

Abstract
Recent studies have demonstrated that transforming growth factor beta (TGF-β1)-induced antiapoptotic factor (TIAF1) is able to form aggregates in the hippocampi of middle-aged normal individuals. The aggregating TIAF1 induces generation of amyloid beta (Aβ) for causing neurodegeneration. Intriguingly, TIAF1 aggregates are shown, together with Smad4 and Aβ, in the cancer stroma and peritumor capsules of many solid tumors. During lung cancer progression, for example, TIAF1 and amyloid fibrils are significantly upregulated in the cancer stroma. Aggregates of TIAF1 and Aβ are shown on the interface between metastatic lung cancer cells and the brain tissues. Conceivably, these peritumor materials are needed for cancer cells to survive. In vitro experiments revealed that TIAF1 is a crucial component for tumor suppressors p53 and WWOX-mediated tumor suppression and apoptosis. While metastatic lung cancer cells are frequently devoid of WWOX and p53, we provide new perspectives regarding the role of TIAF1 in the pathogenesis of lung cancer development, and propose a therapeutic approach for targeting TIAF1.
Electronic supplementary material
The online version of this article (doi:10.1186/2213-0802-1-5) contains supplementary material, which is available to authorized users.
Keywords: TGF-β, TIAF1, Protein aggregation, Lung cancer
Tumor suppressors WWOX and p53
Tumor suppressors p53 and WWOX have been shown to play critical roles in apoptosis and control of cancer progression [1–3]. WW domain-containing oxidoreductase, designated as WWOX, FOR, or WOX1, is encoded by human or mouse WWOX/Wwox gene. The full-length WWOX protein is composed of two N-terminal WW domains and a C-terminal short-chain alcohol dehydrogenase/reductase (SDR) domain (Figure 1) [3]. WWOX may act as an alternative receptor for sex steroid hormones, since its SDR domain possesses a hormone-binding NSYK motif [4, 5]. The first WW domain interacts with proteins possessing a PPxY-motif(s), including AP-2, p73, ErbB4, Ezrin, SIMPLE, c-Jun, RUNX2 and many others [3]. Transiently overexpressed WWOX prevents the relocation of transcription factors AP-2, p73, ErbB4, c-Jun and RUNX2 to accumulate in the nuclei in vitro. However, the observations in vitro do not appear to be true in vivo. Activated Wwox with Tyr33 phosphorylation, along with transcription factors c-Jun, CREB, JNK1, ATF3 and NF-κB, rapidly undergoes relocation to the nuclei in axotomized neurons in sciatic nerve transection in rats [6]. When WWOX undergoes activation via phosphorylation at Tyr33 (probably by SRC kinase), it binds a broad spectrum of proteins without the PPxY motif [3, 5].
Figure 1.
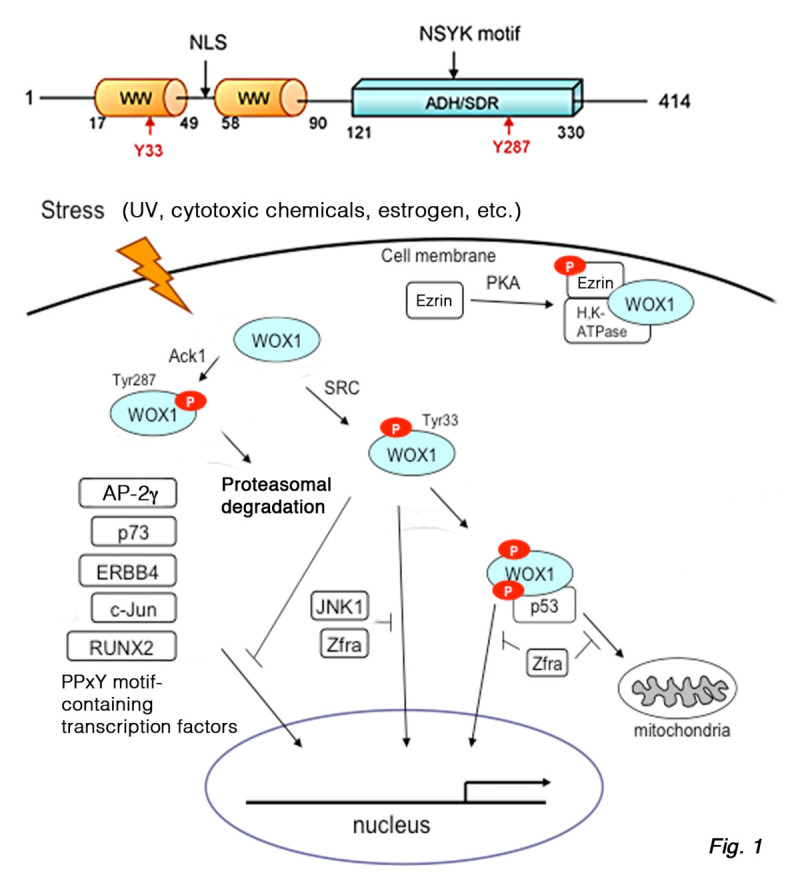
WWOX signaling. The full-length WWOX or WOX1 has two N-terminal WW domains and a C-terminal short-chain alcohol dehydrogenase/reductase (ADH/SDR) domain [1–5]. A nuclear localization signal (NLS) is located between the WW domains. Sex steroid hormones may interact with the NSYK motif in the ADH/SDR domain [4, 5]. Under stress stimuli, tyrosine kinase SRC and probably other kinases induce WWOX activation via Tyr33 phosphorylation. Activated WWOX binds Ser46-phosphorylated p53, and relocates to the mitochondria and nuclei to induce apoptosis. JNK1 and Zfra bind WWOX and counteract its-mediated apoptosis. The first WW domain of WWOX interacts with PPxY motif-containing transcription factors, including AP-2γ, p73, ERBB4, c-Jun and RUNX2. The binding allows transiently overexpressed WWOX to prevent relocation of transcription factors to the nucleus in vitro. However, the event does not work in vivo[6]. Phosphorylated Ezrin binds and anchors WWOX to the membrane/cytoskeleton area. Activated tyrosine kinase ACK1 phosphorylates WWOX at Tyr287 for polyubiquitination and proteosomal degradation.
The domain structure of p53 includes a natively unfolded N-terminal transactivation domains TAD1 and TAD2, a proline-rich region (PRR), a central DNA-binding domain, and a C-terminal tetramerization domain [2, 7]. Activated p53 causes cell cycle arrest, initiates DNA repair, and may lead to apoptosis [7]. p53 transactivates cyclin-dependent kinase inhibitor p21 or microRNA miR34 to induce cell cycle arrest [7]. For apoptosis, p53 transactivates proapoptotic genes such as BAX, PUMA, SCOTIN, and FAS, and inhibits the antiapoptotic gene BCL2 [7]. Under stress conditions (e.g. UV irradiation and chemotherapeutic drugs), activated WWOX physically binds Ser46-phosphorylated p53. This binding allows WWOX to stabilize p53, and both proteins act together in causing apoptosis [3, 5] (Figure 1).
Role of p53 and WWOX in blocking cancer initiation and progression
Cancer initiation and progression is generally considered as a consequence of gene mutation or epigenetic inactivation of tumor suppressors. It is believed that mutations in 2 tumor suppressor genes cause cancer – the so-called “2-hit hypothesis” [8]. Recent development revealed that even partial inactivation of tumor suppressors critically contributes to tumorigenesis [8]. Despite these, it is not surprising to find that many tumor suppressor proteins, e.g. p53, WWOX, Smad4, and others, are significantly upregulated during the early stage of cancer progression [3, 9, 10]. Functional significance of these proteins in blocking cancer progression at the early stage is largely unknown. However, a good possibility is that these proteins are functionally inactivated.
Proapoptotic p53, for example, is functionally inactivated by fortilin, an anti-apoptotic protein [11, 12]. Fortilin physically interacts with the sequence-specific DNA binding domain of p53. Oncogenic monocarboxylic acid transporter 1 (MCT-1) abolishes the p53 function by enhancing its degradation via the ubiquitin/proteasome system [13]. Under stress conditions, NF-κB acts as an oncoprotein to promote cell division and survival and block the proapoptotic function of p53 [14]. Mdm2 and Mdmx abolish the stability of p53 [15]. Mdm2 is an E3 ubiquitin ligase that causes p53 ubiquitination and proteasomal degradation. In contrast, Mdmx does not induce p53 degradation, but inhibits p53 by masking its transcriptional activation domain. When WWOX undergoes Tyr33 phosphorylation, the activated protein binds and stabilizes Ser46-phosphorylated p53 [3, 16]. cJun N-terminal kinase (JNK1) binds and functionally counteracts with WWOX in apoptosis [17–20]. When JNK1 is blocked by chemical inhibitors (e.g. SP600125), WWOX-mediated apoptosis is enhanced. Zfra (zinc finger-like protein that regulates apoptosis) participates in the mitochondrial apoptosis and binds WWOX to its N-terminal first WW domain and C-terminal SDR domain, which results in functional antagonism in causing apoptosis [21, 22]. Together, functional antagonism is well known among proapoptotic and apoptotic proteins. It is still not clear whether there is a direct functional antagonism between defined tumor suppressors.
Silencing WWOX gene promoter by hypermethylation
Promoter hypermethylation is believed to block protein expression, and epigenetic control can touch over a large region of chromosomes in a coordinated manner [23, 24]. Promoters for tumor suppressor genes WWOX and Fhit are frequently hypermethylated and silenced in metastatic cancers [25, 26]. Alternatively, tumor suppressors are inactivated due to gene mutation, fragmentation, or interchromosomal recombination at the later cancer progression and metastasis stages. Nonetheless, expression of tumor suppressor WWOX is increased during the early stages of cancer initiation and growth, and is then dramatically reduced at the late stages of differentiation in skin cancer both in humans and mice [3, 10]. Translational blockade is shown to be involved in the downregulation of WWOX/Wwox protein in humans and mice [10].
Lack of WWOX and increased migration in metastatic cancers
In most cases, metastatic cancer cells do not have functional tumor suppressors such as WWOX and p53 [3, 4, 25, 26]. How tumor suppressors affect normal cell migration and cancer cell metastasis is largely unknown. Ectopic WWOX affects ovarian cell attachment and migration on fibronectin-enriched extracellular matrix, and, conversely, loss of WWOX facilitates ovarian cell migration [27]. While epithelial-mesenchymal transition is an essential step toward cancer cell metastasis, it appears that many signal pathways converge on several transcription factors, including zinc finger proteins Snail and Slug, Twist, ZEB 1/2, and Smads, and the event results in the morphological transition and metastasis [28].
Cancer cell metastasis is affected by the surrounding tumor microenvironments. Metastatic lung cancer cells appear to be able to detect alterations in the environmental cues, so that they successfully migrate to target organs and settle properly [29, 30]. Non-small-cell lung carcinomas (NSCLC) frequently migrate to the contra-lateral lung, the brain and to organs such as adrenal glands, liver and bones [29]. Whether lung cancer cells specifically select susceptible sites in the endothelial lining of blood vessels for docking and penetration into organs is largely unknown.
Many types of metastatic cancers utilize the CXCR4/CXCL12 signaling axis to reach the targets [31–33]. High levels of functional CXCR4 receptors are expressed in small-cell lung cancer (SCLC) cells. Binding of chemokine stromal derived factor-1 (SDF-1/CXCL12) to the CXCR4 receptors increases the expression of integrin-mediated adhesion in SCLC to target cells.
TIAF1-p53-WWOX is an axis of tumor suppression
TIAF1 is a transforming growth beta (TGF-β)-induced 12-kDa protein [34–38]. This protein was originally shown to participate in the regulation of the tumor necrosis factor (TNF) pathway. TIAF1 also controls the signaling of TNF receptor adaptor proteins such as TRADD (TNFR1-associated death domain protein), FADD (FAS-associated death domain-containing protein), and RIP (receptor-interacting serine-threonine kinase) [38]. As a TGF-β-induced protein, TIAF1 acts similarly to that of TGF-β1 [37, 38]. For example, TGF-β1 and transiently overexpressed TIAF1 support the growth of fibroblasts [37]. However, epithelial and monocytic cells, as well as many non-fibroblasts, are sensitive to the growth suppression and apoptosis by TGF-β1 and transiently overexpressed TIAF1. In monocytic U937 cells, for example, ectopic TIAF1 increases the expression of p53 and Cip1/p21 and suppresses ERK phosphorylation, which leads to cell growth inhibition and apoptosis [36, 37].
TIAF1 is essential for cell death caused by tumor suppressors p53 and WWOX and dominant-negative JNK1 [36]. When TIAF1 expression is knocked down by small interfering RNA (siRNA), UV irradiation-mediated p53 activation, via Ser15 phosphorylation and nuclear accumulation, is blocked [36]. Importantly, transiently overexpressed TIAF1, p53 and WWOX work synergistically in suppressing anchorage-independent growth, blocking cell migration, and causing apoptosis in vitro [34]. When one of the components is missing, the apoptotic function of the TIAF1-p53-WWOX trio is significantly reduced, suggesting that the complex is an axis of tumor suppression [34].
p53, WWOX and TIAF1 in the TGF-β signaling and SMAD-responsive promoter activation
p53, WWOX and TIAF1 participate in the pathway of TGF-β/Smad signaling. In the canonical signaling, membrane type II TGF-β receptor (TβRII) is responsible for binding extracellular TGF-β1, β2 or β3, followed by recruiting TβRI and subsequent activation and complex formation of Smad2, 3 and 4. TβRII is a constitutively active serine/threonine kinase. Upon stimulation with TGF-β, TβRII phosphorylates and binds the serine/threonine kinase TβRI. Then, the receptor complex, containing two TβRII and two TβRI, phosphorylates Smad2 and 3 for subsequent recruiting Smad4. The Smad2/3/4 complex translocates to the nucleus to mediate gene transcription [39] (Figure 2). Mutant p53 binds T βRII promoter to block the expression of TβRII [40]. However, wild type p53 and Smad proteins interact with AFP and Mix2 promoters for either enhancing or suppressing the promoter activity [40]. We reported that TGF-β1 binds membrane hyaluronidase Hyal-2, and the binding leads to complex formation of Hyal-2 with WWOX and Smad4 for regulating the SMAD-responsive promoter activation [41] (Figure 2). Excessive SMAD-regulated promoter activation induces cell death [41]. Most recently, we determined that TGF-β1 signals the binding of TIAF1 with Smad4, and the complex relocates to the nucleus to modulate gene transcription governed by Smad4 [35]. Overall, TGF-β1-initiated signal pathways are likely to converge to the complex formation of WWOX, Hyal-2, TIAF1, p53, and Smad4 for regulating the promoter activation governed by SMAD (Figure 2).
Figure 2.
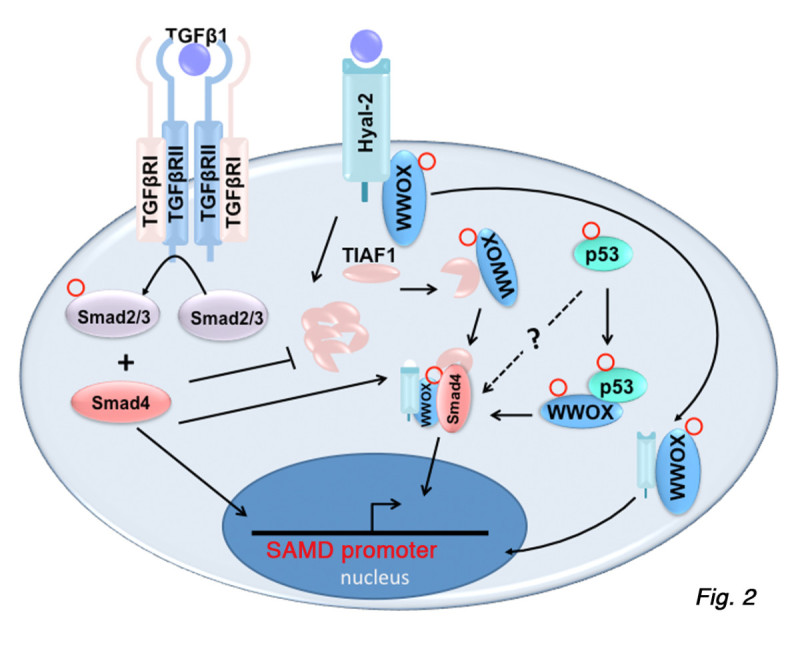
TGF- β1-initiated signal pathways that converge to WWOX, TIAF1, p53, Hyal-2, and Smad4. TGF-β1 initiates several signal pathways: i) In the canonical signaling, extracellular TGF-β1 binds TβRII, followed by recruiting TβRI for subsequent activation and complex formation of Smad2, 3 and 4. The Smad2/3/4 complex mediates gene transcription in the nucleus [39]. ii) Alternatively, TGF-β1 utilizes membrane hyaluronidase Hyal-2 as a receptor for signaling the complex formation of Hyal-2 with WWOX and Smad4 to control SMAD-responsive promoter activation [41]. iii) Additionally, TGF-β1 induces the complex formation of TIAF1 with Smad4 to regulate the SMAD-responsive promoter activation [35]. iv) WWOX binds and prevents p53 degradation and both proteins exert apoptosis synergistically [2, 3]. p53-WWOX-TIAF1 is an axis of tumor suppression [34, 36]. TGF-β1 is likely to converge the TGF-β1-initiated signal pathways to the complex formation of WWOX, TIAF1, p53, Hyal-2, and Smad4. TIAF1 undergoes aggregation during aberrant TGF-β signaling [35]. WWOX: blue; Hyal-2: light blue; TIAF1: pink; p53: light green; Smad4: orange. Red circle: phosphorylation.
TIAF1 binds Smad4 of the TGF-β/Smad signaling, and that transiently overexpressed TIAF1 blocks SMAD-dependent promoter activation [34]. When TIAF1 is knocked down by siRNA, spontaneous activation of Smad proteins occurs as their accumulation in the nucleus [34]. In parallel, when TIAF1 level is low, spontaneous activation of the promoter governed by the SMAD complex occurs [34]. Most intriguingly, in the absence of endogenous p53, TIAF1 undergoes self-aggregation and simultaneously activates the SMAD-regulated promoter [35]. In parallel with the protein expression of WWOX [10], TIAF1 is upregulated in developing tumors, but may disappear in established metastatic cancer cells [34]. As an axis of tumor suppression, the absence of WWOX-p53-TIAF1 trio favors the growth of metastatic cancer cells.
TIAF1 does not directly interact with p53 [34, 36]. However, TIAF1 is needed for UV irradiation-induced p53 activation (with Ser15 phosphorylation) and nuclear accumulation [36]. Without TIAF1, no p53 activation occurs. WWOX physically binds p53 and stabilizes its function [10]. Data from both yeast two-hybrid and Förster resonance energy transfer (FRET) analyses revealed that WWOX acts as a bridge for the indirect interaction between p53 and TIAF1 (Chang et al., unpublished).
TGF-β1 induces the differentiation of regulatory T cells (Treg), and that TIAF1 expression is significantly increased during cell differentiation [42]. TIAF1 is considered as one of the Treg signature proteins [42]. TIAF1 is also involved in allograft rejection, as its level is significantly increased in activated Th2 helper T lymphocytes in patients with chronic kidney and liver allograft rejection [43]. In addition, TIAF1 is associated with Hirschsprung's disease, a congenital complex disorder of intestinal innervation [44].
TGF-β, microenvironment and cell-cell contacts affect TIAF1 aggregation
In response to TGF-β1 or -β2, TIAF1 undergoes self-aggregation in the cultured cells [34]. The event can occur independently of the canonical signaling from TβRII/RI/Smads [34]. TIAF1 aggregation occurs probably as a result of an aberrant TGF-β signaling in cancer cells, as well as in the hippocampus [34, 35]. Also, when cells are exposed to environmental stress, TIAF1 undergoes self-aggregation, which exhibits as a ladder-like protein polymerization pattern in SDS-PAGE [34]. The overly aggregated TIAF1 induces degradation of amyloid precursor protein (APP), super-induction of Aβ, and ultimately cell death in vitro [34]. Caspase inhibitors block the cell death, suggesting that caspases participate in the TIAF1-induced apoptosis.
Microenvironment and cell-cell contacts affect the TGF-β1 induction of endogenous TIAF1 expression and self-aggregation. Also, TGF-β regulates cancer cell and stroma interactions, and that controls the progression of cancer growth [45]. When lung cancer cells are grown on an extracellular matrix (ECM) derived from another cell type, the lung cells have apparently received the environmental stress that leads to the generation of endogenous TIAF1 and Aβ aggregates [34]. No ubiquitination is involved in the TIAF1 polymerization. Intriguingly, the polymerizing TIAF1 increases the expression of tumor suppressors Smad4 and WWOX [34]. And, WWOX in turn upregulates the TIAF1 expression [34]. Smad4 remains a monomer, whereas WWOX may become polymerized as well. Dominant negative TIAF1 is able to block WWOX expression [34].
Furthermore, when fibroblasts and neuroblastoma cells were co-cultured on the ECM of prostate DU145 cells for at least 48 hr, endogenous TIAF1 expression and aggregation were demonstrated in both cells [34]. No TIAF1 aggregate formation was observed by culturing fibroblasts or neuroblastomas alone on the ECM [34]. Additionally, TIAF1 aggregates are present on the interface between metastatic lung cancer cells and the brain cells in vivo [34]. Thus, when two distinct types of cells encounter each other in an unfavorable microenvironment, endogenous TIAF1 aggregation occurs.
TIAF1 aggregation occurs in tumor progression and metastasis in vivo
TIAF1 participates in the initiation of Alzheimer’s disease [35]. We have determined that hippocampal TIAF1 aggregation is shown at ages 40–70, and this event occurs prior to the generation of Aβ plaques in Alzheimer’s disease at 75–90 years old [35]. In parallel with this finding, in vitro analysis showed that aggregating TIAF1 causes APP degradation and generation of Aβ [35]. Whether aggregating TIAF1 induces caspase activation for leading to APP degradation is unknown. Upregulation of TIAF1 is also shown during cancer progression and metastasis [34]. When metastatic U87-MG glioma cells, for instance, were subcutaneously inoculated in both flanks of nude mice, the cells were metastatic to the lung. Expression of TIAF1 and amyloid fibrils is shown in the growing solid tumor (Figure 3).
Figure 3.
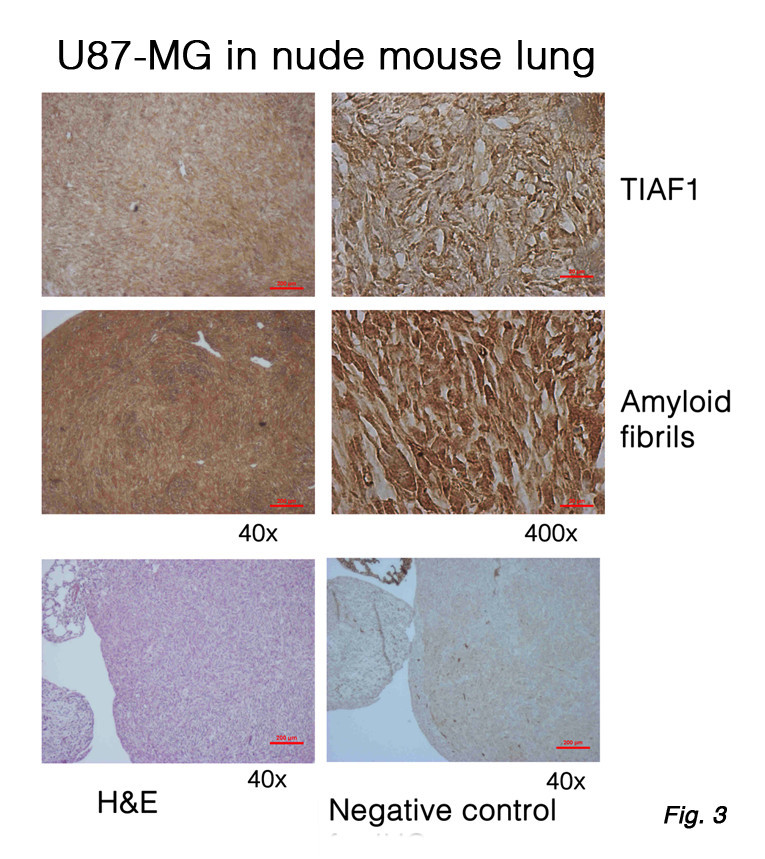
Expression of TIAF1 and amyloid fibrils in U87-MG glioma cells in the lung. U87-MG glioma cells were inoculated in both flanks of nude mice. Two months later, the mice were sacrificed. Shown is the U87-MG cell metastatic to the lung. Each solid tumor lesion has increased expression of TIAF1 and amyloid fibrils, compared to normal mice (data not shown). Both proteins appear to localize intracellularly. A homemade TIAF1 antibody and commercial amyloid fibril antibody were used, as described [34, 35].
TIAF1 aggregates in the metastatic lung cancer cells in the brain
It appears that cancer cells could tolerate more microenvironmental stress than normal cells, as cancer cells tend to utilize local or autocrine TGF-β in building up their own niche. Conceivably, upon reaching and selectively attaching to the docking site in the brain, a single metastatic lung cancer cell must face the challenge of survival. The cell needs to divide and proliferate quickly, and build itself own niche by utilizing available sources from the brain tissue or its own TGF-β. The concern is whether the brain tissue recognizes the cancer cell as foreign. The local glial cells or immune cells may come to attack the invading cancer cell. A successfully built solid tumor normally possesses a peritumor capsule. For example, when metastatic small-cell lung cancer cells migrate to the brain, TIAF1 and Aβ aggregates are shown on the border between cancer and brain cells. Or, the protein aggregates are present in the tumor mass [34].
During solid lung tumor formation in the brain, ECM proteins continue to accumulate and they can organize orderly in the matrix or become aggregated in an irregular manner [34]. The ECM aggregates are located inside the tumor mass, or may be present on the borderline between brain cells and the metastatic lung cancer [34]. Quite frequently, TIAFI and Aβ are present in these protein aggregates. Self-aggregation of TIAF1 has been shown to induce phosphorylation of APP, followed by generation of Aβ and amyloid fibrils [35]. The resulting Aβ fails to cause cancer cell death, whereas it is likely to make damage to neurons or initiation of neurodegeneration. Indeed, no apoptotic lung cancer cells have been shown within the peritumor protein aggregates [34], suggesting that these protein aggregates act as a “protective shield” for the progression of lung cancer cells in the brain.
Intriguingly, when metastatic cancer cells have well established their niche, intracellular TIAF1 disappears significantly in the tumor masses [34]. However, TIAF1, Smad4, Aβ, and many other proteins are components of the fibrous aggregates in the peritumor capsules. The extracellular aggregated TIAF1 and Smad4 are probably functionally inactive.
Concluding remarks
TGF-β1 induces TIAF1 self-aggregation, and the aggregating proteins bind Smad4 and cause Aβ generation. These protein aggregates are likely to induce apoptosis via activation of caspases, as caspase inhibitors can block the cell death [35]. However, cancer cells appear to successfully survive from the stress. The TIAF1-Smad4-Aβ protein aggregates are frequently seen in the peritumor capsules, which are crucial for solid tumor growth and protection. However, the relative concentrations of TIAF1 and Smad4 must be balanced. If cells have a greater amount of TIAF1 than Smad4, they survive upon challenge with TGF-β1. However, when cells express a greater amount of Smad4 than TIAF1, they are highly sensitive to TGF-β1-induced apoptosis [35].
During cancer progression, the expression profiles for WWOX and TIAF1 appear similarly. Both proteins are significantly upregulated during the early phases of benign tumor formation. When tumors acquire their metastatic potential, both proteins are dramatically reduced. WWOX induces the expression of TIAF1, and enhances the transcriptional activation of promoters governed by SMAD and NF-κB [34, 35]. TIAF1 in turn induces the expression of Smad4 and Aβ, depending upon the status of TIAF1 self-aggregation. Intriguingly, under p53-free environment, TIAF1 starts to undergo self-aggregation and the aggregating TIAF1 polymer activates the SMAD-governed promoter [35].
TIAF1 gene participates in human embryonic stem cell self-renewal and pluripotency [46], and is also involved in melanogenesis [47] and genome stability [48]. Given its potential role in cancer progression, TIAF1 can be an ideal target for therapeutic drugs. Transiently overexpressed TIAF1 induces aggregation of monocytic cells and other cancer cells in vitro [37] (Figure 4). It appears that the overexpressed cell surface TIAF1 serves as a molecular glue, plus integrins and matrix proteins, to allow cancer cells to adhere to each other. Thus, it is reasonable to postulate that blocking TIAF1 expression by appropriate chemicals, siRNA, or dominant negative TIAF1 could result in suppression of lung cancer stem cell growth and progression.
Figure 4.
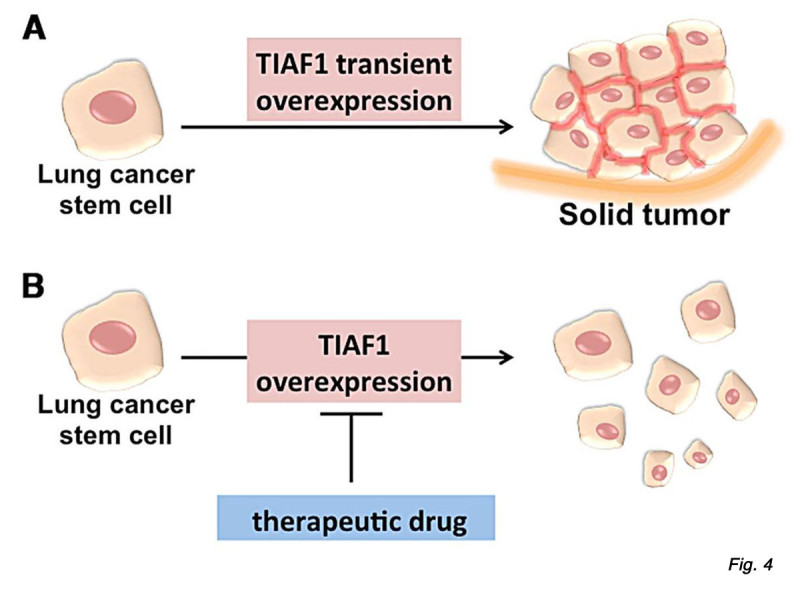
TIAF1 is a potential target for lung cancer therapy. A. When cancer cells are transiently overexpressed with TIAF1, a portion of the expressed TIAF1 localizes on the cell membrane/cytoskeleton area or intercellular junctions (marked in red) [37], which probably assists cells to stick to each other that leads to solid tumor formation. TIAF1 is associated with stem cell pluripotency [46]. It is reasonable to postulate that when a lung cancer stem cell starts to grow, it probably needs the expression of TIAF1 to allow cells to stick to each other in order to proliferate. B. Therefore, blocking TIAF1 expression by appropriate chemicals, siRNA, or dominant negatives could result in suppression of lung cancer stem cell proliferation.
Acknowledgements
This research was supported, in part, by the Department of Defense USA (DAMD17-03-1-0736 and W81XWH-08-1-0682), the Research the National Science Council, Taiwan, ROC (NSC96-2320-B-006-014, 98-2628-B-006-045-MY3, 98-2628-B-006-041-MY3, and 99-2320-B-006-012-MY3), the National Health Research Institute, Taiwan, ROC (NHRI-EX99-9704BI), the National Cheng Kung University Landmark Projects (C0167) and the Department of Health, Taiwan, ROC (DOH101-TD-PB-111-TM010) (to NS Chang).
Abbreviations
- TGF-β
Transforming growth factor beta
- TIAF1
TGF-β-induced antiapoptotic factor
- WWOX
WW domain-containing oxidoreductase
- Smad4
Mothers against decapentaplegic homolog 4
- MCT-1
Oncogenic monocarboxylic acid transporter 1
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Mdm2
p53 E3 ubiquitin protein ligase homolog
- Mdmx
Mdm4 p53 binding protein homolog
- JNK1
cJun N-terminal kinase
- Zfra
Zinc finger-like protein that regulates apoptosis
- NSCLC
Non-small-cell lung carcinomas
- SCLC
Small-cell lung cancer
- CXCR4
Chemokine (C-X-C motif) receptor 4
- SDF-1CXCL21
Chemokine stromal derived factor-1/chemokine (C-X-C motif) ligand 12
- ECM
Extracellular matrix
- Aβ
Amyloid beta
- APP
Amyloid precursor protein
- siRNA
Small interfering RNA
- TREG
Regulatory T cells
- TβRII
Type II TGF-β receptor
- TNF
Tumor necrosis factor.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Footnotes
Qunying Hong, Li-Jin Hsu, Pei-Yi Chou contributed equally to this work.
Competing interests
The authors declare no conflict of interest.
Authors’ contributions
QH, LJH, and YAC: conceived ideas and proof reading; YAC and CYL: worked on graphic arts and immunohistochemistry; YTC, prepared tissue sections; NSC: wrote the manuscript, conceived ideas, and performed microscopy. All authors read and approved the final manuscript.
Contributor Information
Qunying Hong, Email: qyhong68@hotmail.com.
Li-Jin Hsu, Email: lijin.hsu@gmail.com.
Pei-Yi Chou, Email: 3695409@nckualumni.org.tw.
Ying-Tsen Chou, Email: 3695409@nckualumni.org.tw.
Chen-Yu Lu, Email: jllsh99@hotmail.com.
Yu-An Chen, Email: momofish4@hotmail.com.
Nan-Shan Chang, Email: nschang13827@gmail.com.
References
- 1.Yang J, Zhang W. WWOX tumor suppressor gene. Histol Histopathol. 2008;23:877–882. doi: 10.14670/HH-23.877. [DOI] [PubMed] [Google Scholar]
- 2.Chang NS. A potential role of p53 and WOX1 in mitochondrial apoptosis (review) Int J Mol Med. 2002;9:19–24. [PubMed] [Google Scholar]
- 3.Chang NS, Hsu LJ, Lin YS, Lai FJ, Sheu HM. WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol Med. 2007;13:12–22. doi: 10.1016/j.molmed.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Chang NS, Schultz L, Hsu LJ, Lewis J, Su M, Sze CI. 17beta-Estradiol upregulates and activates WOX1/WWOXv1 and WOX2/WWOXv2 in vitro: potential role in cancerous progression of breast and prostate to a premetastatic state in vivo. Oncogene. 2005;24:714–723. doi: 10.1038/sj.onc.1208124. [DOI] [PubMed] [Google Scholar]
- 5.Su WP, Chen SH, Chen SJ, Chou PY, Huang CC, Chang NS. WW Domain-Containing Oxidoreductase is a potential receptor for sex steroid hormones. In: Raghvendra D, editor. Sex Hormones. Crotia: InTech; 2012. pp. 333–352. [Google Scholar]
- 6.Li MY, Lai FJ, Hsu LJ, Lo CP, Cheng CL, Lin SR, Lee MH, Chang JY, Subhan D, Tsai MS, Sze CI, Pugazhenthi S, Chang NS, Chen ST. Dramatic co-activation of WWOX/WOX1 with CREB and NF-kappaB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS One. 2009;4:e7820. doi: 10.1371/journal.pone.0007820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J Biomed Biotechnol. 2012;2012:170325. doi: 10.1155/2012/170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476:163–169. doi: 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe A, Hippo Y, Taniguchi H, Iwanari H, Yashiro M, Hirakawa K, Kodama T, Aburatani H. An opposing view on WWOX protein function as a tumor suppressor. Cancer Res. 2003;63:8629–8633. [PubMed] [Google Scholar]
- 10.Lai FJ, Cheng CL, Chen ST, Wu CH, Hsu LJ, Lee JY, Chao SC, Sheen MC, Shen CL, Chang NS, Sheu HM. WOX1 is essential for UVB irradiation-induced apoptosis and down-regulated via translational blockade in UVB-induced cutaneous squamous cell carcinoma in vivo. Clin Cancer Res. 2005;11:5769–5777. doi: 10.1158/1078-0432.CCR-04-2274. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Fujita T, Zhang D, Doan H, Pinkaew D, Liu Z, Wu J, Koide Y, Chiu A, Lin CC, Chang JY, Ruan KH, Fujise K. Physical and functional antagonism between tumor suppressor protein p53 and fortilin, an anti-apoptotic protein. J Biol Chem. 2011;286:32575–32585. doi: 10.1074/jbc.M110.217836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Zhang D, Fujise K. Characterization of fortilin, a novel antiapoptotic protein. J Biol Chem. 2001;276:47542–47549. doi: 10.1074/jbc.M108954200. [DOI] [PubMed] [Google Scholar]
- 13.Kasiappan R, Shih HJ, Wu MH, Choy C, Lin TD, Chen L, Hsu HL. The antagonism between MCT-1 and p53 affects the tumorigenic outcomes. Mol Cancer. 2010;9:311. doi: 10.1186/1476-4598-9-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ak P, Levine AJ. p53 and NF-κB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24:3643–5362. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 15.Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012;3:228–235. doi: 10.18632/oncotarget.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang NS, Doherty J, Ensign A, Schultz L, Hsu LJ, Hong Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J Biol Chem. 2005;280:43100–43108. doi: 10.1074/jbc.M505590200. [DOI] [PubMed] [Google Scholar]
- 17.Chang NS, Doherty J, Ensign A. JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J Biol Chem. 2003;278:9195–9202. doi: 10.1074/jbc.M208373200. [DOI] [PubMed] [Google Scholar]
- 18.Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, Schultz L, Chang NS. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer's disease. J Biol Chem. 2004;279:30498–30506. doi: 10.1074/jbc.M401399200. [DOI] [PubMed] [Google Scholar]
- 19.Aderca I, Moser CD, Veerasamy M, Bani-Hani AH, Bonilla-Guerrero R, Ahmed K, Shire A, Cazanave SC, Montoya DP, Mettler TA, Burgart LJ, Nagorney DM, Thibodeau SN, Cunningham JM, Lai JP, Roberts LR. The JNK inhibitor SP600129 enhances apoptosis of HCC cells induced by the tumor suppressor WWOX. J Hepatol. 2008;49:373–383. doi: 10.1016/j.jhep.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jamshidiha M, Habibollahi P, Ostad SN, Ghahremani MH. Primary WWOX phosphorylation and JNK activation during etoposide induced cytotoxicity in HEK293 cells. Daru. 2010;18:141–145. [PMC free article] [PubMed] [Google Scholar]
- 21.Dudekula S, Lee MH, Hsu LJ, Chen SJ, Chang NS. Zfra is a small wizard in the mitochondrial apoptosis. Aging (Albany NY) 2010;2:1023–1029. doi: 10.18632/aging.100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong Q, Hsu LJ, Schultz L, Pratt N, Mattison J, Chang NS. Zfra affects TNF-mediated cell death by interacting with death domain protein TRADD and negatively regulates the activation of NF-kappaB, JNK1, p53 and WOX1 during stress response. BMC Mol Biol. 2007;8:50. doi: 10.1186/1471-2199-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet. 2007;16:R88–R95. doi: 10.1093/hmg/ddm051. [DOI] [PubMed] [Google Scholar]
- 24.Palii SS, Robertson KD. Epigenetic control of tumor suppression. Crit Rev Eukaryot Gene Expr. 2007;17:295–316. doi: 10.1615/CritRevEukarGeneExpr.v17.i4.40. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama S, Semba S, Maeda N, Matsushita M, Kuroda Y, Yokozaki H. Hypermethylation-mediated reduction of WWOX expression in intraductal papillary mucinous neoplasms of the pancreas. Br J Cancer. 2009;100:1438–1443. doi: 10.1038/sj.bjc.6604986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iliopoulos D, Guler G, Han SY, Johnston D, Druck T, McCorkell KA, Palazzo J, McCue PA, Baffa R, Huebner K. Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene. 2005;24:1625–1633. doi: 10.1038/sj.onc.1208398. [DOI] [PubMed] [Google Scholar]
- 27.Gourley C, Paige AJ, Taylor KJ, Ward C, Kuske B, Zhang J, Sun M, Janczar S, Harrison DJ, Muir M, Smyth JF, Gabra H. WWOX gene expression abolishes ovarian cancer tumorigenicity in vivo and decreases attachment to fibronectin via integrin alpha3. Cancer Res. 2009;69:4835–4842. doi: 10.1158/0008-5472.CAN-08-2974. [DOI] [PubMed] [Google Scholar]
- 28.Talbot LJ, Bhattacharya SD, Kuo PC. Epithelial-mesenchymal transition, the tumor microenvironment, and metastatic behavior of epithelial malignancies. Int J Biochem Mol Biol. 2012;3:117–136. [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 30.Han N, Dol Z, Vasieva O, Hyde R, Liloglou T, Raji O, Brambilla E, Brambilla C, Martinet Y, Sozzi G, Risch A, Montuenga LM, Brass A, Field JK, Brass A, Field JK, The EUELC Consortium Progressive lung cancer determined by expression profiling and transcriptional regulation. Int J Oncol. 2012;41:242–252. doi: 10.3892/ijo.2012.1421. [DOI] [PubMed] [Google Scholar]
- 31.Gangadhar T, Nandi S, Salgia R. The role of chemokine receptor CXCR4 in lung cancer. Cancer Biol Ther. 2010;9:409–416. doi: 10.4161/cbt.9.6.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raffaghello L, Cocco C, Corrias MV, Airoldi I, Pistoia V. Chemokines in neuroectodermal tumour progression and metastasis. Semin Cancer Biol. 2009;19:97–102. doi: 10.1016/j.semcancer.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Hartmann TN, Burger M, Burger JA. The role of adhesion molecules and chemokine receptor CXCR4 (CD184) in small cell lung cancer. J Biol Regul Homeost Agents. 2004;18:126–130. [PubMed] [Google Scholar]
- 34.Chang JY, Chiang MF, Lin SR, Lee MH, He H, Chou PY, Chen SJ, Chen YA, Yang LY, Lai FJ, Hsieh CC, Hsieh TH, Sheu HM, Sze CI, Chang NS. TIAF1 self-aggregation in peritumor capsule formation, spontaneous activation of SMAD-responsive promoter in p53-deficient environment, and cell death. Cell Death Dis. 2012;3:e302. doi: 10.1038/cddis.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MH, Lin SR, Chang JY, Schultz L, Heath J, Hsu LJ, Kuo YM, Hong Q, Chiang MF, Gong CX, Sze CI, Chang NS. TGF-β induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid β plaques in Alzheimer's disease. Cell Death Dis. 2010;1:e110. doi: 10.1038/cddis.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schultz L, Khera S, Sleve D, Heath J, Chang NS. TIAF1 and p53 functionally interact in mediating apoptosis and silencing of TIAF1 abolishes nuclear translocation of serine 15-phosphorylated p53. DNA Cell Biol. 2004;23:67–74. doi: 10.1089/104454904322745943. [DOI] [PubMed] [Google Scholar]
- 37.Khera S, Chang NS. TIAF1 participates in the transforming growth factor beta1–mediated growth regulation. Ann N Y Acad Sci. 2003;995:11–21. doi: 10.1111/j.1749-6632.2003.tb03206.x. [DOI] [PubMed] [Google Scholar]
- 38.Chang NS, Mattison J, Cao H, Pratt N, Zhao Y, Lee C. Cloning and characterization of a novel transforming growth factor-beta1-induced TIAF1 protein that inhibits tumor necrosis factor cytotoxicity. Biochem Biophys Res Commun. 1998;253:743–749. doi: 10.1006/bbrc.1998.9846. [DOI] [PubMed] [Google Scholar]
- 39.Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 40.Atfi A, Baron R. p53 brings a new twist to the Smad signaling network. Sci Signal. 2008;1:pe33. doi: 10.1126/scisignal.126pe33. [DOI] [PubMed] [Google Scholar]
- 41.Hsu LJ, Schultz L, Hong Q, Van Moer K, Heath J, Li MY, Lai FJ, Lin SR, Lee MH, Lo CP, Lin YS, Chen ST, Chang NS. Transforming growth factor beta1 signaling via interaction with cell surface Hyal-2 and recruitment of WWOX/WOX1. J Biol Chem. 2009;284:16049–16059. doi: 10.1074/jbc.M806688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfoertner S, Jeron A, Probst-Kepper M, Guzman CA, Hansen W, Westendorf AM, Toepfer T, Schrader AJ, Franzke A, Buer J, Geffers R. Signatures of human regulatory T cells: an encounter with old friends and new players. Genome Biol. 2006;7:R54. doi: 10.1186/gb-2006-7-7-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Leij J, van den Berg A, Albrecht EW, Blokzijl T, Roozendaal R, Gouw AS, de Jong KP, Stegeman CA, van Goor H, Chang NS, Poppema S. High expression of TIAF-1 in chronic kidney and liver allograft rejection and in activated T-helper cells. Transplantation. 2003;75:2076–2082. doi: 10.1097/01.TP.0000069829.71088.88. [DOI] [PubMed] [Google Scholar]
- 44.Griseri P, Vos Y, Giorda R, Gimelli S, Beri S, Santamaria G, Mognato G, Hofstra RM, Gimelli G, Ceccherini I. Complex pathogenesis of Hirschsprung's disease in a patient with hydrocephalus, vesico-ureteral reflux and a balanced translocation t(3;17)(p12;q11) Eur J Hum Genet. 2009;17:483–490. doi: 10.1038/ejhg.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology. 2010;51:523–534. doi: 10.1002/hep.23285. [DOI] [PubMed] [Google Scholar]
- 46.Chia NY, Chan YS, Feng B, Lu X, Orlov YL, Moreau D, Kumar P, Yang L, Jiang J, Lau MS, Huss M, Soh BS, Kraus P, Li P, Lufkin T, Lim B, Clarke ND, Bard F, Ng HH. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature. 2010;468:316–320. doi: 10.1038/nature09531. [DOI] [PubMed] [Google Scholar]
- 47.Ganesan AK, Ho H, Bodemann B, Petersen S, Aruri J, Koshy S, Richardson Z, Le LQ, Krasieva T, Roth MG, Farmer P, White MA. Genome-wide siRNA-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS Genet. 2008;4:e1000298. doi: 10.1371/journal.pgen.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, Meyer T, Cimprich KA. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]