

Abstract
22q11.2 Deletion Syndrome (22q11DS) is a multisystem disorder caused by a hemizygous deletion within 22q11.2. Patients with the deletion display a wide range of cognitive deficits. The gene catechol-O-methyl-transferase (COMT) resides in the typically deleted region of 22q11.2 and is rendered hemizygous in individuals affected by the 22q11DS. COMT is a critical enzyme in the degradation of catecholamine neurotransmitters in the brain. A functional polymorphism, Val158Met, has been associated with a variety of neurocognitive outcomes. In this study, 159 patients with 22q11DS were analyzed for a potential association between intelligence quotient (IQ) and COMT genotype. We performed a univariate analysis for overall influence and modified our analysis to focus on possible differences between average, borderline, and intellectually impaired patients. No correlation between COMT genotype and IQ performance was found.
Keywords: 22q11.2 deletion syndrome, velocardiofacial syndrome, COMT
Introduction
The 22q11.2 Deletion Syndrome (22q11DS), now known to be the cause of the majority of cases of DiGeorge syndrome, velocardiofacial syndrome (VCFS), and Conotruncal Anomaly Face Syndrome, is the most common condition caused by a chromosomal microdeletion [McDonald-McGinn et al., 2015]. It occurs in approximately 1 out of every 2,000–4,000 live births and 1 in 1,000 fetuses [Tezenas Du Montcel et al., 1996; Devriendt et al., 1998; Goodship et al., 1998; McDonald-McGinn et al., 1999, 2001; Oskarsdottir et al., 2004; Grati et al., 2015]. Clinical features of 22q11DS include a variety of congenital anomalies and medical problems such as: palatal abnormalities, congenital heart disease, immunodeficiencies, endocrine problems, gastrointestinal abnormalities, renal anomalies and developmental, cognitive and behavioral differences, including speech and language disorders, learning deficits, and psychiatric illness [Moss et al., 1999; McDonald-McGinn et al., 2015]. Approximately 70–90% of patients with the deletion suffer from a mild-to-moderate cognitive learning disability, with most of individuals attaining borderline intellectual functioning [Pagon et al., 1993; Moss et al., 1999].
The 22q11.2 deletion is the result of non-allelic homologous recombination between chromosome-specific low-copy repeats located in 22q11.2. The majority of patients have a typical deletion of three megabases (Mb) in one copy of the chromosome, within the 22q11.2 region. A typical, nested deletions can also lead to overlapping phenotypes. The typical 3Mb deletion results in hemizygosity for as many as 50 genes including a gene postulated to be responsible for the neuropsychological manifestations of 22q11DS which codes for the enzyme catechol-O-methyltransferase (COMT) [Bearden et al., 2004; McDonald-McGinn et al., 2015]. COMT plays a critical role in the metabolism of dopamine and norepinephrine, particularly in the prefrontal cortex [Lachman et al., 1996; Bearden et al., 2004].
The COMT gene contains a functional polymorphism, Val158Met, which dramatically alters the activity level of the enzyme. Tissues homozygous for the low activity allele (Met) show one-fourth the enzymatic activity of those that are homozygous for the high activity allele (Val) [Lachman et al., 1996]. A decrease in enzyme activity leads to an increase in dopamine levels in the prefrontal cortex, which regulates multiple cognitive and behavioral systems including emotional control and metacognitive executive function such as working memory, impulse control, organization and planning, and sustained attention [Egan et al., 2001; Drabant et al., 2006; Shashi et al., 2006]. In addition to the already decreased enzyme activity in individuals who carry the Met allele, studies have shown an approximate 50% reduction in overall mRNA and activity of the COMT enzyme in patients with 22q11DS [Gothelf et al., 2014].
Association studies have examined the possible correlation between COMT genotype and intelligence quotient (IQ) in patients with 22q11DS, with varying conclusions. In their study of 21 children and adolescents with 22q11DS, Shashi et al. [2006] found the Met allele advantageous for intelligence and measures of prefrontal cognition. Conversely, Baker et al. [2005] reported a more than 10-point decrement of IQ in 25 patients with the low activity allele compared to those with the high activity allele. Meanwhile, Glaser et al. [2006] found no association between the COMT genotype and intelligence in 34 patients with 22q11DS.
Notably, the samples in these studies were relatively small. Here, we report on a large population of 22q11DS patients evaluated to assess the association between COMT alleles and cognitive abilities. The aim was to capture a homogenous look at the possible effects of COMT on IQ in the largest 22q11DS cohort studied to date.
Materials and Methods
Eligibility Criteria
To control for potential genomic variation, we chose to limit our study population to Caucasian patients with the typical 3 Mb deletion. Population genetic statistics show that the COMT allele frequency in the European population is 48% Val and 52% Met, while the overall world population is 61% Val and 39% Met [Kersey et al., 2014]. Variation in allele frequency dependent on variation in ancestry, or population stratification, is known to introduce a bias in genetic association studies and may increase the false positive rate. Additionally, The Children's Hospital of Philadelphia's 22q and You Center, where these patients were evaluated, sees a disproportionately lower number of African American, Asian, and Hispanic patients compared to Caucasian individuals [McDonald-McGinn et al., 2005]. We also chose to exclude all 22q11.2 deletions that did not span the whole 3 Mb of the typical or most frequent deletion. Based on these criteria, a total of 143 subjects were excluded from the final analysis. Ninety-nine patients were excluded from analysis based on race only, 34 were excluded based on deletion size only, and 10 were excluded based on both race and deletion size.
Recruitment and Participants
All subjects were recruited and consented with prior Institutional Review Board (IRB) approval through the Children's Hospital of Philadelphia's 22q and You Center. 159 children and adolescents (87 females, 72 males; (ages 6 years 0 months to 19 years 1 month; mean age: 9.85 ± 2.92 years) with a typical 3 Mb 22q11.2 deletion (LCR22A-LCR22D) and at least one measure of full scale intelligence quotient (FSIQ) performed after the age of 6 years were recruited (Table I). Deletion status and size were confirmed with the use of multiplex ligation dependent probe amplification (MLPA) assays.
Table I. Population Demographics.
| Males | Females | |
|---|---|---|
| Met | 39 | 39 |
| Val | 33 | 48 |
| Mean age | 9.84 ± 3.01 | 9.86 ± 2.87 |
DNA Extraction and Genotyping
Blood or saliva samples were collected from each individual. DNA was extracted using a Genotek or Oragene DNA extraction kit according to the manufacturer's protocol. Samples were genotyped for the COMT Val5Met polymorphism (rs4680) with the Taqman genotyping platform using both an Applied Biosystems 7900 HT and 7500 Real-time PCR Systems. The target sequence was CCAGCGGATGGTG-GATTTCGCTGGC[A/G]TGAAGGACAAGGTGTGCATGCCTGA.
Cognitive Testing
IQ testing was conducted or supervised by licensed psychologists. Depending on the participant's age, current versions of the age-appropriate Wechsler IQ test battery were administered: Wechsler Preschool and Primary Scale of Intelligence (WPPSI; subjects up to 6 years and 6 months of age), Wechsler Intelligence Scale for Children (WISC; subjects ages 6 years and 7 months through 16 years and 11 months), or Wechsler Adult Intelligence Scale (WAIS; subjects ages 17 years and older). Standard calculations of Verbal, Performance, and Full Scale IQ scores were conducted for all subjects, as well as additional routine calculations of Verbal Comprehension, Perceptual Reasoning/Perceptual Organization, Working Memory, and Processing Speed index scores for subjects assessed via the WISC or WAIS. In the event that multiple IQ tests were conducted on a single patient, the first test administered after the age of 6 years was used for the final analysis; however, all IQ tests were initially considered individually to evaluate the presence or absence of unusual variation in the outcome. While full scale IQ was obtained for each participant, other composite subdomains may not be represented in the full sample due to age-specific variations in the structure of the different IQ test batteries. Additional analyses were conducted to examine possible correlations between average, borderline, and impaired levels of intelligence with COMT genotype, using the following standard criteria: average intelligence (IQ≥80), borderline intelligence (IQ = 70–79), and intellectual impairment (IQ < 70).
Statistical Analysis
Data analysis were conducted using IBM SPSS Statistics for Windows version 22 (Armonk, NY: IBM Corp.). To allow for comparison with similar studies, the effect of COMT genotype on IQ was tested using linear regression analysis with COMT genotype and sex as fixed variables, age as a covariate, and IQ as the dependent variable [Bearden et al., 2004; Carmel et al., 2014]. Each individual IQ composite (Verbal IQ, Performance IQ, Full-Scale IQ, etc.) was evaluated separately to calculate the significance of the COMT genotype for that test. A main effect, an interaction analysis between sex, COMT genotype, and age, and an interaction analysis between any two of the three variates were evaluated. P-values <0.05 were considered statistically significant.
Results
We examined a total of 159 patients. The allele frequency in this cohort aligned with the known European population frequency, with 49.06% Met and 50.94% Val alleles. None of the seven possible IQ domains tested showed a significant difference between the subjects with the Met versus Val alleles (Table II). Similarly, none of the interactions tested were significant.
Table II. COMT Genotype Versus IQ.
| Met-hemizygous | Val-hemizygous | Analysis | |||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|||||
| Measure | n | Mean | SD | N | Mean | SD | P |
| Full-scale IQ | 78 | 79.17 | 12.03 | 81 | 75.99 | 12.81 | 0.116 |
| Verbal IQ | 48 | 81.13 | 12.45 | 58 | 79.91 | 14.40 | 0.692 |
| Performance IQ | 48 | 76.71 | 12.84 | 58 | 73.86 | 11.62 | 0.265 |
| Verbal comprehension index | 68 | 85.10 | 11.79 | 65 | 83.69 | 12.79 | 0.484 |
| Perceptual reasoning index | 68 | 79.34 | 13.13 | 65 | 76.12 | 13.59 | 0.188 |
| Working memory index | 66 | 84.23 | 13.96 | 65 | 81.60 | 14.32 | 0.262 |
| Processing speed index | 61 | 86.72 | 15.36 | 63 | 82.08 | 14.40 | 0.081 |
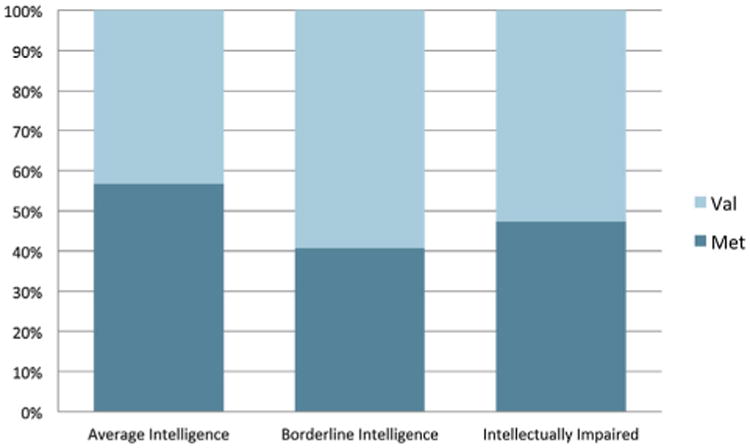
In patients with average intelligence (N = 67), borderline intelligence (N = 54), and intellectual impairment (N = 38), we found no significant differences in the proportion of the Met and Val alleles (Fig. 1).
Fig. 1. Genotype frequency distribution by IQ levels. Frequency of each genotype based on IQ level. The y-axis in the figure is the percentage of individuals in a given IQ class by genotype. Average intelligence is defined as an IQ IQ ≥ 80, borderline intelligence as IQ = 70–79, and intellectually impaired as IQ < 70.
Discussion
We evaluated 159 patients diagnosed with 22q11.2 DS. When we looked at the relationship between COMT genotype and IQ no significant results were seen. Power analysis indicated that we would be able to detect a significant effect if the difference between the two groups was as large as 5.3 points for the maximum sample size of 159 (full scale IQ), and as large as 7.2 for the minimum sample size of 106 (verbal and performance IQ). Similar studies, albeit with fewer participants, on the whole have yielded inconclusive results. In a study with 44 patients (mean age of 11.1 years), Bearden et al. [2004] reported that Val allele carriers tend to have a higher full scale IQ compared to Met allele carriers. Although not significant, our data shows a trend that Met allele carriers actually have a slightly higher mean full-scale IQ. Shashi et al. [2006] found the Met allele advantageous in Full-Scale and Verbal IQ. Although negative findings have been previously reported by Glaser et al. [2006] in a study with 34 patients, and by van Amelsvoort et al. [2008] in a study with 26 patients, our study is the largest reporting negative findings to date, with 159 patients evaluated.
The participants in this study were relatively young (mean age: 9.85 ± 2.92 years). It has been suggested that COMT is expressed from childhood through adolescence and adulthood, with the most optimum levels expressed in early adulthood [Kates et al., 2006; van Amelsvoort et al., 2008]. It is therefore possible that a correlation will occur later, in adolescence or early adulthood, when the VIQ has been reported to decline. In their longitudinal study of 24 patients with 22q11.2DS, Gothelf et al. [2005] reported a significant decrease in Verbal IQ from childhood to late adolescence or early adulthood in participants with the low enzymatic activity Met allele. Conversely, Bassett et al. [2007] studied 73 adults with 22q11.2DS and found no significant association between COMT genotype and IQ. As described earlier, several of our participants have had more than one IQ test administered; however, widely varying time spans between repeated tests did not allow for accurate prediction of possible age related changes. Ideally, a future study would have a significant number of participants with consecutive IQ data points spanning childhood, adolescence, and adulthood in order to address this question of age as a potential mediator of the effects of the COMT genotype on intelligence and other cognitive abilities [Bassett et al., 2007].
Acknowledgments
The authors of this paper would like to thank Kathryn Dormans and Eric Rappaport, PhD for their technical assistance. These studies were supported in part by funding from the NIH/NICHD (HD70454, HD026979, MH87636, MH087626, and MH101719) and the Charles E.H. Upham chair in Pediatrics (B.S.E.).
Grant sponsor: NIH/NICHD; Grant numbers: HD70454, HD026979, MH87636, MH087626, MH101719; Grant sponsor: Charles E.H. Upham Chair in Pediatrics.
References
- Baker K, Baldeweg T, Sivagnanasundaram S, Scambler P, Skuse D. COMT Val108/158 Met modifies mismatch negativity and cognitive function in 22q11 deletion syndrome. Biol Psychiatry. 2005;58(1):23–31. doi: 10.1016/j.biopsych.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Caluseriu O, Weksberg R, Young DA, Chow EW. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biol Psychiatry. 2007;61(10):1135–1140. doi: 10.1016/j.biopsych.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, Jawad AF, Lynch DR, Sokol S, Kanes SJ, McDonald-McGinn DM, Saitta SC, et al. Effects of a functional COMT polymorphism on prefrontal cognitive function in patients with 22q11.2 deletion syndrome. Am J Psychiatry. 2004;161(9):1700–1702. doi: 10.1176/appi.ajp.161.9.1700. [DOI] [PubMed] [Google Scholar]
- Carmel M, Zarchi O, Michaelovsky E, Frisch A, Patya M, Green T, Gothelf D, Weizman A. Association of COMT and PRODH gene variants with intelligence quotient (IQ) and executive functions in 22q11.2DS subjects. J Psychiatr Res. 2014;56:28–35. doi: 10.1016/j.jpsychires.2014.04.019. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35(9):789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabant EM, Hariri AR, Meyer-Lindenberg A, Munoz KE, Mattay VS, Kolachana BS, Egan MF, et al. Catechol O-methyltransferase val158met genotype and neural mechanisms related to affective arousal and regulation. Arch Gen Psychiatry. 2006;63(12):1396–1406. doi: 10.1001/archpsyc.63.12.1396. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Gscheidle T, Weirich M, Rawlings R, Hyde TM, Bigelow L, et al. Relative risk for cognitive impairments in siblings of patients with schizophrenia. Biol Psychiatry. 2001;50(2):98–107. doi: 10.1016/s0006-3223(01)01133-7. [DOI] [PubMed] [Google Scholar]
- Glaser B, Debbane M, Hinard C, Morris MA, Dahoun SP, Antonarakis SE, Eliez S. No evidence for an effect of COMT Val158Met genotype on executive function in patients with 22q11 deletion syndrome. Am J Psychiatry. 2006;163(3):537–539. doi: 10.1176/appi.ajp.163.3.537. [DOI] [PubMed] [Google Scholar]
- Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79(4):348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, Kwon H. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci. 2005;8(11):1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Law AJ, Frisch A, Chen J, Zarchi O, Michaelovsky E, Ren-Patterson R, et al. Biological effects of COMT haplotypes and psychosis risk in 22q11.2 deletion syndrome. Biol Psychiatry. 2014;75(5):406–413. doi: 10.1016/j.biopsych.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati FR, Molina Gomes D, Ferreira JC, Dupont C, Alesi V, Gouas L, Horelli-Kuitunen N, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015;35(8):801–809. doi: 10.1002/pd.4613. [DOI] [PubMed] [Google Scholar]
- Kates WR, Antshel KM, Abdulsabur N, Colgan D, Funke B, Fremont W, Higgins AM, et al. A gender-moderated effect of a functional COMT polymorphism on prefrontal brain morphology and function in velo-cardio-facial syndrome (22q11. 2 deletion syndrome) Am J Med Genet B Neuropsychiatr Genet. 2006;141B(3):274–280. doi: 10.1002/ajmg.b.30284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Allen JE, Christensen M, Davis P, Falin LJ, Grabmueller C, Hughes DS, et al. Ensembl Genomes 2013: Scaling up access to genome-wide data. Nucleic Acids Res. 2014;42:D546–D552. doi: 10.1093/nar/gkt979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, Weinshilboum RM. Human catechol-O-methyltransferase pharmacogenetics: Description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. 1996;6(3):243–250. doi: 10.1097/00008571-199606000-00007. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, et al. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genet Couns. 1999;10(1):11–24. [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet Med. 2001;3(1):23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Minugh-Purvis N, Kirschner RE, Jawad A, Tonnesen MK, Catanzaro JR, Goldmuntz E, et al. The 22q11.2 deletion in African-American patients: An underdiagnosed population? Am J Med Genet Part A. 2005;134(3):242–246. doi: 10.1002/ajmg.a.30069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, Zackai EH, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071. doi: 10.1038/nrdp.2015.71. http://www.nature.com/articles/nrdp201571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EM, Batshaw ML, Solot CB, Gerdes M, McDonald-McGinn DM, Driscoll DA, Emanuel BS, et al. Psychoeducational profile of the 22q11.2 microdeletion: A complex pattern. J Pediatr. 1999;134(2):193–198. doi: 10.1016/s0022-3476(99)70415-4. [DOI] [PubMed] [Google Scholar]
- Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: A population-based study in Western Sweden. Arch Dis Child. 2004;89(2):148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. Seattle (WA): University of Washington Seattle; 1993–2016. GeneReviews® [Internet] Available from: http://www.ncbi.nlm.nih.gov/books/NBK1116/ [Google Scholar]
- Shashi V, Keshavan MS, Howard TD, Berry MN, Basehore MJ, Lewandowski E, Kwapil TR. Cognitive correlates of a functional COMT polymorphism in children with 22q11.2 deletion syndrome. Clin Genet. 2006;69(3):234–238. doi: 10.1111/j.1399-0004.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- Tezenas Du Montcel S, Mendizabai H, Ayme S, Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33(8):719. doi: 10.1136/jmg.33.8.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amelsvoort T, Zinkstok J, Figee M, Daly E, Morris R, Owen MJ, Murphy DG, et al. Effects of a functional COMT polymorphism on brain anatomy and cognitive function in adults with velo-cardio-facial syndrome. Psychol Med. 2008;38:89–100. doi: 10.1017/S0033291707000700. [DOI] [PubMed] [Google Scholar]