

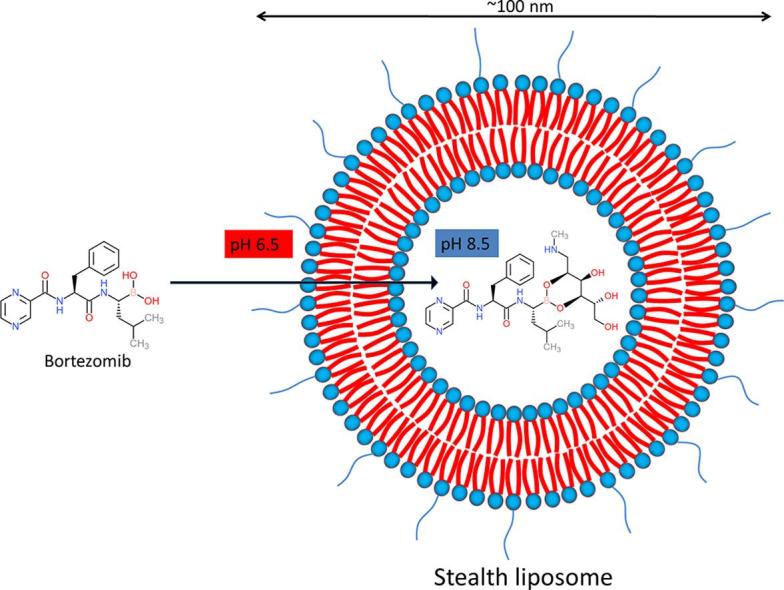
Graphical abstract
Keywords: Liposomes, Bortezomib, Remote loading, Boronate esters, Drug release
Abstract
Bortezomib is a proteasome inhibitor used for the treatment of multiple myeloma. The poor pharmacokinetic profile and off-target adverse effects provide a strong incentive to develop drug delivery systems for bortezomib. In the past, liposomal encapsulation has been proven to improve the therapeutic index of a variety of anti-neoplastic therapeutics. Here, we developed and characterized liposomal bortezomib formulations in order to find the most optimal loading conditions. Polyols were used to entrap bortezomib inside the liposomes as boronate ester via a remote loading strategy. Effect of various polyols, incubation duration, temperature, and total lipid concentration on loading efficiency was examined. Moreover, the effect of drug/lipid ratio on the release kinetics was studied. Loading efficiency was maximal when using meglumine plus mannitol as entrapping agents. Loading at room temperature was better than at 60 °C and loading efficiency was increased with increasing total lipid concentrations. There was a positive correlation between drug/lipid ratio and released amount of bortezomib. In vitro release kinetics in HBS and human plasma showed time dependent release. In HBS, at 4 °C, only 20% of the drug was released in three weeks, whereas at 37 °C 85% of the drug was released in 24 h. In human plasma, 5% of the drug retained after 24 h indicating faster release. Taken together, the most favorable liposomal formulation of bortezomib should be further exploited to study in vitro and in vivo efficacy performance.
1. Introduction
Multiple myeloma (MM) is a malignancy of B lymphocytes characterized by clonal proliferation of a single plasma cell in the bone marrow resulting in monoclonal immunoglobulin production (Hideshima et al., 2007, Kumar et al., 2017). It is the second most common hematological malignancy, accounting for 1% of all cancers and 13% of all hematological cancers (Jones et al., 2012, Deshantri et al., 2018). One of the most promising targets for the treatment of MM has proven to be the ubiquitin proteasome system (UPS) (Manasanch and Orlowski, 2017). UPS degrades defective proteins by recognizing the poly-ubiquitin chain attached to them. Thereby, inhibiting the activity of proteasome results in accumulation of damaged proteins in the cell leading to apoptosis. By blocking UPS, proteasome inhibitors have antitumor and anti-angiogenic properties. Bortezomib (Velcade®), the first-in-class proteasome inhibitor is clinically approved for the treatment of MM and mantle cell lymphoma (Chen et al., 2011). Bortezomib is a boronic acid dipeptide that directly binds to the 26S proteasome inhibiting its chymotrypsin-like activity of proteasome (Mateos and San Miguel, 2012). Despite encouraging clinical results, use of bortezomib has been limited because all cells depend on proteasome activity, leading to toxicity (Chen et al., 2011). The most common adverse reactions to bortezomib include asthenic conditions, diarrhea, nausea, constipation, peripheral neuropathy, vomiting, pyrexia, thrombocytopenia, psychiatric disorders, anorexia and decreased appetite, neutropenia, neuralgia, leukopenia and anemia (Chen et al., 2011, Mateos and San Miguel, 2012). Additionally, bortezomib has pharmaceutical challenges. It shows poor water solubility as a pKa value of 13 (https://www.drugbank.ca/drugs/DB00188; https://pubchem.ncbi.nlm.nih.gov/compound/Bortezomib) and a short half-life in circulation. The physicochemical properties of bortezomib are depicted in Table 1. Both toxicity and poor pharmaceutical properties are major hurdles to its clinical use. Therefore, there is a need for a targeted formulation of bortezomib.
Table 1.
Physicochemical properties of bortezomib.
| Molecular structure | 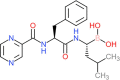 |
| IUPAC name | [(1R)-3-methyl-1-[[(2S)-3-phenyl-2-(pyrazine-2-carbonylamino)propanoyl]amino]butyl]boronic acid |
| Molecular Weight | 384.243 |
| logP | 0.89 (ALOGPS) |
| logP | 1.53 (ChemAxon) |
| pKa (strongest acidic) | 13.04 (ChemAxon) |
| pKa (strongest basic) | −0.7 (ChemAxon) |
| Water solubility | 0.0532 mg/mL |
| Physiological Charge | 0 |
Currently, bortezomib is available in a lyophilized form (Velcade®) for intravenous or subcutaneous injection. Mannitol (10 mg mannitol/mg of bortezomib) is used to improve the solubility upon reconstitution (Velcade(R)). The sugar molecule binds covalently with the boronic acid moiety of bortezomib forming a boronate ester. The ester helps dissolving bortezomib in two ways. Firstly, it prevents formation of trimer of bortezomib, which would have even lower water solubility. Secondly, mannitol is a highly water soluble polyol and shifts the pKa three units below its original pKa, thereby increasing water solubility (Marinaro and Stella, 2006). Fig. 1 depicts the chemical reactions forming bortezomib trimer and boronate ester.
Fig. 1.
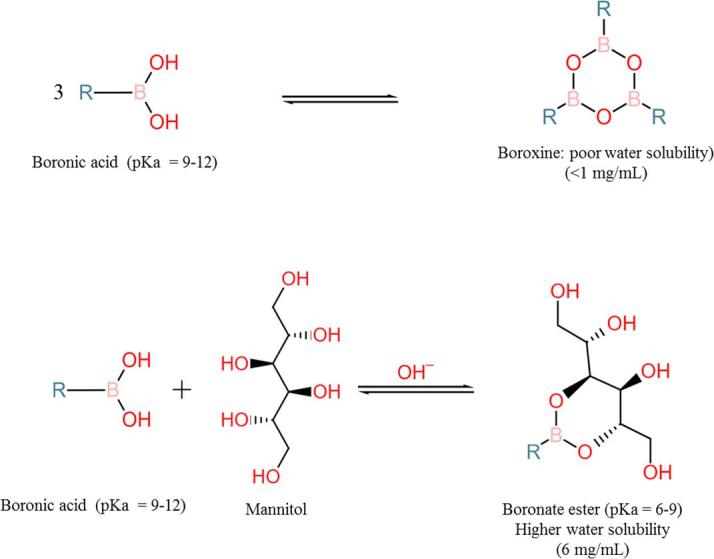
Change in pKa and water solubility by boronate ester formation. Formation of trimer of boronic acid in free from (A). The trimer formation is prevented and pKa is lowered by formation of boronate ester with a polyol (mannitol in this example) thereby increasing solubility of the boronic acid (B).
Liposomal encapsulation is a proven approach to overcome pharmaceutical and toxicity issues of chemotherapeutic drugs (Deshantri et al., 2018). The aim of this work was to develop and characterize liposomal formulations of bortezomib. A remote loading strategy was developed for bortezomib based on various polyols for the entrapment of bortezomib inside the liposomes.
2. Materials and methods
2.1. Materials and instruments
1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (mPEG2000-DSPE) were obtained from Lipoid GmbH, Germany. Cholesterol and Whatman® Anodisc inorganic membranes were obtained from Sigma Aldrich, Germany. Bortezomib was purchased from LC Laboratories, Canada. High-pressure extruder from Lipex, Northern Lipids, Canada was used for liposome resizing. NanoSight NS500 instrument from Malvern Panalytical, UK was used for particle size determination. Ultra-high Performance Liquid Chromatography (UPLC) from Waters Corporation, USA was used for determination of bortezomib concentrations.
2.2. Methods
2.2.1. Passive loading of bortezomib into the aqueous core
Liposomes were prepared as described previously (Deshantri et al., 2016). In brief, appropriate amounts of DPPC, mPEG2000-DSPE, and cholesterol were dissolved in chloroform at a molar ratio of 50:5:45, respectively. A lipid film was prepared under reduced pressure on a rotary evaporator and dried under a stream of nitrogen until complete dryness. The lipid film was then hydrated with bortezomib solution in HEPES buffered saline (HBS) pH 7.4 at 1 mg bortezomib/mL in HBS containing 10% v/v dimethyl sulphoxide (DMSO). DMSO was used to first solubilize bortezomib, which was further diluted in HBS. The liposome dispersion was then extruded through high-pressure extruder 10 times using Whatman® Anodisc inorganic membranes of pore size 100 nm. Non-encapsulated bortezomib was removed by dialysis in HBS 7.4 at 4 °C for 24 h. Particle size was determined by Nanoparticle Tracking Analysis (NTA) using NanoSight NS500. Bortezomib loading was determined by UPLC using a C18 column (ACQUITYUPLC®BEHC18 1.7 µm, 2.1 × 50 mm). Liposomes were dissolved in 35% acetonitrile in HBS. A solution of water/acetonitrile/perchloric acid 65/35/0.1 was used as mobile phase. Absorbance was detected at 270 nm at flow rate of 0.600 mL/min and injection volume of 7.5 μL. Run time was set to 1 min. Percentage encapsulation efficiency (%EE) was calculated by following equation:
2.2.2. Passive loading of bortezomib into the bilayer
Liposomes were prepared as described above. Appropriate amounts of DPPC, mPEG2000-DSPE, and cholesterol were dissolved in chloroform at a molar ratio of 50:5:45, respectively. In addition, bortezomib was added to the lipid solution before preparation of the lipid film, at a concentration of 1 mg/mL. The lipid film was hydrated with HBS pH 7.4. The liposome dispersion was then extruded 10 times using Whatman® Anodisc inorganic membranes of pore size 100 nm. Particle size was determined by NTA and the encapsulated amount of bortezomib was measured by UPLC.
2.2.3. Remote loading of bortezomib
For remote loading, a lipid film was prepared as described above and hydrated with appropriated hydration buffer containing the indicated polyol(s) as entrapping agent at pH 8.5. The liposome dispersion was then extruded 10–15 times using Whatman® Anodisc inorganic membranes of 100 nm pore size. To establish a pH gradient, dialysis was performed against 140 mM NaCl and 20 mM HEPES buffer pH at 6.5, using a 10 kD MWCO dialysis membrane at 4 °C for 24 h. Size distribution of liposomes was determined by using NTA. For remote loading, 1 mL of 3 mg/mL bortezomib solution in HBS, containing 10 or 20% DMSO, was added to 2 mL of liposomes and incubated for indicated time and temperature. Alternatively, appropriate amounts of bortezomib solution in DMSO were directly added to the liposomes resulting final DMSO concentration 0.25%. Non-encapsulated bortezomib was removed by dialysis against HBS pH 7.4 for 24 h at 4 °C. Bortezomib loading was determined by UPLC.
2.2.4. Loading of bortezomib into poly(vinyl alcohol) coated liposomes
Liposomes were prepared as described in previous section containing 100 mM of total lipids, and were coated with poly(vinyl alcohol) MW 6 kD (PVA) as described previously (Mu and Zhong, 2006). In brief, after resizing by extrusion, liposome suspension was mixed with equal volume of aqueous solution of 2% PVA, and incubated at 4 °C for 1 h on a roller bench. Unbound PVA was removed by dialysis against HBS using a dialysis cassette MWCO 20 kD at 4 °C for 24 h. A control batch of liposomes was prepared by adding the same volume of HBS instead of PVA to the liposomes, followed by 1 h incubation at 4 °C. A 3 mg/mL bortezomib solution in 20% DMSO was added to 2 mL of liposomes and incubated overnight at RT for remote loading. Non-encapsulated bortezomib was removed by dialysis against HBS pH 7.4 for 24 h at 4 °C. Encapsulation efficiency (EE) was determined by UPLC. To compare the EE into PVA-coated to PVA-loaded liposomes, a separate batch was prepared by hydrating the lipid film with 2% PVA and treated similarly as the coated liposomes for bortezomib loading.
2.2.5. Effect of entrapping agents on remote loading
To evaluate the effect of various entrapping agents on EE by the aforementioned remote loading method, liposomes (100 mM total lipids) were prepared by hydrating lipid films with solutions containing either sorbitol, mannitol, meglumine (each 10%), an aqueous solution of meglumine and acetic acid (300 mM each), or a solution of mannitol, meglumine, and acetic and HEPES at 200 mM, 50 mM, 30 mM and 20 mM, respectively. Remote loading was performed after direct addition of bortezomib in DMSO solution to the liposomes dispersions. The final concentration of bortezomib was 2 mg/mL and DMSO was 0.25% v/v. External buffer was 140 mM NaCl and 20 mM HEPES. A pH gradient was set to pH 8.5 inside/pH 6.5 outside. Remote loading was performed by overnight incubation at room temperature (RT). Non-encapsulated drug was removed by size exclusion using PD-10 desalting columns (GE Healthcare). Bortezomib concentrations were measure by UPLC.
2.2.6. Effect of incubation duration and temperature on remote loading
Effect of incubation temperature on remote loading was tested in liposomes containing 100 mM total lipids and meglumine plus acetic acid (300 mM each) as entrapping agents. Bortezomib solution in DMSO was added to the liposomes after pH gradient was set to pH 8.5 inside/pH 6.5 outside. Remote loading was performed at four temperature conditions: (i) at 60 °C for 10–20 min followed by overnight incubation at RT, (ii) at 60 °C for 30 min followed by overnight incubaton at RT, (iii) at 60 °C for 2 h followed by overnight incubation at RT, and (iv) at RT for approximately 24 h. Non-encapsulated drug was removed by size exclusion using PD-10 desalting columns. EE was estimated by UPLC measurement of loaded bortezomib.
2.2.7. Effect of total lipid and bortezomib concentration on remote loading
The effect of total lipid concentration and initial bortezomib concentration on remote loading was evaluated in liposomes containing mannitol plus meglumine and acetic acid and HEPES. Liposomes with total lipid concentrations 25, 50, and 100 mM were prepared and remotely loaded with bortezomib. With each batch of liposomes, bortezomib was added to get four different final concentrations of 0.5, 1, 1.5, or 2 mg/mL. Bortezomib solutions in DMSO were added to the liposomes resulting final DMSO concentration 0.25% for each condition. After remote loading, non-encapsulated drug was removed by size exclusion using PD-10 desalting column. Encapsulated amount of bortezomib was determined by UPLC. EE was calculated for each condition.
2.2.8. Effect of drug/lipid ratio on release kinetics
Liposomes were prepared as described above with various bortezomib/lipid ratios. Release kinetics was performed in HBS at pH 7.4 and 4 °C or 37 °C. Bortezomib loaded liposomes with drug/lipid ratio 0.01, 0.04, and 0.08 were dialyzed in a dialysis cassette with MWCO 10 kD. 3 mL of bortezomib-loaded liposomes were placed in the dialysis cassette. The cassette was suspended into dialysis buffer containing 140 mM NaCl and 20 mM HEPES at pH 7.4. Dialysis was performed at 4 °C or 37 °C for 96 h and 24 h, respectively. A 250 µL aliquot from dialysis buffer was sampled just before the start of dialysis (0 h), at 0.5, 1, 2, 4, 8, and 24 h at 4 °C and 37 °C, and subsequently every 24 h at 4 °C. Released amounts of bortezomib were measured by UPLC and percent of encapsulated drug was calculated.
2.2.9. Release kinetics in HBS and human plasma
Liposomes containing mannitol + meglumine + acetic acid + HEPES were prepared at 100 mM total lipids, and were loaded with 100 µg/mL of bortezomib. Immediately after loading, the liposomes were diluted with an equal volume of plasma inside a Float-A-lyzer with a MWCO 300 kD. Dialysis was performed in a volumetric tube at 37 °C against 50% plasma in the dialysis buffer pH 7.4. For release in HBS, undiluted liposomes loaded with approximately 1.2 mg bortezomib/mL were dialyzed in a 10 kD MWCO dialysis cassette at 4 °C, RT (22 °C), and 37 °C against HBS pH 7.4. 50 µL aliquots were sampled from inside the cassette at various time points (0, 0.5, 1, 2, 4, 6, 8 and 24 h at 37 °C, and subsequently upto 1 week and 3 weeks at RT and 4 °C respectively), and stored at −80 °C until further analysis. Bortezomib was extracted from plasma and analyzed by UPLC. For extraction, samples were diluted with 450 µL of 0.01% formic acid in acetonitrile in order to precipitate the plasma proteins and lipid components of liposomes. Subsequently, samples were vortexed for 15 min and centrifuged at 16,000g for 15 min. Then, the supernatant was collected and evaporated with a Christ vacuum concentrator at 50 °C for 2 h. Finally, samples were re-suspended in 100 µL acetonitrile, vortexed, and centrifuged at 16,000g for 15 min. 50 µL of the supernatant was used for bortezomib determination by UPLC. A calibration curve was prepared using spiked concentrations of bortezomib in plasma. Samples from bortezomib release in HBS were processed as described in Section 2.1 and analyzed by UPLC for bortezomib concentrations.
3. Results and discussion
Bortezomib is a potent proteasome inhibiting drug used for the treatment of MM and mental cell lymphoma (Chen et al., 2011). Recent reports support a remarkable efficacy of bortezomib for other indications including hematological malignancies such as chronic myeloid leukemia (Yang et al., 2016), and solid tumors such as neuroblastoma (Zuccari et al., 2015) and breast cancer (Deshantri et al., 2016, Shen et al., 2015). The clinical use of bortezomib, however, is limited due to pharmaceutical issues such as short half-life and off target adverse effects (Chen et al., 2011). Moreover, resistance to bortezomib remains a major obstacle. Liposomal encapsulation is a proven method to improve the pharmacokinetic and pharmacodynamic properties of chemotherapeutic drugs. A few recent publications describe liposomal formulations of bortezomib by using various encapsulation methods. In a recent report, an attempt was made to load bortezomib into the liposome bilayer (Joshi et al., 2017). The loading efficiency, however, was substantially low for these liposomes ranging from 17 to 36%. This may be due to the rapid leakage of bortezomib from the bilayer.
Remote loading of small molecules into the liposomes is an attractive strategy and could be a much more efficient method to reach sufficient drug concentrations inside liposomes. As much as 100% EE can be achieved depending on the physicochemical properties of the drug molecule. A classical method to actively load compounds into the aqueous compartment of liposomes, is based on ion encapsulation creating a gradient across the lipid bilayer. Amphipathic weak acids can, for example, be successfully loaded by creating a gradient with calcium acetate. Similarly, amphipathic weak bases can be loaded by introducing an ammonium sulfate gradient (Cern et al., 2012). Doxil® is a well-known example of remote loading of doxorubicin by making use of such an ammonium sulfate gradient. Although the pH and/or ion gradient strategy works efficiently for many compounds, it is not always straightforward. Bortezomib is an amphiphilic molecule with poor water solubility (https://www.drugbank.ca/drugs/DB00188; https://pubchem.ncbi.nlm.nih.gov/compound/Bortezomib). Moreover, the relatively high pKa of bortezomib limits the applicability of using a pH gradient. The boronic acid moiety seems to be the most feasible opportunity for remote loading of bortezomib. Boronic acids make cyclic esters by reacting to polyols. Earlier reports also describe development of liposomal formulation of bortezomib by formation of boronate esters. Liposomes containing a boronate ester prodrug of bortezomib were developed by attaching bortezomib to the liposome surface via a series of linker molecules (Ashley et al., 2014). The stability of the boronate esters turned out, however, very low with the maximum half-life being 190 min. In other reports, the remote loading of bortezomib was performed by using polyols inside the liposomes as entrapping agents (Huang et al., 2007, Zalipsky and Martin, 2004). Fig. 2 depicts remote loading scheme using polyol as entrapping agent.
Fig. 2.

Schematic presentation of remote loading of bortezomib. Remote loading of bortezomib in PEGylated liposomes is depicted. Higher inside/lower outside pH gradient is created to facilitate bortezomib permeation into the liposome. Once inside, bortezomib encounters an entrapping agent (mannitol in this example), forming a boronate ester of bortezomib. The reaction is favored by basic pH inside the liposome.
In the present work, we comprehensively developed and characterized liposomal formulations of bortezomib by various loading methods, including passive and remote loading.
3.1. Passive vs active loading of bortezomib
We tested passive loading of bortezomib in the aqueous core and into the bilayer of PEGylated liposomes. For loading into the aqueous core, the lipid film was hydrated with bortezomib solution in HBS containing 10 or 20% DMSO to increase solubility. UPLC results indicate a very low EE from approximately 3–4%. To load bortezomib into the bilayer, it was mixed with the lipids to form a lipid film. Next, the lipid film was hydrated with HBS, and finally liposomes were sized followed by purification to remove non-encapsulated drug. The loaded amount of drug, when this loading method was applied, was 150 µg/mL with an EE of 15% (Table 2).
Table 2.
Overview of various batches of bortezomib-encapsulated liposomes by passive and remote loading methods.
| Loading method | Entrapping agent | Particle size |
Bortezomib concentration µg/mL (%EE) |
||
|---|---|---|---|---|---|
| Mean ± SD | Mode ± SD | 10% DMSO | 20% DMSO | ||
| Passive loading into aqueous core | NA | 97 ± 0.8 | 85 ± 1.0 | 39 (3.9%) | 31 (3.1%) |
| Passive loading into bilayer | NA | 125 ± 3.9 | 112 ± 7.0 | 150 (15%) | |
| Remote loading | Sorbitol 10% | 109 ± 0.8 | 95 ± 0.7 | 17 (1.7%), 10 (1%) | |
| Sorbitol 20% | 99 ± 0.6 | 86 ± 1.2 | 59 (5.9%) | ||
| Sorbitol 30% | 122 ± 1.0 | 109 ± 1.2 | 8.6 (0.9%), 19 (2%) | ||
| Sorbitol 40% | 120 ± 0.9 | 109 ± 1.4 | 6.6 (0.7%) | ||
| PVA 6 K (1% w/v) | n.d. | 60 (6%) | |||
| PVA 6 K (5% w/v) | 125 ± 2.7 | 111 ± 0.7 | 147 (14.7%) | ||
| PVA 6 K (10% w/v) | 124.1 ± 3.0 (PDI = 0.04 ± 0.04)* |
20 (2%), 172 (17.2%) | |||
| PVA 6 K (0.2% w/v) | 113 ± 1.6 | 95 ± 2.2 | 124 (12.4%), 94 (9.4%) | ||
| PVA 6 K (0.7% w/v) | 112 ± 4.0 | 97 ± 5.0 | 653 (65.3%), 357 (35.7%), 334 (33.4%) | ||
| PVA 6 K (2% w/v) | 107 ± 1.9 | 95 ± 3.8 | 381 (38.1%), 559 (55.9%) | ||
| Loading to PVA coated liposomes | PVA 6 K (2%) | 94 ± 2.1 | 85 ± 4.8 | 326 (32.6%), 445 (44.5%) | |
NA = not applicable; n.d. = not done; %EE = percentage encapsulation efficiency; PDI = polydispersity index; PVA, poly(vinyl alcohol).
Measured by dynamic light scattering (DLS).
In order to increase EE, we employed remote loading strategies by entrapping the polyols sorbitol (a low molecular weight polyol) and poly(vinyl alcohol) MW 6 kD (PVA) (a high molecular weight polyol). Aqueous solutions of increasing concentrations of either entrapping agent were used to hydrate the lipid film. Sorbitol was used at 10, 20, 30 or 40% w/v. PVA was used at 1, 5 or 10% w/v. After sizing by extrusion, a higher inside/lower outside pH gradient was created. Remote loading was performed at RT by mixing bortezomib solution in HBS containing 10% DMSO. The results show considerable variability in the EEs ranging from approximately 0.7 to 6% in case of sorbitol. The EE was not concentration dependent. In case of PVA, EE was higher than that of sorbitol. At 1% PVA, EE was 6%, which was increase to approximately 15% for 5% PVA. At 10% PVA, substantial variability was observed with EE% ranging from 2% and 17% in two independent experiments (Table 2).
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.ijpx.2019.100011.
We hypothesized that the variability may be due to poor aqueous solubility of bortezomib. Therefore, in order to increase solubility, we dissolved bortezomib in HBS containing 20% DMSO. It was confirmed that 20% DMSO did not compromise liposome integrity in terms of size distribution (Supplemental Table 1). Remote loading was performed by using PVA as entrapping agent at concentrations 0.2, 0.7, and 2% w/v. Interestingly, EE was increased to approximately 9–12%, 33–65%, and 38–56% in case of 0.2, 0.7, and 2% PVA respectively (Table 2).
Supplementary Table 1.
It has been reported that PVA can be coated on the external surface of liposomes by simple incubation in an aqueous solution of the polymer (Mu and Zhong, 2006). This raises a question on the loading method, using PVA-liposomes. During lipid film hydration and extrusion steps, liposomes were suspended into the PVA solution, which might result in coating on the liposome surface. The loading of bortezomib could therefore occur on the external surface rather than the internal core. To test this, we performed remote loading in liposomes loaded or coated with PVA. Surprisingly, EE in PVA coated liposomes was found to be approximately 33–45%, which is comparable to PVA-loaded liposomes (Table 2). This indicates that the remote loading for PVA-loaded liposomes likely resulted in bortezomib on the liposomes’ surface as PVA-boronate ester.
3.2. Effect of entrapping agents on remote loading
Because sorbitol produced poor EE and high molecular weight polyols resulted in aspecific surface coating, we tested several small polyols as sorbitol alternative. To evaluate the effect of different entrapping agents on EE, remote loading was performed using liposomes loaded with mannitol, sorbitol, meglumine, meglumine plus acetic acid, or meglumine plus mannitol and acetic acid in HEPES. After extrusion and establishing pH gradient, bortezomib solution in DMSO was added to the liposomes followed by incubation at RT overnight. UPLC revealed that liposome batches containing meglumine showed higher EE compared to mannitol and sorbitol. Interestingly, the highest EE was observed in liposomes containing the combination of meglumine + mannitol + acetic acid + HEPES resulting in nearly 100%. EE was 20 ± 2%, 20 ± 7%, 62 ± 3%, 85 ± 7%, and 94 ± 3% in case of mannitol, sorbitol, meglumine, meglumine plus acetic acid, and mannitol plus meglumine plus acetic acid plus HEPES, respectively (Fig. 3).
Fig. 3.

Effect of entrapping agents on remote loading. Liposomes were prepared by loading the entrapping agents. Remote loading was performed overnight at room temperature. Bortezomib concentrations were measured in the liposomes after removing the non-encapsulated drug. Man, mannitol; Sorb, sorbitol; Meg, meglumine; Meg + AA, meglumine plus acetic acid (300 mM each); Man + Meg + AA, mannitol plus meglumine plus acetic acid (200, 50, and 30 mM respectively) in 20 mM HEPES. Data is presented as mean ± SD of n = 3 or 4.
3.3. Effect of incubation duration and temperature on remote loading
Liposomes loaded with meglumine plus acetic acid were used to evaluate the effect of incubation temperature on remote loading. After adding bortezomib, liposomes were incubated at RT for 24 h with or without prior incubation at 60 °C. The results showed that incubation at 60 °C reduced the EE. Interestingly, a negative correlation was observed between duration of incubation at 60 °C and EE (Table 3). EE was approximately 30%, 15%, and 10% when liposomes were incubated at 60 °C for 10–20 min, 30 min, and 2 h, respectively prior to RT. Highest EE, approximately 80%, was seen at RT without prior incubation at 60 °C. Earlier reports describe remote loading of bortezomib at temperatures higher than the phase transition temperature (Tm) of the phospholipids (41 °C for DPPC, in our case) (Zalipsky and Martin, 2004). In contrast, another report showed that loading at temperatures below the Tm results in higher EE for bortezomib (Huang et al., 2007). Also in our case, incubation below Tm was found to have higher EE.
Table 3.
Effect of incubation duration and temperature on remote loading.
| Batch# | Incubation temperature and duration |
Bortezomib concentration (mg/mL) |
% Encapsulation efficiency | ||
|---|---|---|---|---|---|
| 60 °C | 22 °C | Added | Encapsulated | ||
| Batch 1 | 10–20 min | 14–18 h | 1.88 | 0.515 | 27.4 |
| Batch 2 | 10–20 min | 14–18 h | 1.88 | 0.615 | 32.7 |
| Batch 3 | 30 min | 14–18 h | 1.88 | 0.280 | 14.9 |
| Batch 4 | 2 h | 14–18 h | 1.88 | 0.189 | 10.0 |
| Batch 5 | n.d. | 22 h | 1.5 | 1.192 | 79.4 |
| Batch 6 | n.d. | 24 h | 1.5 | 1.199 | 79.9 |
| Batch 7 | n.d. | 23 h | 1.5 | 1.210 | 80.7 |
n.d. = not done.
3.4. Effect of total lipids and bortezomib concentration on remote loading
To test the effect of total lipid and bortezomib concentrations on remote loading, liposomes were prepared with 25, 50, and 100 mM total lipid concentration. Bortezomib was added into the liposomes at increasing concentrations for remote loading. UPLC results show that the absolute concentration of encapsulated bortezomib increased with the total lipid concentration, being highest for the 100 mM concentration. The increase was seen at all bortezomib concentrations tested (Fig. 4A). Interestingly, percentage EE was found to be decreased for 25 and 50 mM total lipids. The EE decreased from approximately 55 to 33% and from 85 to 70% for 25 and 50 mM total lipids respectively (Fig. 4B). At 100 mM total lipids, 0.5, 1, and 1.5 mg/mL bortezomib resulted in similar EE (approx. 80–85%). At the highest bortezomib concentration (2 mg/mL), a decrease in EE was observed to approximately 70%. This suggests a saturation of intra-liposomal drug already observed at the 25 and 50 mM total lipid concentration for lower concentrations of bortezomib and for 100 mM total lipid concentration for 2 mg/mL bortezomib.
Fig. 4.

Effect of total lipids and bortezomib concentration on remote loading. Liposomes containing mannitol + meglumine + acetic acid (200, 50, and 30 mM, respectively) in 20 mM HEPES were prepared at 100 mM total lipids concentration and further diluted to get 25 and 50 mM total lipids. Bortezomib was added at increasing concentrations followed incubation at room temperature for remote loading. Encapsulated amount of bortezomib was measured by UPLC (A). Encapsulation efficiency was calculated (B). Data is presented as mean ± SD of two independent experiments.
3.5. Effect of drug/lipid ratio on release kinetics
Since the EE of bortezomib vary depending upon total lipid concentration in liposomes as well as the initial amount of drug added for remote loading, the loaded amount of drug may also have impact on release from the liposomes. The drug/lipid ratio is therefore an important parameter affecting the release kinetics of the encapsulated drug. Indeed, we saw that the higher intra-liposomal concentration of the drug triggered faster release. We tested release of liposomal bortezomib at a drug/lipid ratio 0.01, 0.04 and 0.08. Release was seen over time at 4 °C and 37 °C for liposomes at all conditions. The formulation with the lowest drug/lipid ratio 0.01 was the most stable formulation. At 4 °C there was still approximately 90% of the initial drug retained even after 96 h. For formulations with 0.04 and 0.08 drug/lipid ratio, the retained drug was approximately 50 and 25%, respectively, showing a strong correlation between drug/lipid ratio and release (Fig. 5A). At 37 °C, the release was faster for all three ratios. Also here, the formulation with 0.01 drug/lipid ratio showed slower release compared two 0.04 and 0.08 (Fig. 5B). The reason why bortezomib is easily released from the liposomes is likely its amphiphilic nature. Bortezomib is encapsulated as boronate ester in the liposomes. The ester bond is reversible, and once bortezomib is in the free form, it can cross the bilayer. The higher concentration in the liposome core creates higher inside/lower outside gradient, resulting in faster release through passive diffusion across the bilayer.
Fig. 5.

Effect of drug/lipid ratio on release kinetics. Liposomes containing mannitol plus meglumine and acetic acid (200, 50, and 30 mM respectively) in 20 mM HEPES were loaded at various bortezomib/total lipid ratios. Release was performed by dialysis in HBS at 4 °C (A) and 37 °C (B). Released amount of bortezomib was measured in the dialysis buffer. Percentage of initially encapsulation bortezomib was estimated. Data is presented as mean ± SD of two independent experiments.
3.6. In vitro release kinetics
Release kinetics was investigated for liposomal bortezomib formulation in HBS and human plasma. For release in HBS, concentration of bortezomib released in the external HBS was measured by UPLC. Bortezomib release was found to be faster at 37 °C compared to 4 °C and 22 °C. At 4 °C, approximately 80% of the drug was still loaded after a period of three weeks (504 h). At 22 °C, approximately 40% of drug was loaded after one week. Only about 15% of the initial drug was found into the liposomes after 24 h at 37 °C (Fig. 6A). Release kinetics in human plasma were measured at 37 °C. Liposomes loaded with 100 µg bortezomib/mL were diluted in an equal volume of plasma. Dialysis was performed in 50% plasma. UPLC results show that approximately 60% of the drug was released after 8 h. At 24 h, nearly all drug was released. Only 7% of the initial drug was encapsulated after 24 h, indicating a rapid release (Fig. 6B).
Fig. 6.

In vitro release kinetics of liposomal bortezomib. Liposomes were prepared by hydrating the lipid film with a solution of mannitol plus meglumine and acetic acid (200, 50, and 30 mM respectively) in 20 mM HEPES. After remote loading, release kinetics was performed by dialysis in HBS (A) or in 50% human plasma (B). Sampling at indicated time points was performed from inside the dialysis cassette. Plasma samples were processed for extraction.
4. Summary
A liposomal formulation would benefit the clinical performance of bortezomib by improving its pharmaceutical and pharmacokinetic characteristics. Moreover, since bortezomib is already widely used in clinic as an important part of multiple myeloma and mantle cell lymphoma treatment regimens, the clinical translation of liposomal formulation could be fast. Here we evaluated various strategies for bortezomib loading into long circulating liposomes. Earlier reports describe remote loading of bortezomib as boronate ester into liposomes by using polyols. We show that remote loading at RT outperforms remote loading compared to elevated temperature. Moreover, our results indicate that remote loading by using a combination of polyols mannitol and meglumine was found to be the most efficient. Due to the fact that the intra-liposomally formed boronate ester is reversible, entrapment was not stable. Reasonable stability was obtained for bortezomib-liposomes stored in buffer at 4 °C, allowing future in vivo studies to be performed. The formulation could be evaluated pre-clinically for anti-tumor activity in multiple myeloma and other (hematological) malignancies. Improved stability may be obtained by liposome lyophilization. The fast release at 37 °C in both buffer and plasma, appears to be too fast to take full advantage of the passive targeting of tumors by long-circulating liposomes. These liposomes will arrive at the target site mostly empty. Additional screening of polyols (and combinations) might reveal compounds that can better retain bortezomib.
Acknowledgments
Acknowledgements
This work was funded by Netherlands Organization for Scientific Research (NWO) High Tech Systems & Materials grant.
Conflict of interest
AKD and SNM are employed by Sun Pharma. JMM is CEO at Enceladus Pharma.
References
- Ashley J.D. Liposomal bortezomib nanoparticles via boronic ester prodrug formulation for improved therapeutic efficacy in vivo. J. Med. Chem. 2014;57(12):5282–5292. doi: 10.1021/jm500352v. [DOI] [PubMed] [Google Scholar]
- Cern A. Quantitative structure-property relationship modeling of remote liposome loading of drugs. J. Control. Release. 2012;160(2):147–157. doi: 10.1016/j.jconrel.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets. 2011;11(3):239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshantri A.K. Liposomal prednisolone inhibits tumor growth in a spontaneous mouse mammary carcinoma model. J. Control. Release. 2016;243:243–249. doi: 10.1016/j.jconrel.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Deshantri A.K. Nanomedicines for the treatment of hematological malignancies. J. Control. Release. 2018;287:194–215. doi: 10.1016/j.jconrel.2018.08.034. [DOI] [PubMed] [Google Scholar]
- Hideshima T. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer. 2007;7(8):585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- Huang, A., Luo, B., Wang, J., Zhang, Y., 2007. Liposome compositions for in vivo administration of boronic acid compounds. Patent number: US 2009/0092661, A1.
- Jones C.I. Identification of circulating microRNAs as diagnostic biomarkers for use in multiple myeloma. Br. J. Cancer. 2012;107(12):1987–1996. doi: 10.1038/bjc.2012.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi Medha D., Park Gabriel, Pichlik Jennifer, Patel Kavin, Patel Roshni. In vitro evaluation of bortezomib encapsulated in cationic and C6-ceramide liposomes. J. Pharm. Sci. Pharmacol. 2017;3(2):146–154. [Google Scholar]
- Kumar S.K. Multiple myeloma. Nat. Rev. Dis. Primers. 2017;3:17046. doi: 10.1038/nrdp.2017.46. [DOI] [PubMed] [Google Scholar]
- Manasanch E.E., Orlowski R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017;14(7):417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinaro W.A., Stella S.V. Physical and chemical properties of boronic acids. Formulat. Implicat. 2006 [Google Scholar]
- Mateos M.V., San Miguel J.F. Safety and efficacy of subcutaneous formulation of bortezomib versus the conventional intravenous formulation in multiple myeloma. Ther. Adv. Hematol. 2012;3(2):117–124. doi: 10.1177/2040620711432020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X., Zhong Z. Preparation and properties of poly(vinyl alcohol)-stabilized liposomes. Int. J. Pharm. 2006;318(1–2):55–61. doi: 10.1016/j.ijpharm.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Shen S. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J. Control. Release. 2015;208:14–24. doi: 10.1016/j.jconrel.2014.12.043. [DOI] [PubMed] [Google Scholar]
- Velcade(R) (Bortezomib for Injection) prescribing information. https://www.velcade-hcp.com/pdf/VELCADE_PRESCRIBING_INFORMATION.pdf.
- Yang X. Liposomal bortezomib is active against chronic myeloid leukemia by disrupting the Sp1-BCR/ABL axis. Oncotarget. 2016;7(24):36382–36394. doi: 10.18632/oncotarget.8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalipsky S., Martin, F. 2004, Liposomal formulation of bortezomib (ps-341). Patent number: WO2006052733A1.
- Zuccari G. Tumor vascular targeted liposomal-bortezomib minimizes side effects and increases therapeutic activity in human neuroblastoma. J. Control. Release. 2015;211:44–52. doi: 10.1016/j.jconrel.2015.05.286. [DOI] [PubMed] [Google Scholar]