primer Bank | 6753088a1 |
| qRT-PCR primer: mouse Xiap Reverse: GCAATTTGGGGATATTCTCCTGT | Primer Bank | 6753088a1 |
| Software and Algorithms | ||
| Biomark Data Collection Software | Fluidigm | https://www.fluidigm.com/software |
| Biomark qPCR software | Fluidigm | https://www.fluidigm.com/software |
| BioVenn | Hulsen et al., 2008 | http://www.biovenn.nl/index.php |
| BoxPlotR | Spitzer et al., 2014 | http://shiny.chemgrid.org/boxplotr |
| CASAVA 1.8.2 | Illumina | N/A |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/N/Ahtml/DESeq2.html |
| Fiji | Schindelin J et al., 2012 | https://fiji.sc |
| FlowJo | Treestar | https://www.flowjo.com |
| Gene set enrichment analysis (GSEA) software | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| Multi Experiment Viewer (MeV) | Saeed et al., 2003 | http://mev.tm4.org |
| Sequencing Analysis Viewer | Illumina | https://support.illumina.com/sequencing/sequencing_software/sequencing_analysis_viewer_sav/downloads.html |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| R | The R Foundation | http://www.r-project.org |
HIGHLIGHTS.
A TNFα-p65-cIAP2 axis supports HSC survival during inflammation
TNFα drives PU.1 upregulation and myeloid regeneration from HSCs
TNFα prevents HSC necroptosis and induces T cell suppression activity
The HSC-specific TNFα signature is upregulated in aged and malignant HSCs
ACKNOWLEDGEMENTS
We thank Dr. A. Warren (WEHI) for Mlkl−/− mice, Dr. K. Newton (Genentech) for Ripk1KD mice, Dr. A. Ma (UCSF) for Tnf−/− and Ripk3−/− mice, Dr. S. Hedrick (UCSD) for Casp8f/f mice, Dr. S. Zinkel (Vanderbilt University) for Bid−/− mice, Dr. R. Schmid (Munich University) for p65f/f mice, Dr. C. Jobin (University of North Carolina at Chapel Hill) for NF-κB-eGFP mice, Genentech for recombinant TNFα, GlaxoSmithKline for GSK’963 compound, M. Lee (UCSF) and M. Kissner (Columbia University) for management of our flow cytometry core facilities, UCLA Clinical Microarray Core for RNA sequencing, J. Pollack, K. Shinoda and P. Dellorusso for bioinformatics analyses, R. Kratchmarov for help with T cell studies, G. Perez, S.Y. Zhang and T. Su for technical assistance, E. Verovskaya and T. Ho for RNA-seq analyses of aged HSCs, L. Kohli for initiating these studies, and all members of the Passegué laboratory for insights and suggestions. M.Y. was supported by a JSPS overseas research fellowship. This work was supported in part through the NIH/NCI Cancer Center Support Grant P30CA013696 and by NIH 2R01HL092471, R01HL111266 and 1R35HL135763 grants, and an LLS Scholar Award to E.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Algul H, Treiber M, Lesina M, Nakhai H, Saur D, Geisler F, Pfeifer A, Paxian S, and Schmid RM (2007). Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest 117, 1490–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisner DR, Ch’en IL, Kolla RV, Hoffmann A, and Hedrick SM (2005). Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J Immunol 175, 3469–3473. [DOI] [PubMed] [Google Scholar]
- Berger SB, Harris P, Nagilla R, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Ouellette M, et al. (2015). Characterization of GSK’963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov 1, 15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D, Blaser H, and Mak TW (2015). Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15, 362–374. [DOI] [PubMed] [Google Scholar]
- Broxmeyer HE, Williams DE, Lu L, Cooper S, Anderson SL, Beyer GS, Hoffman R, and Rubin BY (1986). The suppressive influences of human tumor necrosis factors on bone marrow hematopoietic progenitor cells from normal donors and patients with leukemia: synergism of tumor necrosis factor and interferon-gamma. J Immunol 136, 4487–4495. [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. [DOI] [PubMed] [Google Scholar]
- Caux C, Saeland S, Favre C, Duvert V, Mannoni P, and Banchereau J (1990). Tumor necrosis factor-alpha strongly potentiates interleukin-3 and granulocyte-macrophage colony-stimulating factor-induced proliferation of human CD34+ hematopoietic progenitor cells. Blood 75, 2292–2298. [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, and Goodell MA (2007). Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 5, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day R (2002). Adverse reactions to TNF-alpha inhibitors in rheumatoid arthritis. Lancet 359, 540–541. [DOI] [PubMed] [Google Scholar]
- Docke WD, Prosch S, Fietze E, Kimel V, Zuckermann H, Klug C, Syrbe U, Kruger DH, von Baehr R, and Volk HD (1994). Cytomegalovirus reactivation and tumour necrosis factor. Lancet 343, 268–269. [DOI] [PubMed] [Google Scholar]
- Espin-Palazon R, Weijts B, Mulero V, and Traver D (2018). Proinflammatory Signals as Fuel for the Fire of Hematopoietic Stem Cell Emergence. Trends Cell Biol 28, 58–66. [DOI] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, and Trumpp A (2009). IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908. [DOI] [PubMed] [Google Scholar]
- Etzrodt M, Ahmed N, Hoppe PS, Loeffler D, Skylaki S, Hilsenbeck O, Kokkaliaris KD, Kaltenbach HM, Stelling J, Nerlov C, et al. (2018). Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood. [DOI] [PubMed] [Google Scholar]
- Fulda S, and Vucic D (2012). Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 11, 109–124. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, Plevritis SK, Majeti R, and Alizadeh AA (2010). Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA 304, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM, and Jordan CT (2001). Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98, 2301–2307. [DOI] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, and Wang X (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. [DOI] [PubMed] [Google Scholar]
- Herault A, Binnewies M, Leong S, Calero-Nieto FJ, Zhang SY, Kang YA, Wang X, Pietras EM, Chu SH, Barry-Holson K, et al. (2017). Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 544, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Suzuki S, Lai CY, Yamazaki S, Kakuta S, Iwakura Y, Nojima M, Takeuchi Y, Higashihara M, Nakauchi H, et al. (2017). Pre-Transplantation Blockade of TNF-alpha-Mediated Oxygen Species Accumulation Protects Hematopoietic Stem Cells. Stem Cells 35, 989–1002. [DOI] [PubMed] [Google Scholar]
- Kondo K, Kaneshima H, and Mocarski ES (1994). Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci U S A 91, 11879–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtonyuk LV, Fritsch K, Feng X, Manz MG, and Takizawa H (2016). Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front Immunol 7, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EW, Seong D, Seo J, Jeong M, Lee HK, and Song J (2015). USP11-dependent selective cIAP2 deubiquitylation and stabilization determine sensitivity to Smac mimetics. Cell Death Differ 22, 1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, and Jobin C (2004). In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol 173, 1561–1570. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, and Passegué E (2010). Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 7, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Naik S, Larsen SB, Cowley CJ, and Fuchs E (2018). Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease. Cell 175, 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. (2014). Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Chikuma S, Iwai Y, Fagarasan S, and Honjo T (2013). A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol 14, 1212–1218. [DOI] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Alexopoulou L, Episkopou V, and Kollias G (1996). Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med 184, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl-Yafe M, Mizrahi K, Stein J, Yolcu ES, Kaplan O, Shirwan H, Yaniv I, and Askenasy N (2010). Tumor necrosis factor receptors support murine hematopoietic progenitor function in the early stages of engraftment. Stem Cells 28, 1270–1280. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, and Passegué E (2014). Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med 211, 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJ, Veiby OP, Bryder D, and Jacobsen SE (2011). Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med 208, 1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, et al. (2014). RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188. [DOI] [PubMed] [Google Scholar]
- Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, and Kelliher MA (2014). Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci U S A 111, 14436–14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadik CD, Kim ND, and Luster AD (2011). Neutrophils cascading their way to inflammation. Trends Immunol 32, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, and Hartland EL (2013). Masters, marionettes and modulators: intersection of pathogen virulence factors and mammalian death receptor signaling. Curr Opin Immunol 25, 436–440. [DOI] [PubMed] [Google Scholar]
- Staber PB, Zhang P, Ye M, Welner RS, Nombela-Arrieta C, Bach C, Kerenyi M, Bartholdy BA, Zhang H, Alberich-Jorda M, et al. (2013). Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell 49, 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, and Green DR (2016). Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352, aaf2154. [DOI] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, and Passegué E (2013). FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, Erlandsson R, Ngara M, Anderson K, Deng Q, et al. (2014). Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 25, 794–808. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, and Nakauchi H (2013). Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126. [DOI] [PubMed] [Google Scholar]
- Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, and Korsmeyer SJ (1999). Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400, 886–891. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Lenardo MJ, and Baltimore D (2017). 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 168, 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Umikawa M, Zhang S, Huynh H, Silvany R, Chen BP, Chen L, and Zhang CC (2011). Ex vivo expanded hematopoietic stem cells overcome the MHC barrier in allogeneic transplantation. Cell Stem Cell 9, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Genes upregulated in HSCs and GMPs after TNFα treatment, related to Figure 4
List of upregulated genes (FDR < 0.1, fold change > 3) in HSCs or GMPs treated with TNFα under each condition (in vitro for 3h and 12h and in vivo for 3h) relative to −TNFα control. Genes are ranked based on fold change, and P value, FDR and normalized read count of each biological replicate are also displayed. Related Venn diagrams are shown in Figure 4.
Table S6. GO analysis of TNFα signature genes, related to Figure 4
List of GO terms enriched in each TNFα signature, enrichment P value and contributing genes are displayed. GO terms are ranked based on P value.
Data Availability Statement
RNA-seq data used to identify TNFα signature genes have been deposited online with the Gene Expression Omnibus (GEO) under accession number GSE115403. RNA-seq data used for GSEA analysis between mouse old vs. young HSCs have been deposited to GEO under accession number GSE127522. Source Data for all the figures are provided with the paper. All other data are available from the corresponding author upon reasonable request.