

Abstract
Objective
The mechanism by which G-protein-coupled receptor 40 (GPR40) signaling amplifies glucose-stimulated insulin secretion through activation of protein kinase C (PKC) is unknown. We examined whether a GPR40 agonist, GW9508, could stimulate conventional and novel isoforms of PKC at two glucose concentrations (3 mM and 20 mM) in INS-1D cells.
Methods
Using epifluorescence microscopy, we monitored relative changes in the cytosolic fluorescence intensity of Fura2 as a marker of change in intracellular Ca2+ ([Ca2+]i) and relative increases in green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) as a marker of PKC activation in response to GW9508 at 3 mM and 20 mM glucose. To assess the activation of the two PKC isoforms, relative increases in membrane fluorescence intensity of PKCα-GFP and PKCε-GFP were measured by total internal reflection fluorescence microscopy. Specific inhibitors of each PKC isotype were constructed and synthesized as peptide fusions with the third α-helix of the homeodomain of Antennapedia.
Results
At 3 mM glucose, GW9508 induced sustained MARCKS-GFP translocation to the cytosol, irrespective of changes in [Ca2+]i. At 20 mM glucose, GW9508 induced sustained MARCKS-GFP translocation but also transient translocation that followed sharp increases in [Ca2+]i. Although PKCα translocation was rarely observed, PKCε translocation to the plasma membrane was sustained by GW9508 at 3 mM glucose. At 20 mM glucose, GW9508 induced transient translocation of PKCα and sustained translocation as well as transient translocation of PKCε. While the inhibitors (75 μM) of each PKC isotype reduced GW9508-potentiated, glucose-stimulated insulin secretion in INS-1D cells, the PKCε inhibitor had a more potent effect.
Conclusion
GW9508 activated PKCε but not PKCα at a substimulatory concentration of glucose. Both PKC isotypes were activated at a stimulatory concentration of glucose and contributed to glucose-stimulated insulin secretion in insulin-producing cells.
Introduction
Diabetes is reaching pandemic proportions and is expected to affect over 592 million people worldwide by 2035 [1]. The most common form of this disease, type 2 diabetes, develops when pancreatic β-cells are unable to secrete an adequate amount of insulin to compensate for peripheral insulin resistance [2]. Insulin secretion is regulated chiefly by glucose through intracellular metabolism and is also modulated by non-glucose secretagogues, such as incretin hormones and fatty acids, and muscarinic stimulation via G protein-coupled receptors (GPCRs) [3]. Because activation of these receptors augments insulin secretion only when glucose levels are elevated, they can be targeted therapeutically to raise insulin levels with minimal risk of iatrogenic hypoglycemia.
Glucagon-like peptide-1 (GLP-1) potentiates glucose-stimulated insulin secretion (GSIS) by binding to its GPCRs in β-cells [4,5]. GLP-1 receptor agonists are clinically available and pose little risk of iatrogenic hypoglycemia. GLP-1 potentiates GSIS by increasing cyclic adenosine monophosphate (cAMP) levels, leading to activation of protein kinase A (PKA) [6]. Conversely, in our previous study and a recent report from another group, GLP-1 also stimulated insulin secretion by activating protein kinase C (PKC) [7,8].
Ten isoforms of PKC have been identified and divided into three major classes, based on their requirements for activators. Conventional PKC (cPKC; PKCα, PKCβI, PKCβII, and PKCγ) is activated by both Ca2+ and diacylglycerol (DAG); novel PKC (nPKC; PKCδ, PKCε, PKCη, and PKCθ) requires only DAG; and atypical PKC (aPKC; PKCζ and PKCλ) binds neither Ca2+ nor DAG [9]. DAG, which is essential for the activation of cPKC and nPKC, is mainly derived from the hydrolysis of plasma membrane phosphatidylinositol 4,5-bisphosphate (PIP2). This hydrolysis is due to activation of phospholipase C (PLC), following the binding of an agonist to a GPCR. Previously, we showed that GLP-1 activated an nPKC (PKCε) predominantly via PLC activation, rather than a cPKC (PKCα) in INS-1 cells, an insulin-secreting cell line [7]. However, it was insufficient to assess how PLC/PKC signaling alone was involved in GSIS, because GLP-1 must activate not only PLC/PKC signaling but also cAMP/PKA signaling.
G-protein-coupled receptor 40 (GPR40) belongs to the free fatty acid receptor family [10–12]. Several reports have shown that GPR40 couples mainly to Gαq/Gα11, which activates PLC, resulting in the formation of inositol 1,4,5-trisphosphate (IP3) and DAG via PIP2 hydrolysis [13]. IP3 induces Ca2+ release from the endoplasmic reticulum (ER), and DAG activates PKC [14]. The natural ligands of GPR40 include medium- and long-chain free fatty acids [15,16]. GPR40 is preferentially expressed in pancreatic islets and β-cell lines [15,17], and its expression in human pancreatic islets appears to be higher than that of sulfonylurea receptor type 1 (SUR1) and GLP-1 receptor (GLP-1R) [17,18]. GPR40 contributes to approximately half of the insulin secretory response to fatty acids [19]. Activation of GPR40 significantly enhances GSIS from rodent pancreatic islets and β-cell lines [15,20–23]. However, reports have never been clear that PKC is involved in the increase in GSIS via GPR40 signaling, nor has GPR40-mediated PKC activation been examined at the isoform level.
We evaluated the role of the PKC pathway in the enhancement of GSIS by GPR40 activation. To analyze this, we chose GW9508, a selective and potent small-molecule agonist of GPR40 [21]. Among the multiple PKC isoforms that are expressed in pancreatic β-cells, PKCα and PKCε are likely to have dominant functions in GSIS [24,25]. The roles of these two proteins in GW9508-potentiated GSIS were also determined. To minimize the interference of glucose against GW9508-induced signal transduction, we conducted this study in INS-1 cells, which secrete less insulin in response to glucose stimulation than primary β-cells.
Materials and methods
Plasmid construction
PKCα-pEGFP was obtained from Clontech (Palo Alto, CA, USA). The plasmids encoding PKCε-GFP and MARCKS-GFP were prepared as described previously [26,27].
Cell culture and transfection
INS-1D cells were a gift from Dr. Sekine (Tokyo University) [28]. The cells were grown in 60-mm culture dishes at 37°C and 5% CO2 in a humidified atmosphere. The culture medium was RPMI 1640 (Sigma, St. Louis, MO, USA) supplemented with 10 mM glucose, 10% fetal bovine serum, 1 mM sodium pyruvate, 1 mM L-glutamine, and 50 μM 2-mercaptoethanol. For fluorescence imaging, the cells were cultured in a 35-mm glass-bottom dish (AGC Techno Glass Co., Ltd., Shizuoka, Japan) at 50% confluence 2 days before transfection. A plasmid encoding the GFP-tagged proteins was transfected into the cells using Lipofectamine 2000 (Invitrogen, Burlington, ON, Canada). Experiments were performed within 2 days of transient transfection. We established stable transfectants from parental INS-1 cells expressing myristoylated alanine-rich C kinase substrate (MARCKS)-GFP or PKCα-GFP by G418 selection and cloning.
Solutions
The standard extracellular solution contained 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 3 mM glucose, and 10 mM Hepes-NaOH (pH 7.3). The solution for membrane depolarization contained 105 mM NaCl, 40 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 3 mM glucose, and 10 mM Hepes-NaOH (pH 7.3). For the high-glucose condition, 20 mM glucose replaced 3 mM glucose in the extracellular solution. The cells, placed in a glass-bottom dish, were perfused continuously from a gravity-fed system. Experiments were performed in the standard extracellular solution at room temperature, unless otherwise noted. Krebs-Ringer buffer (KRB) contained 119 mM NaCl, 4.6 mM KCl, 1 mM MgSO4, 0.15 mM Na2HPO4, 0.4 mM KH2PO4, 25 mM NaHCO3, 2 mM CaCl2, and 20 mM Hepes-NaOH (pH 7.3).
Reagents
GW9508 was purchased from Focus Biomolecules (Plymouth Meeting, PA, USA). Gö 6976 and bisindolylmaleimide I (BIS I) were from Calbiochem (La Jolla, CA, USA). Fura2 (acetoxymethyl form, AM) was obtained from Invitrogen. All other chemicals were acquired from Sigma. A PKCα inhibitory peptide, antennapedia-PKC-(19–31) (antp-PKCα; RRMKW KKRFA RKGAL RQKNV), and a PKCε inhibitory peptide, antennapedia-PKC-(149–164) (antp-PKCε; RRMKW KKERM RPRKR QGAVR RRV), were synthesized by Medical and Biological Laboratories Co., Ltd. (Nagoya, Japan). These inhibitory peptides are synthesized in tandem and comprise amino acids from the third α-helix of the homeodomain of Antennapedia (antp; residues 52–58, which regulate penetration) [29][30] and the PKCα (residues 19–31) or PKCε (residues 149–164) pseudosubstrate peptides [31][32]. Antennapedia, used as a control, was also synthesized by Medical and Biological Laboratories Co., Ltd.
Imaging experiments
Epifluorescence microscopy
Fluorescence images were captured at 5-s intervals using a Nikon inverted microscope (60×/1.45 numerical aperture oil immersion objective) that was equipped with a cooled (−85°C) charge-coupled-device digital camera, and recorded and analyzed on a NIS-Elements imaging station (Nikon Corporation, Tokyo, Japan). The excitation light source was a 150-watt xenon lamp with a high-speed scanning polychromatic light source. GFP fluorescence was excited at 488 nm, and the emitted light was collected through a 535/45-nm bandpass filter with a 505-nm dichroic mirror. We measured the fluorescence intensity of the GFP-tagged proteins in the cytosol, excluding the nucleus, as markers of translocation. These values (F) were normalized to each initial value (F0), and the relative fluorescence change was referred to as F/F0. The cells expressing GFP-tagged proteins were loaded with 2 μM Fura2 for the measurement of intracellular Ca2+ concentration [Ca2+]i in the standard extracellular solution for 30 min at room temperature. The cells were washed twice and used within 2 h. Fura2 was excited at wavelengths alternating between 340 and 380 nm, and emissions were collected using the same bandpass filter used for the GFP fluorescence. A shortpass filter of 330–495 nm was used to reduce the background fluorescence between the dichroic mirror and the emission filter, which allowed for simultaneous measurements of GFP and Fura2 fluorescence. We previously determined that GFP and Ca2+ signals were distinguishable under these experimental conditions [26].
Total internal reflection fluorescence microscopy, or evanescent wave microscopy
To obtain a high signal-to-noise ratio as compared with conventional epifluorescence microscopy, we installed a total internal reflection fluorescence microscopy (TIRFM) unit (Olympus Corp., Tokyo, Japan) on an Olympus inverted microscope (60x/1.45 numerical aperture oil immersion objective) that was equipped with an automatic focus device (ZDC2) and a digital complementary metal oxide semiconductor (CMOS) camera (ORCA-Flash4.0, C11440, Hamamatsu Photonics, Hamamatsu, Japan). Incidental light was introduced from the objective lens for TIRFM to generate the electromagnetic zone, or the so-called “evanescent field.” The evanescent wave selectively excites fluorophores within 100 nm of the glass–water interface, which enabled us to monitor fluorescent proteins at and beneath the plasma membrane of a cell. GFP was excited by a 488-nm laser with a 1.3 neutral density filter (Edmond Optics, Tokyo, Japan), and emissions were collected through a 520/35-nm bandpass filter (Semrock, Rochester, NY, USA). HCIMAGE software (Hamamatsu Photonics) was used to capture fluorescence images. The fluorescence intensity of a range of interest (ROI) in individual cells was measured and analyzed on an Aquacosmos imaging station (Hamamatsu Photonics).
Measurement of insulin secretion
Insulin secretion from INS-1D cells was measured in a static incubation system as described previously [33]. INS-1 cells were subcultured in 35-mm dishes and grown to 80–90% confluence for 3–4 days. INS-1 cells were preincubated in KRB buffer containing 3 mM glucose at 37°C in a humidified incubator for 1 h. The solution was then replaced with KRB alone or KRB containing various test agents. Antennapedia, antp-PKCα, and antp-PKCε were added 1 h prior to the insulin secretion experiment. The stimulation time was carefully adjusted to standardize the time required for solution changes and sample collection. The experiments were terminated by withdrawing the supernatant solution after 1 h of incubation. The supernatant was then placed in an ice bath. Samples were kept at −20°C until further analysis. Insulin concentration was measured using an insulin enzyme-linked immunosorbent assay kit (Morinaga Institute of Biological Science, Kanagawa, Japan). All samples were assayed in triplicate.
Statistical analysis
Data are given as means ± standard error. Statistical significance was evaluated using student’s t-test for paired observations. Multiple comparisons were examined by one-way analysis of variance with post hoc Fisher’s LSD test. A p value < 0.05 was considered to be statistically significant. Data were analyzed using BellCurve for Excel (Social Survey Research Information Co., Ltd., Tokyo, Japan).
Results
GW9508 enhances glucose-stimulated insulin secretion from INS-1D cells
First, we examined GSIS from INS-1D cells in the presence or absence of GW9508. As expected, 10 μM GW9508 enhanced insulin secretion at a stimulatory concentration (20 mM) of glucose (Fig 1). At the substimulatory concentration (3 mM) of glucose, GW9508 did not significantly increase insulin secretion (Fig 1).
Fig 1. GW9508 enhances insulin secretion only at 20 mM glucose in INS-1D cells.
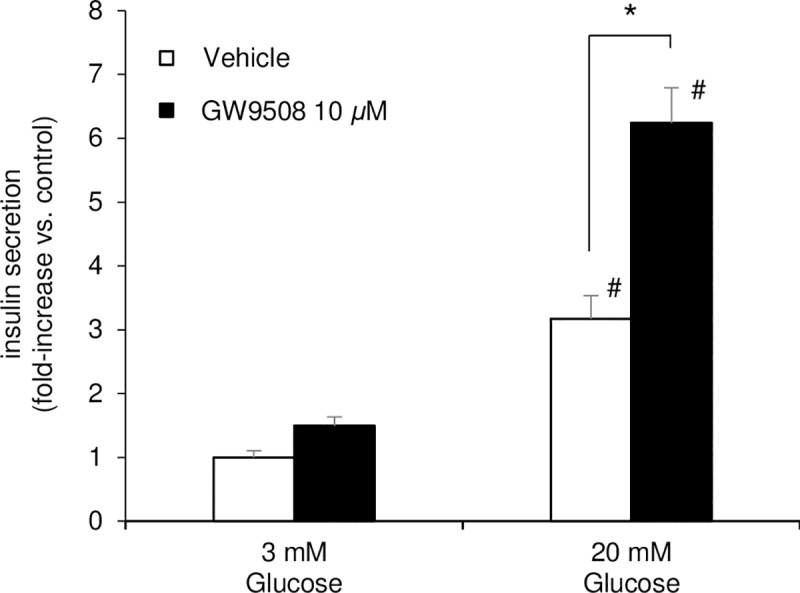
INS-1D cells were incubated for 1 h with Krebs-Ringer buffer (KRB) containing 3 mM or 20 mM glucose in the presence or absence of GW9508, an agonist of G-protein coupled receptor 40 (GPR40). Data shown are mean ± standard error of the mean of three independent experiments with triplicate samples in each group. *p < 0.05 vs. absence of GW9508; #p < 0.05 vs. 3 mM glucose.
GW9508 translocates MARCKS-GFP from the plasma membrane to the cytosol at a substimulatory concentration of glucose
We used GFP-tagged MARCKS, a putative substrate for PKC [34], as a marker of PKC activation to examine the mechanism of GW9508 activation of PKC in living cells. When activated PKC phosphorylates plasma membrane-anchored MARCKS, the phosphorylated MARCKS is translocated from the plasma membrane to the cytosol [35]. This translocation can be identified by reciprocal changes in the fluorescence intensity of MARCKS-GFP between the cytosol and the plasma membrane [26]. Thus, we measured the relative fluorescence change in MARCKS-GFP in the cytosol. MARCKS translocation and [Ca2+]i levels in INS-1 cells stably expressing MARCKS-GFP were monitored simultaneously. To reduce the effects of glucose on GW9508-induced signal transduction as much as possible, we used a standard extracellular solution containing 3 mM glucose, which is substimulatory in terms of electrical activity and insulin secretion. These conditions were used to evaluate PKC activation by GW9508 in real time; Fig 2 shows a representative experiment (n = 81).
Fig 2. GW9508 induces sustained MARCKS-GFP translocation at 3 mM glucose.
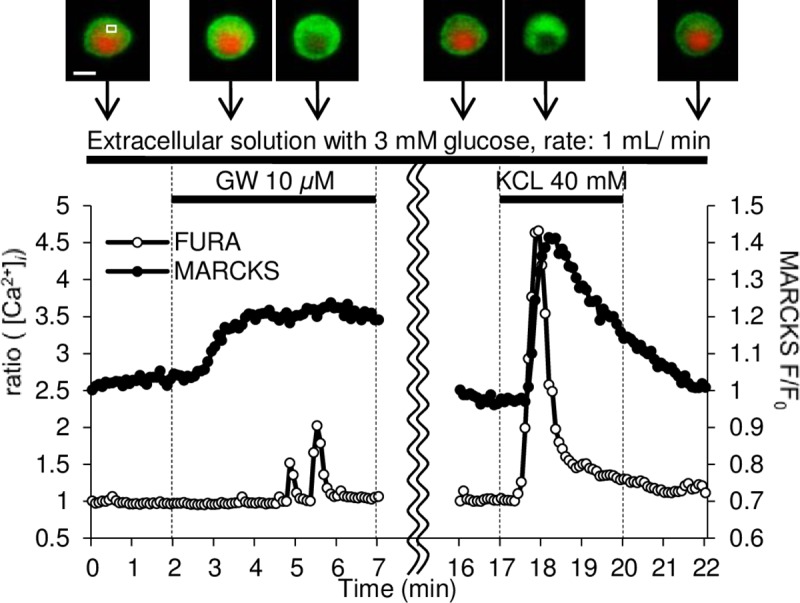
Experiments were performed using epifluorescence microscopy. Simultaneous monitoring of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP; filled circles) translocation (as a marker of PKC activation) and intracellular Ca2+ concentration ([Ca2+]i) (open circles). The intensity of Fura2 for [Ca2+]i measurement is represented as the 340/380 nm ratio. These values and the fluorescence intensity (F) of MARCKS-GFP were normalized to each initial value (F0; ratio [Ca2+]i and MARCKS F/F0). INS-1D cells were perfused with extracellular solution containing 3 mM glucose at 1 mL per minute from 5 minutes before the start of the experiment to the end. To avoid light-induced cell damage, monitoring was paused from 7 to 16 minutes after starting the experiment. At 16 minutes, the distribution of MARCKS-GFP in cells was similar to that observed just after starting the experiment. Images were taken at the times indicated by the arrows. The bar represents 10 μm. The regions of interest in the cytosol are indicated by a white box. When MARCKS-GFP (green) was translocated from the plasma membrane to the cytosol, MARCKS F/F0 in the cytosol increased. When the fluorescence intensity at 380 nm (red) declined, [Ca2+]i, represented as the 340/380 nm ratio, increased (eight independent experiments, 81 cells).
At 10 μM GW9508, we noted sustained translocation of MARCKS-GFP to the cytosol (Fig 2). In contrast to the transient MARCKS-GFP translocation that followed a change in [Ca2+]i induced by a depolarizing concentration of potassium (40 mM KCL), GW9508-induced translocation of MARCKS-GFP was not affected by changes in [Ca2+]i, indicating that GW9508 activated PKC in a Ca2+-independent manner (Fig 2).
Stimulatory glucose concentration changes GW9508-induced translocation of MARCKS-GFP from a Ca2+-independent to a Ca2+-dependent mechanism
We demonstrated that GW9508 increased GSIS and did not amplify insulin secretion at 3 mM glucose (Fig 1). Next, we compared GW9508-induced MARCKS translocation at 3 mM and 20 mM glucose. The application of GW9508 resulted in sustained translocation of MARCKS-GFP to the cytosol, as well as multiple transient translocations of MARCKS-GFP that occurred just following sharp increases in [Ca2+]i, at 20 mM glucose (Fig 3B). We then plotted the [Ca2+]i-related increases in the F/F0 of MARCKS-GFP in the cytosol against sharp elevations in [Ca2+]i during the 5-min application of GW9508 at 3 mM or 20 mM glucose. The correlation between the increase in MARCKS and [Ca2+]i elevation was weak at 3 mM glucose (r = 0.349; p < 0.01), but stronger at 20 mM glucose (r = 0.752; p < 0.01) (Fig 3C and 3D). A strong correlation between [Ca2+]i elevation and the increase in MARCKS also existed below a 1.5 elevation of the [Ca2+]i ratio at 20 mM glucose (r = 0.572; p < 0.01) (Fig 3E and 3F). These observations suggest that GW9508-evoked Ca2+ signals induced the activation of PKC more robustly at a stimulatory concentration of glucose.
Fig 3. GW9508 evokes transient translocation of MARCKS-GFP following Ca2+ oscillations at 20 mM glucose.

(A, B): Representative data showing the translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) and intracellular Ca2+ concentration [Ca2+]i under stimulation by GW9508 in an extracellular solution containing (A) 3 mM glucose (the same data as in Fig 2 but with different scales; eight independent experiments, 81 cells) or (B) 20 mM glucose (four independent experiments, 80 cells). Biphasic translocation of MARCKS-GFP occurred after the second sharp increase in Ca2+ at 20 mM glucose. Scatterplots of the ratio of [Ca2+]i-related increases in the relative fluorescence intensity of MARCKS-GFP in the cytosol versus the elevation in ratio [Ca2+]i during GW9508 application at (C) 3 mM glucose (eight independent experiments, 56 cells) or (D) 20 mM glucose (four independent experiments, 63 cells). Cells that did not respond to GW9508 were excluded from the calculation. To count the cells that responded to GW9508, only cells that were almost unchanged in fluorescence of MARCKS-GFP before the application of GW9508 were chosen for study at 3 mM glucose. (E, F) To equalize the [Ca2+]i elevation, only cells with an [Ca2+]i ratio elevated by 1.5 were selected for analysis from C (all 56 cells) and D (47 cells) (dashed box in C and D).
Profiles of PKCα and PKCε translocation in response to GW9508 at substimulatory and stimulatory concentrations of glucose
The observations above prompted us to investigate whether there were differences in the activation of PKC isoforms between substimulatory and stimulatory concentrations of glucose. We examined the GW9508-evoked translocation of PKCα-GFP and PKCε-GFP in transfected INS-1D cells using TIRFM. Only 17% of experimental cells showed transient translocation of PKCα in response to GW9508 at 3 mM glucose (Fig 4A, Table 1). At 20 mM glucose, more than twice the number of cells responded to GW9508, i.e. 35% of cells showed transient PKCα translocation (Fig 4B, Table 1). Conversely, 51% of cells showed transient or sustained translocation of PKCε at 3 mM glucose (Fig 4C, Table 1). Interestingly, in addition to sustained translocation of PKCε, GW9508 also elicited transient translocation of PKCε from a higher percentage of cells at 20 mM (51%) compared with 3 mM (20%) glucose (p < 0.01) (Fig 4D, Table 1). However, the response time for translocation of both PKCα and PKCε did not differ significantly between 3 mM glucose and 20 mM glucose (Table 1). These results suggest that PKCε played a dominant role in GW9508-induced MARCKS activation at substimulatory and stimulatory concentrations of glucose.
Fig 4. Temporal profile of PKCα and PKCε translocation at 3 or 20 mM using total internal reflection fluorescence microscopy.

Representative data of the relative change in fluorescence intensity (F/F0) of PKCα-GFP (A, B) or PKCε-GFP (C, D) at the plasma membrane. Images a-f were taken at the times indicated in each graph. Scale bars = 10 μm. When green fluorescent protein (GFP)-tagged PKCα or PKCε is translocated from the cytosol to the plasma membrane, F/F0 increases. (A, B) Translocation of PKCα-GFP evoked by GW9508 at 20 mM glucose (B; 15 independent experiments, 165 cells) was higher than that at 3 mM glucose (A; 19 independent experiments, 283 cells), but lower than that induced by 40 mM KCL. (C, D) Translocation of PKCε-GFP induced by GW9508 at 20 mM glucose (D; 15 independent experiments, 57 cells) was biphasic, higher than that at 3 mM glucose (C; 18 independent experiments, 47 cells), and comparable with that induced by 40 mM KCL.
Table 1. Effect of GW9508 on PKCα and PKCε activation at 3 mM and 20 mM glucose.
| 3 mM Glucose | 20 mM Glucose | |
|---|---|---|
| PKCα | ||
| Fraction of INS-1 cells responding to GW9508 | 47 of 283 (17%) | 58 of 165 (35%)* |
| Lag time (sec.) | 131.4 ± 11.9 | 141.8 ± 10.7 |
| PKCε | ||
| Fraction of INS-1 cells responding to GW9508 | 25 of 49 (51%) | 38 of 57 (67%) |
| Fraction of cells with a transient response | 10 of 49 (20%) | 29 of 57 (51%)* |
| Fraction of cells with a sustained response | 21 of 49 (43%) | 28 of 57 (49%) |
| Lag time (sec.) | 56.6 ± 5.3 | 77.6 ± 7.8 |
Only cells exhibiting an increase in the F/F0 of ≥ 0.1 over the baseline level were counted as responding to GW9508. The response to GW9508 was further categorized into cell fractions with a sustained or transient translocation of PKC. Lag time = response time of PKC, and is shown as mean ± standard error of the mean.
*p < 0.01 vs. GW9508 at 3 mM glucose.
Effect of PKC inhibitors on GW9508-potentiated insulin secretion in INS-1 cells
We tested the isoform-specific roles of the two PKCs in GW9508-potentiated insulin secretion in INS-1D cells using antp-PKCα and antp-PKCε. GW9508-induced insulin secretion at 20 mM glucose was significantly reduced by 75 μM antp-PKCα (p < 0.05) and 75 μM antp-PKCε (p < 0.01) (Fig 5). Antp-PKCε inhibited insulin secretion more potently than antp-PKCα at 20 mM glucose (p < 0.05) (Fig 5). Double inhibition with antp-PKCα and ε did not decrease insulin secretion below the level inhibited by antp-PKCε alone (Fig 5). To strengthen these results, we also tested Gö 6976, an inhibitor of conventional PKC, and BIS I, a broad PKC inhibitor. While both Gö 6976 and BIS I significantly reduced GW9508-induced insulin secretion, BIS I had a stronger effect (Fig 5). These results agreed with those using antp-PKCα and antp-PKCε. Taken together, the results of the PKC inhibitor experiments suggest that both PKC isoforms, but PKCε in particular, were responsible for GW9508-potentiated insulin secretion in INS-1D cells.
Fig 5. PKC inhibitors suppress GW9508-induced insulin secretion in INS-1D cells.

INS-1D cells were incubated for 1 h in Krebs-Ringer buffer containing 3 mM or 20 mM glucose with stimulation by GW9508 in the absence or presence of 75 μM antennapedia (antp), 75 μM antp-PKCα (antpα), 75 μM antp-PKCε (antpε), both antpα and antpε, 1 μM Gö 6976, or 1 μM bisindolylmaleimide I (BIS I). Data are shown as mean ± standard error of the mean of 12 independent experiments with triplicate samples in each group. *p < 0.05; **p < 0.01; #p < 0.05 vs. GW9508 at 20 mM glucose; ##p < 0.01 vs. GW9508 at 20 mM glucose.
Discussion
We demonstrated in this study that: 1) GW9508-induced activation of PKC was mainly characterized by sustained PKCε, which was inconsistent with changes in [Ca2+]i at a substimulatory concentration of glucose (Figs 2, 3A, 3C, 4A and 4C); 2) a stimulatory concentration of glucose enabled GW9508-induced transient translocation of PKCα and PKCε that followed changes in [Ca2+]i (Figs 3B, 3D, 4B and 4D); and 3) GW9508-potentiated GSIS was disrupted by two PKC inhibitory peptides, with more marked inhibition of PKCε, in INS-1D cells (Fig 5). These results indicate that PKCε is directly activated by GW9508, independent of glucose concentration, and suggest that the shift in PKCα and PKCε activation from a stable, sustained mode to a transient mode is involved in the potentiation of GSIS.
In a previous report, we demonstrated that Ca2+ influx via voltage-dependent Ca2+ channels (VDCCs) can activate PKC [26], and that GLP-1-induced PKC activation is transient and Ca2+-dependent [7]. Nevertheless, in this study PKC activation by GW9508 was independent of elevations in [Ca2+]i and sustained during the application of GW9508 at a substimulatory concentration of glucose (Figs 2 and 3A). GPR40 signaling generates IP3 and DAG via PLC activation [13]. IP3 induces Ca2+ release from the ER, and DAG activates PKC directly [14,36]. Thus, GPR40-mediated DAG, but not IP3, could play a key role in sustained PKC activation that is induced by GW9508 at a substimulatory concentration of glucose. However, a significant but weak correlation between PKC activation and [Ca2+]i elevation was confirmed, which could represent a population of INS-1 cells that allowed Ca2+ influx through VDCCs (Fig 3C and 3E). The TIRFM imaging experiments showed that GW9508 induced sustained PKCε activation, but not sustained PKCα activation, at a substimulatory concentration of glucose (Fig 4A and 4C). These results suggest that IP3-induced Ca2+ release was insufficient for activation of the conventional PKCα isotype in response to GW9508 at a substimulatory concentration of glucose in INS-1D cells, whereas DAG was sufficient for activation of the novel PKCε isotype.
We have shown here that a stimulatory concentration of glucose altered GW9508-induced PKC activation from a Ca2+-independent to a Ca2+-dependent mechanism, despite the amplitudes of induced [Ca2+]i elevations over the entire cell being similar between the stimulatory and substimulatory concentrations of glucose (Fig 3A–3F). On the other hand, nifedipine, a blocking agent of VDCCs, attenuated the transient translocation of MARCKS-GFP that was induced by GW9508 at a stimulatory concentration of glucose (S1 Fig). The TIRFM imaging experiments showed that GW9508 increased the fraction of cells that underwent transient PKCα activation at a stimulatory concentration of glucose (Fig 4B, Table 1). High concentrations of glucose are known to stimulate insulin secretion through an intracellular pathway involving an increase in the intracellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio and closure of KATP channels, followed by membrane depolarization, which leads to the activation of VDCCs and a rise in [Ca2+]i [37–40]. In our previous report, we demonstrated that Ca2+ influx was a much stronger stimulus of PKCα translocation than Ca2+ mobilization from intracellular stores in INS-1D cells [26]. In light of that result, it could be interpreted that the Ca2+ mobilization that was induced by GW9508-generated IP3 in the current study failed to translocate PKCα at the substimulatory concentration of glucose, and that KATP-induced Ca2+ influx through VDCCs activated PKCα at the stimulatory concentration of glucose. However, INS-1D cells are known to exhibit a strong electrical and insulin response to KCl stimulation and a less potent response to glucose stimulation. This could explain the lack of differentiation in the response time of PKCα between the substimulatory and stimulatory concentrations of glucose (Table 1).
TIRFM imaging also showed that GW9508 induced transient activation of PKCε, in addition to the sustained activation, at a stimulatory concentration of glucose (Fig 4D, Table 1). We demonstrated in a previous report that Ca2+ influx via VDCCs could activate PLC [26]. Among all known PLC isoforms, the PLCδ isoforms are the most sensitive to Ca2+ [41]. Thus, Ca2+ influx via VDCCs would be expected to activate PLC, and PLCδ, in particular, leading to the transient activation of PKCε that we observed at the stimulatory concentration of glucose. Another recent report concluded that the rapid activation of PKCε in the plasma membrane is due to exocytotic release of ATP, with autocrine feedback activation of P2Y1 purinoceptors, which in turn induces DAG via PLC activation [42,43]. Thus, glucose-stimulated Ca2+ signaling and autocrine signaling could be sufficient to trigger the rapid activation of PLC, which activates PKCε by generating DAG.
The exact mechanism of GPR40-potentiated GSIS in β-cells remains unclear. Here, we observed that both PKCα and, to a greater degree, PKCε, were involved in GW9508-potentiated insulin secretion, but only at a stimulatory concentration of glucose (Figs 1 and 5). However, 12-O-tetradecanoylphorbol 13-acetate (TPA), which binds the diacylglycerol site to potently activate PKC, has been reported to induce insulin secretion at substimulatory as well as stimulatory concentrations of glucose in insulin-producing cells [44–46]. This discrepancy could be explained by a requirement for PKC activation to exceed a threshold value for GPR40-mediated insulin secretion to occur. We also observed a larger amplitude of [Ca2+]i increase in GW9508-mediated INS-1D cells compared with vehicle at a stimulatory concentration of glucose (S2 Fig). A recent report showed that fasiglifam, another GPR40 agonist, enhanced GSIS through both IP3-mediated amplification of Ca2+ oscillations and DAG-mediated augmentation of downstream secretory mechanisms independent of Ca2+ oscillations [47]. Thus, IP3 might be involved in insulin secretion only at a stimulatory concentration of glucose, as shown in this study. Recently, it was reported that GPR40 depolarizes the plasma membrane and increases background current via the transient receptor potential canonical 3 (TRPC3) channel at a substimulatory concentration of glucose in pancreatic β-cells [48]. TRPC3 is a class of nonselective cation channels that is activated by PLC/PKC signaling, not Ca2+ from the ER, resulting in the potentiation of GSIS [48]. It has also been reported that physiological concentrations of GLP-1 stimulate insulin secretion through the PKC-dependent activation of transient receptor potential melastatin 4 (TRPM4) and TRPM5, which are Na+-permeable cation channels [8]. Thus, the GW9508-induced PKCε activation at substimulatory concentrations of glucose that we observed here might have involved TRPC3 and TRPM activation, which potentiate Ca2+ influx at stimulatory concentrations of glucose.
PKCε played a dominant role over that of PKCα in GW9508-induced insulin secretion by INS-1D cells (Fig 5), consistent with evidence of the dominant translocation of PKCε induced by GW9508 (Fig 4A–4D). PKCε is involved in GSIS, and several studies have shown that the inhibition of the function of PKCε is associated with reduced GSIS [24,25,49]. Activated PKCε has been shown to localize to insulin granules, enhance biosynthetic pathways of proinsulin, and induce the processing of proinsulin to mature insulin [24,49]. Another recent study showed that novel PKCs stimulated mitochondrial ATP production via ERK1/2 signaling [50], which increased the cytosolic ATP/ADP ratio [51]. These mechanisms may contribute to the GW9508-enhanced insulin response to a stimulatory concentration of glucose via PKCε activation, as observed in our study. In contrast, the contribution of PKCα activation to GSIS remains a subject of debate. Inconsistencies in the data may be explained in part by the different effects of PKCα on the initial and late phases of secretion [52].
We also investigated the effect of γ-linolenic acid (γ-LA), a natural ligand of GPR40, and found that it elicited insulin secretion not only at the stimulatory but also at the substimulatory concentration of glucose (S5 Fig). At a substimulatory concentration of glucose, γ-LA elicited sustained PKC activation, whereas at the stimulatory concentration of glucose, PKC exhibited not only sustained activation but also transient activation (S3 and S4 Figs, S1 Table). Unlike GW9508, however, γ-LA-stimulated insulin secretion was not affected by antp-PKCα or antp-PKCε (S5 Fig). These results suggest that receptor-independent pathways rather than the GPR40 pathway are also involved in γ-LA–evoked insulin secretion, and could include the malonyl-CoA/long-chain acyl-CoA pathway and triglyceride/free fatty acid cycling via the intracellular metabolism of fatty acids [53]. Our results using GW9508, however, suggest that PKC-dependent pathways are the sole signaling pathways for GPR40-dependent insulin secretion.
In conclusion, the GPR40 agonist GW9508 induced the sustained activation of the novel isoform PKCε at substimulatory concentrations of glucose, and evoked the transient activation of the conventional isoform PKCα and PKCε following increases in [Ca2+]i via VDCCs at stimulatory concentrations of glucose. This activation, which was especially potent for PKCε, was involved in GW9508-potentiated GSIS. GPR40 agonists have the potential to be key drugs for increasing insulin levels with minimal risk of iatrogenic hypoglycemia in patients with type 2 diabetes.
Supporting information
INS-1D cells were perfused with extracellular solution with 10 μM nifedipine containing 20 mM glucose at 1 mL per minute from 5 minutes before the start of the experiment to the end. Representative data of translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) by GW9508 (four independent experiments, 80 cells).
(TIF)
Elevation of the ratio of the intracellular Ca2+ concentration ([Ca2+]i) during GW9508 application at 3 mM (eight independent experiments, 56 cells) or 20 mM glucose (four independent experiments, 63 cells). #p < 0.01 vs. GW9508 at 3 mM glucose.
(TIF)
Representative epifluorescence microscopy showing the translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) by γ-linolenic acid (γ-LA) (seven independent experiments, 98 cells). INS-1D cells were perfused with extracellular solution containing 3 mM glucose at 1 mL per minute from 5 minutes before the start of the experiment to the end. MARCKS-GFP was monitored at the cytosol. Fluorescence intensity (F) values were normalized to the initial value (F0; MARCKS F/F0). To avoid light-induced cell damage, monitoring was paused from 7 to 16 minutes after the start of each experiment. At 16 minutes, the distribution of MARCKS-GFP in cells was similar to that observed just after starting the experiment.
(TIF)
Representative epifluorescence microscopy showing the translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) by γ-linolenic acid (γ-LA) in an extracellular solution containing 3 mM glucose (A; the same data as shown in S3 Fig) or 20 mM glucose (B; four independent experiments, 80 cells).
(TIF)
INS-1D cells were incubated for 1 h in Krebs-Ringer buffer (KRB) containing 3 mM or 20 mM glucose with stimulation by γ-linolenic acid (γ-LA) in the presence or absence of 75 μM antennapedia (antp), 75 μM antp-PKCα, 75 μM antp-PKCε, or both antp-PKCα and antp-PKCε. Data are shown as mean ± standard error of the mean of three independent experiments with triplicate samples in each group. *p < 0.05; **p < 0.01.
(TIF)
The response to GW9508 was further categorized into cell fractions with a sustained or transient translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP). Lag time = the response time of MARCKS-GFP, and is shown as mean ± standard error of the mean. *p < 0.05 vs. GW9508 at 3 mM glucose; **p < 0.01 vs. GW9508 at 3 mM glucose.
(DOCX)
Acknowledgments
We thank H. Nakamura for valuable technical assistance. We thank Michelle Kahmeyer-Gabbe, PhD, from Edanz Group (www.edanz.com/ac) for editing a draft of this manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Guariguata L, Whiting DR, Hambleton I, Beagley J. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. Elsevier Ireland Ltd; 2014;103: 137–149. 10.1016/j.diabres.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 2.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116: 1802–1812. 10.1172/JCI29103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newsholme P, Gaudel C MN. Nutrient regulation of insulin secretion and beta-cell functional integrity. Adv Exp Med Biol. 2010;654: 91–114. 10.1007/978-90-481-3271-3_6 [DOI] [PubMed] [Google Scholar]
- 4.Drucker DJ. Incretin action in the pancreas: Potential promise, possible perils, and pathological pitfalls. Diabetes. 2013;62: 3316–3323. 10.2337/db13-0822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8: 369–385. 10.1038/nrd2782 [DOI] [PubMed] [Google Scholar]
- 6.Holz GG. Epac: A New cAMP-Binding Protein in Support of Glucagon-like Peptide-1 Receptor-Mediated Signal Transduction in the Pancreatic β-Cell. Diabetes. 2004;53: 5–13. 10.2337/diabetes.53.1.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki Y, Zhang H, Saito N, Kojima I, Urano T, Mogami H. Glucagon-like peptide 1 activates protein kinase C through Ca2+-dependent activation of phospholipase C in insulin-secreting cells. J Biol Chem. 2006;281: 28499–28507. 10.1074/jbc.M604291200 [DOI] [PubMed] [Google Scholar]
- 8.Shigeto M, Ramracheya R, Tarasov AI, Cha CY, Chibalina M V, Hastoy B, et al. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J Clin Invest. 2015;125: 4714–4728. 10.1172/JCI81975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332: 281–292. 10.1042/bj3320281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rayasam GV, Tulasi VK, Davis JA BV. Fatty acid receptors as new therapeutic targets for diabetes. Expert Opin Ther Targets. 2007;11: 661–671. 10.1517/14728222.11.5.661 [DOI] [PubMed] [Google Scholar]
- 11.Milligan G, Stoddart LA, Brown AJ. G protein-coupled receptors for free fatty acids. Cell Signal. 2006;18: 1360–1365. 10.1016/j.cellsig.2006.03.011 [DOI] [PubMed] [Google Scholar]
- 12.Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008;60: 405–417. 10.1124/pr.108.00802 [DOI] [PubMed] [Google Scholar]
- 13.Fujiwara K, Maekawa F, Yada T, Maekawa F, Oleic TY, Chain L, et al. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab. 2005;289: E670–E677. 10.1152/ajpendo.00035.2005 [DOI] [PubMed] [Google Scholar]
- 14.MJ B, RF. I. Inositol phosphates and cell signalling. Nature. 1989;341: 197–205. 10.1038/341197a0 [DOI] [PubMed] [Google Scholar]
- 15.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422: 173–176. 10.1038/nature01478 [DOI] [PubMed] [Google Scholar]
- 16.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278: 11303–11311. 10.1074/jbc.M211495200 [DOI] [PubMed] [Google Scholar]
- 17.Tomita T, Masuzaki H, Iwakura H, Fujikura J, Noguchi M, Tanaka T, et al. Expression of the gene for a membrane-bound fatty acid receptor in the pancreas and islet cell tumours in humans: evidence for GPR40 expression in pancreatic beta cells and implications for insulin secretion. Diabetologia. 2006;49: 962–968. 10.1007/s00125-006-0193-8 [DOI] [PubMed] [Google Scholar]
- 18.Tomita T, Masuzaki H, Noguchi M, Iwakura H, Fujikura J, Tanaka T, et al. GPR40 gene expression in human pancreas and insulinoma. Biochem Biophys Res Commun. 2005;338: 1788–1790. 10.1016/j.bbrc.2005.10.161 [DOI] [PubMed] [Google Scholar]
- 19.Latour MG, Alquier T, Oseid E, Tremblay C, Thomas L, Luo J, et al. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56: 1087–1094. 10.2337/db06-1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan CP, Feng Y, Zhou Y, Eiermann GJ, Petrov A, Zhou C, et al. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes. 2008;57: 2211–2219. 10.2337/db08-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briscoe CP, Peat AJ, Mckeown SC, Corbett DF, Goetz AS, Littleton TR, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148: 619–628. 10.1038/sj.bjp.0706770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salehi A, Flodgren E, Nilsson NE, Jimenez-Feltstrom J, Miyazaki J, Owman C OB. Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res. 2005;322: 207–215. 10.1007/s00441-005-0017-z [DOI] [PubMed] [Google Scholar]
- 23.Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD. Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem Biophys Res Commun. 2005;335: 97–104. 10.1016/j.bbrc.2005.07.042 [DOI] [PubMed] [Google Scholar]
- 24.Mendez CF, Leibiger IB, Leibiger B, Høy M, Gromada J, Berggren P, et al. Rapid association of protein kinase C-epsilon with insulin granules is essential for insulin exocytosis. J Biol Chem. 2003;278: 44753–44757. 10.1074/jbc.M308664200 [DOI] [PubMed] [Google Scholar]
- 25.Yedovitzky M, Mochly-rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, et al. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic beta-cells. J Biol Chem. 1997;272: 1417–1420. 10.1074/jbc.272.3.1417 [DOI] [PubMed] [Google Scholar]
- 26.Mogami H, Zhang H, Suzuki Y, Urano T, Saito N, Kojima I, et al. Decoding of short-lived Ca2+ influx signals into long term substrate phosphorylation through activation of two distinct classes of protein kinase C. J Biol Chem. 2003;278: 9896–9904. 10.1074/jbc.M210653200 [DOI] [PubMed] [Google Scholar]
- 27.Shirai Y, Kashiwagi K, Yagi K, Sakai N, Saito N. Distinct effects of fatty acids on translocation of gamma- and epsilon-subspecies of protein kinase C. J Cell Biol. 1998;143: 511–521. 10.1083/jcb.143.2.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asfari M, Janjic D, Meda P, Li G, Halban P, Wollheim C. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130: 167–178. 10.1210/endo.130.1.1370150 [DOI] [PubMed] [Google Scholar]
- 29.Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8: 84–87. [PubMed] [Google Scholar]
- 30.Fischer PM, Zhelev NZ, Wang S, Melville JE, Fåhraeus R LD. Structure-activity relationship of truncated and substituted analogues of the intracellular delivery vector Penetratin. J Pept Res. 2000;55: 163–172. 10.1034/j.1399-3011.2000.00163.x [DOI] [PubMed] [Google Scholar]
- 31.Harris TE, Persaud SJ, Jones PM. Pseudosubstrate peptide inhibitors of beta-cell protein kinases: altered selectivity after myristoylation. Mol Cell Endocrinol. 1999;155: 61–68. 10.1016/s0303-7207(99)00114-8 [DOI] [PubMed] [Google Scholar]
- 32.Zoukhri D, Hodges RR, Sergheraert C, Toker A, Dartt DA. Lacrimal gland PKC isoforms are differentially involved in agonist-induced protein secretion. Am J Physiol. 1997;272: C263–C269. 10.1152/ajpcell.1997.272.1.C263 [DOI] [PubMed] [Google Scholar]
- 33.Yamada S, Komatsu M, Sato Y, Yamauchi K, Kojima I, Aizawa T, et al. Time-dependent stimulation of insulin exocytosis by 3’,5’-cyclic adenosine monophosphate in the rat islet beta-cell. Endocrinology. 2002;143: 4203–4209. 10.1210/en.2002-220368 [DOI] [PubMed] [Google Scholar]
- 34.Blackshear PJ. The MARCKS family of cellular protein kinase C substrates. J Biol Chem. 1993;268: 1501–1504. [PubMed] [Google Scholar]
- 35.Papers JBC, Doi M, Ohmori S, Sakai N, Shirai Y, Yamamoto H, et al. Importance of protein kinase C targeting for the phosphorylation of its substrate, myristoylated alanine-rich C-kinase substrate. J Biol Chem. 2000;275: 26449–26457. 10.1074/jbc.M003588200 [DOI] [PubMed] [Google Scholar]
- 36.Clapham DE. Calcium signaling. Cell. 1995;80: 259–268. 10.1016/0092-8674(95)90408-5 [DOI] [PubMed] [Google Scholar]
- 37.Barg S. Mechanisms of exocytosis in insulin-secreting B-cells and glucagon-secreting A-cells. Pharmacol Toxicol. 2003;92: 3–13. [DOI] [PubMed] [Google Scholar]
- 38.Proks P, Lippiat JD. Membrane ion channels and diabetes. Curr Pharm Des. 2006;12: 485–501. 10.2174/138161206775474431 [DOI] [PubMed] [Google Scholar]
- 39.Rendell M. The role of sulphonylureas in the management of type 2 diabetes mellitus. Drugs. 2004;64: 1339–1358. 10.2165/00003495-200464120-00006 [DOI] [PubMed] [Google Scholar]
- 40.Mears D. Regulation of Insulin Secretion in Islets of Langerhans by Ca2+Channels. J Membr Biol. 2004;200: 57–66. 10.1007/s00232-004-0692-9 [DOI] [PubMed] [Google Scholar]
- 41.Hanoune J DN. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41: 145–174. 10.1146/annurev.pharmtox.41.1.145 [DOI] [PubMed] [Google Scholar]
- 42.Wuttke A, Idevall-hagren O, Tengholm A. P2Y1receptor-dependent diacylglycerol signaling microdomains in β cells promote insulin secretion. FASEB J. 2013;27: 1610–1620. 10.1096/fj.12-221499 [DOI] [PubMed] [Google Scholar]
- 43.Wuttke A, Yu Q, Tengholm A. Autocrine Signaling Underlies Fast Repetitive Plasma Membrane Translocation of Conventional and Novel Protein Kinase C Isoforms in β Cells. J Biol Chem. Nature Publishing Group; 2016;291: 14986–14995. 10.1074/jbc.M115.698456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malaisse WJ, Sener A, Herchuelz A, Poloczek P, Carpinelli AR, Winand J, et al. Insulinotropic Effect of the Tumor Promoter 12-O-Tetradecanoylphorbol-13- Acetate in Rat Pancreatic Islets. Cancer Res. 1980;40: 3827–3832. [PubMed] [Google Scholar]
- 45.Bozem M, NenQuin M, Henquin JC. The Ionic, Electrical, and Secretory Effects of Protein Kinase C Activation in Mouse Pancreatic B-Cells: Studies with a Phorbol Ester. Endocrinology. 1987;121: 1025–1033. 10.1210/endo-121-3-1025 [DOI] [PubMed] [Google Scholar]
- 46.Easom RA, Hughes JH, Landt M, Wolf BA, Turk J, Mcdaniel ML. Comparison of effects of phorbol esters and glucose on protein kinase C activation and insulin secretion in pancreatic islets. Biochem J. 1989;264: 27–33. 10.1042/bj2640027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakuma K, Yabuki C, Maruyama M, Abiru A, Komatsu H, Negoro N, et al. Fasiglifam (TAK-875) has dual potentiating mechanisms via Gαq-GPR40/FFAR1 signaling branches on glucose-dependent insulin secretion. Pharmacol Res Perspect. 2016;4: e00237 10.1002/prp2.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamada H, Yoshida M, Ito K, Dezaki K, Yada T, Ishikawa S, et al. Potentiation of Glucose-stimulated Insulin Secretion by the GPR40 –PLC–TRPC Pathway in Pancreatic β -Cells. Sientific Reports. Nature Publishing Group; 2016;6: 25912 10.1038/srep25912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warwar N, Dov A, Abramovitch E, Wu R, Jmoudiak M, Haber E, et al. PKCepsilon mediates glucose-regulated insulin production in pancreatic beta-cells. Biochim Biophys Acta. 2008;1783: 1929–1934. 10.1016/j.bbamcr.2008.04.007 [DOI] [PubMed] [Google Scholar]
- 50.Santo-domingo J, Chareyron I, Dayon L, Galindo AN, Cominetti O, Gimenes MPG, et al. Coordinated activation of mitochondrial respiration and exocytosis mediated by PKC signaling in pancreatic β cells. FASEB J. 2017;31: 1028–1045. 10.1096/fj.201600837R [DOI] [PubMed] [Google Scholar]
- 51.Fransson U, Rosengren AH, Schuit FC, Renström E, Mulder H. Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets. Diabetologia. 2006;49: 1578–1586. 10.1007/s00125-006-0263-y [DOI] [PubMed] [Google Scholar]
- 52.Zhang H, Nagasawa M, Yamada S, Mogami H, Suzuki Y, Kojima I. Bimodal role of conventional protein kinase C in insulin secretion from rat pancreatic beta cells. J Physiol. 2004;561: 133–147. 10.1113/jphysiol.2004.071241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nolan CJ, Madiraju MSR, Delghingaro-Augusto V, Peyot M-L, Prentki M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes. 2006;55: S16–S23. 10.2337/db06-s003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
INS-1D cells were perfused with extracellular solution with 10 μM nifedipine containing 20 mM glucose at 1 mL per minute from 5 minutes before the start of the experiment to the end. Representative data of translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) by GW9508 (four independent experiments, 80 cells).
(TIF)
Elevation of the ratio of the intracellular Ca2+ concentration ([Ca2+]i) during GW9508 application at 3 mM (eight independent experiments, 56 cells) or 20 mM glucose (four independent experiments, 63 cells). #p < 0.01 vs. GW9508 at 3 mM glucose.
(TIF)
Representative epifluorescence microscopy showing the translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) by γ-linolenic acid (γ-LA) (seven independent experiments, 98 cells). INS-1D cells were perfused with extracellular solution containing 3 mM glucose at 1 mL per minute from 5 minutes before the start of the experiment to the end. MARCKS-GFP was monitored at the cytosol. Fluorescence intensity (F) values were normalized to the initial value (F0; MARCKS F/F0). To avoid light-induced cell damage, monitoring was paused from 7 to 16 minutes after the start of each experiment. At 16 minutes, the distribution of MARCKS-GFP in cells was similar to that observed just after starting the experiment.
(TIF)
Representative epifluorescence microscopy showing the translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP) by γ-linolenic acid (γ-LA) in an extracellular solution containing 3 mM glucose (A; the same data as shown in S3 Fig) or 20 mM glucose (B; four independent experiments, 80 cells).
(TIF)
INS-1D cells were incubated for 1 h in Krebs-Ringer buffer (KRB) containing 3 mM or 20 mM glucose with stimulation by γ-linolenic acid (γ-LA) in the presence or absence of 75 μM antennapedia (antp), 75 μM antp-PKCα, 75 μM antp-PKCε, or both antp-PKCα and antp-PKCε. Data are shown as mean ± standard error of the mean of three independent experiments with triplicate samples in each group. *p < 0.05; **p < 0.01.
(TIF)
The response to GW9508 was further categorized into cell fractions with a sustained or transient translocation of green fluorescent protein (GFP)-tagged myristoylated alanine-rich C kinase substrate (MARCKS-GFP). Lag time = the response time of MARCKS-GFP, and is shown as mean ± standard error of the mean. *p < 0.05 vs. GW9508 at 3 mM glucose; **p < 0.01 vs. GW9508 at 3 mM glucose.
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.