

SUMMARY
We examine synaptic connectivity and cocaine-evoked plasticity at specific networks within the nucleus accumbens (NAc). We identify distinct subpopulations of D1+ medium spiny neurons (MSNs) that project to either the ventral pallidum (D1+VP) or the ventral tegmental area (D1+VTA). We show that inputs from the ventral hippocampus (vHPC), but not the basolateral amygdala (BLA), are initially biased onto D1+VTA MSNs. However, repeated cocaine exposure eliminates the bias of vHPC inputs onto D1+VTA MSNs, while strengthening BLA inputs onto D1+VP MSNs. Our results reveal that connectivity and plasticity depend on the specific inputs and outputs of D1+ MSNs and highlight the complexity of cocaine-evoked circuit level adaptations in the NAc.
Graphical Abstract
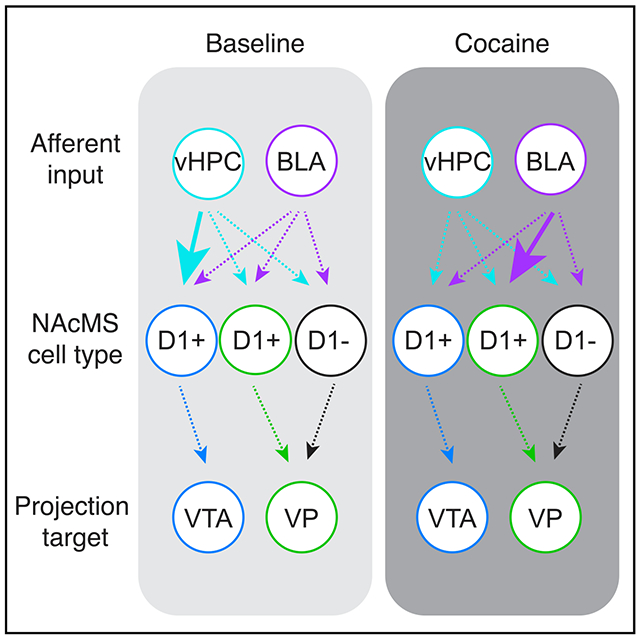
In Brief
Baimel et al. examine how cocaine exposure alters specific circuits in the nucleus accumbens medial shell. They find that D1-expressing (D1+) medium spiny neurons projecting to ventral tegmental area and ventral pallidum are distinct populations. These two cell types differ in both their baseline synaptic connectivity and cocaine-evoked synaptic plasticity.
INTRODUCTION
The nucleus accumbens (NAc) mediates reward-related behaviors and is reorganized by repeated exposure to drugs of abuse (Hyman et al., 2006; Lüscher, 2016). Throughout the striatum, medium spiny neurons (MSNs) segregate into two broad populations, expressing either the D1 or D2 subtype of dopamine receptors (Gerfen et al., 1990; Lobo and Nestler, 2011). Selective activation of D1+ or D2+ MSNs engage distinct patterns of brain wide activity (Lee et al., 2016) and exert opposing influences on motivated behavior (Bock et al., 2013; Calipari et al., 2016; Creed et al., 2016; Kravitz et al., 2010, 2012; Lobo et al., 2010). In the dorsal striatum, D1+ MSNs project via the “direct” pathway to the substantia nigra, whereas D2+ MSNs project via the “indirect” pathway to the globus pallidus (Kravitz et al., 2010; Smith et al., 1998). Recent work highlights additional diversity of D1+ MSNs between (Yang et al., 2018) and within (Al-Hasani et al., 2015; Gibson et al., 2018) different subregions of the NAc. In particular, D1+ MSNs can project to multiple targets (Gibson et al., 2018; Mogenson et al., 1983; O’Connor et al., 2015), including the ventral pallidum (VP) and ventral tegmental area (VTA) (Creed et al., 2016; Kupchik et al., 2015). However, it remains unclear whether distinct subpopulations of D1+ MSNs project to the VP and VTA and if these cells receive and process different synaptic connections.
MSNs in the NAc medial shell (NAcMS) are driven by long-range excitatory inputs, including from the ventral hippocampus (vHPC) and basolateral amygdala (BLA) (Britt et al., 2012; Phillipson and Griffiths, 1985; Sesack and Grace, 2010). These inputs carry distinct motivational signals (Ambroggi et al., 2008; Caine et al., 2001; Goto and Grace, 2008; Stuber et al., 2011) and undergo robust drug-evoked synaptic plasticity (Britt et al., 2012; MacAskill et al., 2014; Pascoli et al., 2014). Changes in synaptic strength contribute to the development of addiction (Britt and Bonci, 2013; Lüscher and Malenka, 2011) and depend on both the presynaptic input (vHPC versus BLA) and postsynaptic cell type (D1+ versus D2+). For example, cocaine exposure primarily rewires connections to D1+ MSNs (Grueter et al., 2013; Kim et al., 2011; Lee et al., 2006; MacAskill et al., 2014; Pascoli et al., 2014), but the mechanisms differ for BLA and vHPC inputs (Britt et al., 2012; MacAskill et al., 2014; Pascoli et al., 2014). However, it is unknown if changes in synaptic strength are homogeneous across D1+ MSNs or also depend on their projection targets (VP versus VTA). For example, does equivalent plasticity occur for all D1+ MSNs, or are particular output pathways strengthened or weakened? Similarly, does the same plasticity occur for vHPC and BLA inputs, or are signals differentially routed to downstream areas? Here we answer these questions in the mouse NAcMS, revealing a critical role for the projection target of MSNs.
RESULTS
Distinct Subpopulations of D1+ MSNs Project to the VP and VTA
To examine the projection targets of MSNs, we first injected anterograde AAV-eGFP into the NAcMS, which labeled axons in both the VP and VTA (Figure S1). To identify projection neurons in the NAcMS, we then injected fluorescent retrograde tracers (cholera toxin subunit B [CTB]-647 and CTB-488) into the VP and VTA of D1-tdTomato mice. We found that VTA projections consist largely of D1+ MSNs, while the VP receives inputs from both D1+ and D1− MSNs, with the latter used as a proxy for D2+ MSNs (Figures 1A and 1B) (Scudder et al., 2018). Overall, retrograde injections in the VP and the VTA labeled equivalent numbers of D1+ MSNs (VP = 225 ± 32 cells per mouse; VTA =170 ± 30 cells per mouse; Mann-Whitney test: U = 7, p = 0.3; n = 5 mice). However, there was minimal co-labeling (dual-labeled cells = 32 ± 6 cells per mouse, 7% of labeled D1+ MSNs; n = 5 mice), indicating that D1+ MSNs targeting the VP (D1+VP) or targeting the VTA (D1+VTA) are largely distinct populations (Figure 1B). We next examined if D1+VP and D1+VTA MSNs could be differentiated on the basis of their morphological or physiological properties. Using whole-cell recordings and two-photon microscopy, we found subtle differences between D1+ and D1− MSNs (Gertler et al., 2008; Kreitzer and Malenka, 2007; Shen et al., 2019), but none between D1+VP and D1+VTA MSNs. All MSN subtypes had similar dendritic lengths (D1− = 1,213 ± 108 μm; D1+VTA = 1,289 ± 114 μm; D1+VP = 1,404 ± 138 μm; Kruskal-Wallis test: H[2] = 1.184, p = 0.6; n = 12–14 cells from 8–10 mice per group) (Figure 1C) and intrinsic properties (Figure S1), but D1− MSNs were more excitable than both D1+VP and D1+VTA MSNs (two-way ANOVA: current step × cell type interaction, F[16, 296] = 6.6, p < 0.0001; Sidak’s multiple comparisons: p = 0.4; n = 12–14 cells from 8–10 mice per group) (Figure 1D). These results reveal two subpopulations of D1+ MSNs in the NAcMS, motivating us to ask how they are contacted by afferents and whether these connections undergo differential synaptic plasticity.
Figure 1. D1+ MSNs Projecting to VP and VTA Are Distinct Cell Populations.

(A) Left: schematic and representative images for injections of CTB-647 into the VP (green) and CTB-488 into the VTA (blue) of D1-tdTomato mice (red) (scale bar, 500 μm). Right: confocal images of retrogradely labeled neurons in the NAcMS (arrows indicate dual-labeled MSNs; scale bar, 100 μm).
(B) Left: quantification of CTB and tdTomato overlap in the NAcMS (total cell count of three slices per mouse, five mice). Right: summary of the number of D1+VTA, D1+VP and dual-projecting MSNs in the NAcMS (average number of labeled cells of three slices per mouse, five mice).
(C) Two-photon images of D1+VTA, D1+VP, and D1− MSNs (left) (scale bar, 20 μm) and quantification of total dendritic length for all reconstructed cells (right), showing no differences.
(D) Examples of physiological responses of D1+VTA, D1+VP, and D1− MSNs to current injections of 250 and −50 pA. Right: summary of current step-evoked firing, showing similar number of action potentials (APs) for D1+VTA and D1+VP MSNs.
Box-and-whisker plots represent median and minimum to maximum. F-I curve is presented as mean ± SEM. *p < 0.05.
See also Figure S1.
Hippocampal Inputs Are Selectively Biased onto D1+VTA MSNs
We next examined how long-range afferents from the vHPC and the BLA contact D1+VTA and D1+VP MSNs in the NAcMS. These inputs differentially target D1+ and D1− MSNs and undergo distinct forms of drug-evoked synaptic plasticity (MacAskill et al., 2014). To study specific connections, we injected AAV-ChR2-eYFP in the vHPC or the BLA and CTB-647 in the VTA or the VP of D1-tdTomato mice. vHPC fibers in the NAc were largely restricted to the NAcMS (Figure 2A; n = 3 mice) (Britt et al., 2012), where they preferentially synapsed onto D1+ MSNs (Figure S2; n = 9 pairs / 5 mice) (MacAskill et al., 2012, 2014; Scudder et al., 2018). To determine if this connectivity depends on projection target, we recorded AMPAR and NMDAR excitatory postsynaptic currents (EPSCs) from pairs of nearby D1+VTA and D1− MSNs or D1+VP and D1− MSNs in the same slice, allowing us to directly compare synaptic strength (MacAskill et al., 2012, 2014; Scudder et al., 2018). We found that vHPC-evoked AMPAR EPSCs were larger onto D1+VTA MSNs relative to neighboring D1− MSNs (AMPAR amplitude: D1− = 245 ± 47 pA, D1+VTA = 493 ± 44 pA; Wilcoxon test: W = 55, p = 0.002) (Figure 2B), with a similar bias found for vHPC-evoked NMDAR EPSCs (Figure S1). We quantified this bias as the EPSC amplitude ratio for each pair of recorded neurons (D1+VTA / D1− ratio: AMPAR = 2.2 [95% CI = 1.5–3.3]; one-sample t test: t(9) = 4.9, p = 0.0009; NMDAR = 1.9 [95% CI = 1.1–3.1]; one-sample t test: t(9) = 2.8, p = 0.02; n = 10 pairs/7 mice) (Figures 2B and S1). Surprisingly, this bias was absent when comparing vHPC-evoked EPSCs at pairs of D1+VP and D1− MSNs (AMPAR amplitude: D1− = 462 ± 79 pA, D1+VP = 398 ± 42 pA; Wilcoxon test: W = −13, p = 0.5) (D1+VP / D1− ratio: AMPAR = 0.9 [95% CI = 0.7–1.2]; one-sample t test: t(8) = 0.7, p = 0.5; NMDAR = 0.8 [95% CI = 0.6–1.2]; one-sample t test: t(8) = 1.1, p = 0.3; n = 9 pairs / 8 mice) (Figure 2C and S1). These findings indicate that preferential targeting of vHPC inputs onto D1+ MSNs is not a universal property but a specific feature of cells that project to the VTA.
Figure 2. vHPC Inputs Are Stronger onto D1+VTA Cells in Naive Mice.

(A) Left: schematic for injections of AAV-ChR2-eYFP into the vHPC and CTB-647 into the VP or the VTA. Right: representative image of viral expression in the vHPC (scale bar, 500 μm) and of dense vHPC axon labeling in the NAcMS (scale bar, 100 μm).
(B) Left: vHPC inputs evoke larger EPSCs at D1+VTA MSNs compared with neighboring D1− MSNs. Middle: summary of the absolute amplitude of vHPC-evoked AMPAR EPSCs at D1− and D1+VTA MSNs, where lines indicate pairs of recorded neurons. Right: summary of D1+VTA/D1− amplitude ratios for AMPAR EPSCs at −70 mV and NMDAR EPSCs at +40 mV.
(C) Similar to (B), showing no bias of vHPC inputs onto pairs of D1+VP and D1− MSNs.
(D) Left: schematic for injections of AAV-ChR2-eYFP into the BLA and CTB-647 into the VP or the VTA. Right: representative image of viral expression in the BLA (scale bar, 500 μm) and of BLA axon labeling in the NAcMS (scale bar, 100 μm).
(E) Left: there is no bias of BLA-evoked EPSCs at D1+VTA MSNs compared to neighboring D1− MSNs. Middle: summary of the absolute amplitude of BLA-evoked AMPAR EPSCs at D1− and D1+VTA MSNs, where lines indicate pairs of recorded neurons. Right: summary of D1+VTA/D1− amplitude ratios for AMPAR EPSCs at −70 mV and NMDAR EPSCs at +40 mV.
(F) Similar to (E) showing no bias of BLA-evoked EPSCs at D1+VP MSNs compared to neighboring D1− MSNs.
Average traces are presented as mean ± SEM. Ratio data are presented as geometric mean with 95% CI on logarithmic axes. *p < 0.05.
See also Figure S1.
BLA fibers are also prominent in the NAcMS (Figure 2D; n = 3 mice) but, unlike vHPC inputs, are unbiased onto D1+ and D1− MSNs (Figure S2; n = 7 pairs/5 mice) (MacAskill et al., 2014). We found that BLA-evoked AMPAR and NMDAR EPSCs were also unbiased at neighboring D1+VTAand D1− MSNs (AMPAR amplitude: D1− = 247 ± 76 pA, D1+VTA = 221 ± 63 pA; Wilcoxon test: W = −8, p = 0.6) (D1+VTA / D1− ratio: AMPAR = 0.9 [95% CI = 0.4–2.0]; one-sample t test: t(7) = 0.4, p = 0.7; NMDAR = 1.1 [95% CI = 0.6–2.0]; one-sample t test: t(7) = 0.3, p = 0.8; n = 8 pairs / 6 mice) (Figures 2E and S1) and pairs of D1+VP and D1− MSNs (AMPAR amplitude: D1− = 242 ± 62 pA, D1+VP = 214 ± 42 pA; Wilcoxon test: W = 0, p > 0.9) (D1+VP / D1− ratio: AMPAR =1.0 [95% CI = 0.6–1.6]; one-sample t test: t(6) = 0.009, p = 1.0; NMDAR = 1.0 [95% CI = 0.6–1.7]; one-sample t test: t(6) = 0.1, p = 0.9; n = 7 pairs / 6 mice) (Figures 2F and S1). Because similar targeting was found for AMPAR and NMDAR EPSCs, there was no difference in the AMPAR/NMDAR ratio across input or cell type (Figure S1). Together, these results suggest that baseline connectivity strongly depends on both the specific input and output of D1+ MSNs.
Cocaine-Evoked Synaptic Plasticity Depends on MSN Projection Target
Cocaine exposure fundamentally rewires circuits in the NAcMS, and these effects occur predominantly at D1+ MSNs (MacAskill et al., 2014; Pascoli et al., 2014; Scudder et al., 2018). However, it remains unknown if drug-evoked synaptic plasticity depends primarily on dopamine receptor expression or projection target. For example, does plasticity occur at all D1+ MSNs, irrespective of downstream target? Or does it occur at only a subset of D1+ MSNs that project to either the VP or VTA? To distinguish between these possibilities, we next examined the impact of cocaine sensitization on synaptic connectivity at the three subtypes of MSNs. Mice were injected with either cocaine or saline for 5 consecutive days, followed by a cocaine challenge after a 24 h withdrawal (Figure 3A). Cocaine-treated mice showed increased locomotion in response to the cocaine challenge, a signature of sensitization (distance traveled: cocaine, 15.3 ± 1.8 m; saline, 8.1 ± 0.7 m; Mann-Whitney test: U = 34, p = 0.03; n = 12 mice per group) (Figure 3B).
Figure 3. Cocaine-Induced Plasticity Depends on Projection Target.

(A) Sensitization protocol, with 3 days of habituation, 5 days of either cocaine or saline injections, and 1 day of withdrawal, followed by either cocaine challenge to assess behavior or slice recordings to study synaptic physiology.
(B) Left: typical movement of saline and cocaine mice to a challenge dose of cocaine on day 6 of the protocol. Middle: summary of distance traveled in the cocaine challenge session. Right: summary of distance traveled per experiment day.
(C) Left: vHPC inputs evoke larger EPSCs at D1+VTA MSNs in saline mice, but this bias is abolished in cocaine mice. Right: summary of D1+VTA/D1− amplitude ratios.
(D) Similar to (C), showing equivalent vHPC-evoked EPSCs at D1+VP and D1− MSNs in both saline and cocaine mice.
(E) Left: BLA-evoked EPSCs are similar at D1+VTA and D1− MSNs in saline and cocaine mice. Right: summary of D1+VTA/D1− amplitude ratios.
(F) Similar to (E) but showing a large increase in EPSCs at D1+VP MSNs in cocaine mice.
Average traces are presented as mean ± SEM. Ratio data are presented as geometric mean with 95% CI on logarithmic axes. *p < 0.05.
See also Figure S3.
To examine cocaine-evoked synaptic plasticity, we used the same protocol but performed acute slice recordings on the challenge day instead of behavior (Figure 3A). We found that cocaine exposure equalized vHPC inputs onto D1+VTA and nearby D1− cells, whereas saline exposure had no effect (D1+VTA/ D1− ratio: AMPAR: cocaine = 1.0 [95% CI = 0.7–1.4]; saline = 2.4 [95% CI = 1.3–4.5]; Mann-Whitney test: U = 9, p = 0.01; NMDAR: cocaine = 0.8 [95% CI = 0.5–1.2]; saline = 2.6 [95% CI, 1.2–5.4]; Mann-Whitney test: U = 6, p = 0.005; n = 8 pairs from 5 or 6 mice per group) (Figures 3C and S3). In contrast, vHPC inputs remained similar onto nearby D1+VP and D1− cells in both cocaine- and saline-treated mice (D1+VP / D1− ratio: AMPAR: cocaine = 1.2 [95% CI = 0.7–1.9]; saline = 1.0 [95% CI = 0.61.5]; Mann-Whitney test: U = 32, p = 0.7; NMDAR: cocaine = 1.5 [95% CI = 0.8–2.6]; saline = 1.0 [95% CI = 0.6–1.5]; Mann-Whitney test: U = 20, p = 0.1; n = 8 or 9 pairs from 7 or 9 mice per group) (Figures 3D and S3). These results agree with previous work showing that cocaine dampens vHPC inputs onto unsorted D1+ MSNs (Figure S4; n = 8 or 9 pairs from 5 or 6 mice per group) (MacAskill et al., 2014; Scudder et al., 2018). However, they further indicate that drug-evoked plasticity of vHPC inputs is highly selective for D1+ MSNs projecting to the VTA.
Last, we performed parallel experiments for BLA inputs. Surprisingly, we found that cocaine exposure did not potentiate BLA inputs onto D1+VTA MSNs, resulting in similar targeting to saline-treated mice (D1+VTA/ D1− ratio: AMPAR: cocaine = 0.8 [95% CI = 0.4–1.7]; saline = 1.0 [95% CI = 0.5–2.0]; Mann-Whitney test: U = 33, p = 0.5; NMDAR: cocaine = 0.8 [95% CI = 0.61.1]; saline = 0.8 [95% CI = 0.4–1.7]; Mann-Whitney test: U = 40, p >0.99; n = 9 pairs/6 or 7 mice per group) (Figures 3E and S3). In contrast, cocaine greatly enhanced BLA inputs onto D1+VP MSNs relative to D1− MSNs, whereas saline exposure had no effect (D1+VP / D1− ratio: AMPAR: cocaine = 3.9 [95% CI = 2.0–7.8]; saline = 1.1 [95% CI = 0.7–2.0]; Mann-Whitney test: U = 15, p = 0.01; NMDAR: cocaine = 2.8 [95% CI = 1.2–6.3]; saline = 0.9 [95% CI = 0.5–1.4]; Mann-Whitney test: U = 13, p = 0.008; n = 10 pairs from 7 or 10 mice per group) (Figures 3F and S3). These findings are consistent with cocaine’s enhancing BLA inputs onto unsorted D1+ cells (Figure S4; n = 12 or 13 pairs from 9 mice per group) (MacAskill et al., 2014). However, they highlight the importance of projection target, showing that drug-evoked plasticity of BLA inputs occurs only at D1+ MSNs projecting to the VP.
DISCUSSION
Our results indicate that synaptic connectivity and cocaine-evoked plasticity depend on the specific afferent, type of dopamine receptor, and projection target of MSNs. We found that D1+ MSNs in the NAcMS consist of two distinct populations that send projections to either the VP or the VTA. Although both subtypes are contacted by inputs from the vHPC and the BLA, vHPC inputs are stronger onto D1+VTA MSNs, whereas BLA inputs show no bias. Cocaine exposure selectively dampens vHPC inputs at D1+VTA MSNs but strengthens BLA inputs at D1+VP MSNs. These findings emphasize the importance of the projection targets of D1+ MSNs and how drug exposure reshapes the NAc circuit in both an input- and output-specific manner.
Recent evidence highlights how diverse projections of the NAc participate in motivated behavior (Creed et al., 2016; Gibson et al., 2018; O’Connor et al., 2015; Pardo-Garcia et al., 2019). However, it has been unclear if these projections consisted of collateralizing axon or discrete populations of neurons. We used dual retrograde labeling to show that both D1+ and D1− MSNs project to the VP, whereas only D1+ MSNs project to the VTA. Moreover, we found that D1+ MSNs projecting to the VP and VTA are two largely separate populations of neurons in the NAcMS. This finding contrasts with a recent report, which concluded that the majority of D1+ MSNs in the NAcMS and NAc core collateralize to both the VP and VTA (Pardo-Garcia et al., 2019). The reason for this discrepancy is unclear but could reflect differences in injection strategies. We targeted the VP at an angle in order to avoid the anterior commissure and potential leak into the NAc itself. Our anatomical findings are consistent with the highly selective synaptic connectivity and drug-evoked plasticity we observed at these two classes of projection neurons. Because D1+ MSNs that project to the VTA and LH also represent different subpopulations of cells (Gibson et al., 2018), our results establish that D1+ MSNs constitute multiple parallel output pathways with distinct connectivity, plasticity, and behavioral implications.
Our findings build on decades of work on drug-evoked plasticity in the NAc (Britt and Bonci, 2013; Lobo and Nestler, 2011; Lüscher, 2016; Lüscher and Malenka, 2011), revealing unexpected complexities in how neural circuits can be modified in the progression to addiction (Grueter et al., 2012; Lüscher, 2016). Early work showed that cocaine exposure dramatically alters dendrite and spine morphology (Dumitriu et al., 2012; Maze et al., 2010; Robinson et al., 2001), which suggested that synaptic connections might be fundamentally reorganized. Cocaine-evoked synaptic plasticity is now well established, with changes in glutamatergic signaling in the NAc closely linked to behavior (Bock et al., 2013; Creed et al., 2015; Pascoli et al., 2014; Scheyer et al., 2016). Recently, this reorganization was shown to depend on both input and cell type (Britt et al., 2012; MacAskill et al., 2014; Pascoli et al., 2014). For example, drug-evoked plasticity throughout the NAc primarily occurs at D1+ MSNs compared with neighboring D2+ or D1− MSNs (Grueter et al., 2013; Kim et al., 2011; Lee et al., 2006; MacAskill et al., 2014; Pascoli et al., 2014). We have previously shown that vHPC and BLA inputs are bidirectionally redistributed after cocaine exposure, with the strength of individual vHPC connections weakened, and the number of BLA connections increased at D1+ MSNs in the NAcMS (MacAskill et al., 2014). Here we extend on these findings by showing that drug-evoked changes critically depend on the specific long-range projection targets of D1+ MSNs. Our findings are parsimonious with previous observations but show how even more specific subnetworks of MSNs are affected by drug exposure. Given that drug-evoked plasticity evolves after withdrawal, with changes in AMPAR and NMDAR responses shifting over days and weeks (Lee et al., 2013; Lüscher, 2016; Pascoli et al., 2014; Thomas et al., 2001), it will be important to assess synaptic changes in these subnetworks of MSNs at other time points after drug exposure and to determine how they contribute to drug-related behaviors.
Our results indicate that cocaine exposure shifts the influence of vHPC and BLA inputs on divergent NAc output to the VP and the VTA. We find that vHPC inputs are initially biased only onto D1+VTA MSNs and that cocaine selectively reduces vHPC inputs only onto these cells. Interestingly, recent work shows that the primary target of NAcMS D1+ terminals in the VTA are medial dopamine neurons that project back to NAcMS (Yang et al., 2018). The selective dampening of vHPC inputs onto D1+VTA MSNs might represent a disinhibitory mechanism, reducing the inhibition onto dopamine neurons in the VTA, and in turn enhancing dopamine release in the NAcMS. This could work in harmony with local disinhibition in the lateral VTA to drive cocaine-induced behaviors (Bocklisch et al., 2013; Yang et al., 2018). In contrast, we found that BLA inputs are initially unbiased onto MSNs, but cocaine selectively enhanced BLA inputs only onto D1+VP MSNs. Because cocaine sensitization is dependent on potentiation of D1+ MSN outputs to the VP (Creed et al., 2016), the plasticity we observe may reflect an upstream circuit mechanism to enhance D1+ terminal activity in the VP. Given the diversity of cell types and projections of the VP (Faget et al., 2018; Mahler et al., 2014; Tooley et al., 2018), in the future it will be important to determine the precise circuit connections of D1+ and D1− MSNs in this region.
Overall, our work highlights that circuitry and drug-evoked plasticity in the NAc are not simply regulated at levels of input (vHPC versus BLA), receptor expression (D1 versus D2), or projection target (VP versus VTA) but rather a complex combination of these factors. Importantly, many of these details would be missed when pooling vHPC and BLA inputs, D1+ and D1− cells, or even D1+VTA and D1+VP cells. Therefore, our results expand the classical binary view of D1+ and D2+ MSNs and highlight the heterogeneity of D1+ MSNs within the NAcMS. An exciting future direction will be to determine if projection identity distinguishes distinct subpopulations of MSNs in other parts of the striatum and if these neurons also undergo differential plasticity in disease states.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Adam Carter (adam.carter@nyu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Physiology experiments were performed in acute slices from D1-tdTomato hemizygous BAC transgenic mice (Ade et al., 2011) crossed with wild-type C57BL/6J mice (originally purchased from Jackson Laboratories). Behavior and anatomy experiments also used wild-type mice. For all experiments both sexes were used, and mice were aged P42-P70. No animals had been involved in previous procedures. Animals were group-housed with same-sex littermates in a dedicated animal care facility and were maintained on a 12-hour light / dark cycle with food and water given ad libitum. All procedures were conducted in accordance with guidelines approved by the New York University animal welfare committee.
METHOD DETAILS
All experiments were replicated in at least 3 animals. For all cocaine experiments, pairs of sex-matched littermates were weaned and housed together. Within a given pair, one mouse was assigned to the cocaine group and the other to the saline group and both mice underwent all surgical, behavioral and recording procedures together to control for age, viral expression time and experimental conditions. Experimenters were not blind to experimental groups. No pre-test analyses were used to estimate sample sizes. No data were excluded from final analyses.
Stereotaxic injections
Stereotaxic injections were performed on P33-P46 mice. Mice were anesthetized with a mixture of ketamine (10 mg/ml) and xylazine (0.1 mg/ml) and head fixed in a stereotax (Kopf Instruments). A small craniotomy was made over the injection site, through which retrograde tracers and viruses were injected. Injection site coordinates were determined relative to bregma (mediolateral axis, dorsoventral axis, and rostrocaudal axis (in mm): NAc = −1.7, −4.4, +1.6 at a 14° angle; vHPC = −3.0, −4.6 and −3.6, −3.0; BLA = −3.1, −5.0, −1.5; VP = −1.4, −5.7, −2.5 at a 27° angle approaching from the caudal end of the skull; VTA= −0.4, −4.5, −3.0). Borosilicate pipettes with 5–10 μm tip diameters were backfilled and 100 – 360 nL was pressure-injected using a Nanoject III (Drummond) with 45 – 60 s inter-injection intervals. Pipettes were left in place for at least 10 minutes after injection before being slowly withdrawn. AAV1-hSyn-GFP (UPenn Vector Core, AV-1-PV1696) and AAV1-DIO-eYFP (UPenn Vector Core, AV-1-27056) were used for Cre-independent and -dependent axon labeling, and optogenetic stimulation was achieved with AAV1-hSyn-ChR2-eYFP (UPenn Vector Core, AV-1-26973P). For retrograde labeling, pipettes were filled with cholera toxin subunit B (CTB) conjugated to Alexa 488 or 647 (Life Technologies). After all injections, animals were returned to their home cages for 10 – 16 days before being used for experiments.
Slice preparation
Mice were anesthetized with an intraperitoneal injection of a lethal dose of ketamine/xylazine and perfused intracardially with an ice-cold cutting solution containing the following (in mM): 65 sucrose, 76 NaCl, 25 NaHCO3,1.4 NaH2PO4, 25 glucose, 2.5 KCl, 7 MgCl2, 0.4 Na-ascorbate, and 2 Na-pyruvate (bubbled with 95% O2/5% CO2). 300 μm coronal sections were cut in this solution and transferred to ACSF containing the following (in mM): 120 NaCl, 25 NaHCO3,1.4 NaH2PO4, 21 glucose, 2.5 KCl, 2 CaCl2, 1 MgCl2, 0.4 Na-ascorbate, and 2 Na-pyruvate (bubbled with 95% O2/5% CO2). Slices recovered for 30 min at 35°C and then were stored for at least 30 min at 24°C prior to recording. All experiments were conducted at 30 – 32°C.
Electrophysiology
Whole-cell recordings were made from fluorescently identified MSNs in the NAcMS, located 300 – 600 μm medial to the anterior commissure. D1+ and D1− MSNs were identified by the presence or lack of tdTomato fluorescence (Scudder et al., 2018). D1+VTA and D1+VP projection neurons were identified by the additional expression of CTB-647. Neurons were recorded in sequential pairs (D1+ x D1−, D1+VTA x D1−, or D1+VP x D1−), where cells were located at the same depth in the slice and within 50 μm of each other (MacAskill et al., 2012, 2014; Scudder et al., 2018). Recording order was varied between cell types within a given pair. For voltage-clamp experiments, borosilicate pipettes (3-5 MΩ) were filled with a Cs-based internal (in mM: 130 Cs-gluconate, 10 HEPES, 10 Na-phosphocreatine, 4 Mg2-ATP, 0.4 NaGTP, 10 TEA, 2 QX-314, and 10 EGTA, pH 7.3 with CsOH). For current-clamp recordings, pipettes were filled with a K-based internal (in mM: 135 K-gluconate, 7 KCl, 10 HEPES, 10 Na-phosphocreatine, 4 Mg2-ATP, 0.3 NaGTP, and 0.5 EGTA, pH 7.3 with KOH) with 30 μM AlexaFluor-594 added to visualize morphology with 2-photon microscopy. All voltage-clamp recordings were made with 10 μM gabazine to block GABAA receptors and with 10 μM D-serine to allow NMDAR activation. Cells were held at −70 mV to measure AMPAR-mediated currents, and at +40 mV from the experimentally-determined excitatory reversal potential to measure NMDAR-mediated currents. Current-clamp recordings were made with 10 μM NBQX to block AMPAR, 10 μM CPP to block NMDAR, and 10 μM gabazine, and current steps were applied to cells from a holding potential set to −80 mV. Input resistance was calculated using a −50 pA current injection. All chemicals were purchased from Tocris Bioscience or Sigma.
Electrophysiological data were collected using a MultiClamp 700B (Axon Instruments), signals were sampled at 10 kHz, and filtered at 2 kHz for voltage-clamp and 10 kHz for current-clamp recordings. Series resistance was monitored, less than 25 MΩ, and not compensated.
Optogenetics
Glutamate release was triggered by activating channelrhodopsin-2 (ChR2) present in presynaptic terminals of vHPC or BLA inputs to the NAc using 1 – 2 ms pulses (unless otherwise indicated) of 473 nm light from a blue light-emitting diode (LED; 473 nm; Thorlabs) through a 10X0.3 NA immersion objective (Olympus) with a power range of 2 – 22 mW measured at the back focal plane of the objective. Within a given pair of neurons, a light intensity was empirically chosen that yielded usable responses (i.e., 50 – 1000 pA EPSC amplitude at −70 mV for voltage-clamp experiments).
Two-photon microscopy
To obtain images of recorded neurons, cells were filled via the patch pipette during whole-cell recording with AlexaFluor-594 dye (30 μM in internal solution) for at least 15 minutes. Two-photon imaging was performed on a custom microscope using a Titanium:Sapphire laser (Coherent) tuned to 810 nm (Carter and Sabatini, 2004; Chalifoux and Carter, 2010). Az-stack was acquired at 512 × 512 resolution through a 60X 1.0NA objective (Olympus). Morphological reconstruction and analysis of two-photon images were conducted using Volume Integration and Alignment System (Vias) and Neuron Studio software (Computational Neurobiology and Imaging Center, Mount Sinai School of Medicine).
Histology and fluorescence microscopy
Mice were anesthetized and perfused intracardially with 0.01 M PBS followed by 4% PFA. Brains were stored in 4% PFA for 12 – 18 hours at 4°C before being washed three times in 0.01 M PBS. Slices were cut on a VT-1000S vibratome (Leica) at 70 μm thickness, or 40 μm for immunostaining, and were either mounted directly onto gel-coated glass slides or processed for immunolabeling. For tdTomato immunolabeling, slices were incubated for 1 hour in blocking solution (1% bovine serum albumin and 0.2% Triton-X in 0.01 M PBS), primary antibody was applied overnight (rabbit anti-RFP (Rockland) at 1:1000 in blocking solution). Slices were then washed in PBS and incubated with secondary antibody (goat anti-rabbit 594 (abcam) at 1:400), in blocking solution) for 1.5 hours. Slices were coverslipped using ProLong Gold anti-fade reagent with DAPI (Invitrogen) or VectaShield with DAPI (Vector Labs). Fluorescent images were taken on an Olympus VS120 microscope, using a 10X 0.25NA objective (Olympus) or a Leica TCS SP8 confocal microscope, using a 20X 0.75NA and 40X 1.3NA objective (Olympus). All images were processed using NIH ImageJ.
Cocaine sensitization
Mice were first habituated to the behavior room for one 2-hour session. The next day, they were placed in a dimly-lit, sound-attenuated behavioral chamber (1 ft x 1 ft) for a 35-min habituation session. On the following day, to acclimate the animals to injections and to obtain a baseline locomotion measure, mice were placed in the chamber for a 15-min baseline period followed by an IP injection of saline and a 20-min post-injection session in the chamber. Animals then underwent 5 days (1 session per day) of cocaine (15 mg/kg) or saline injection sessions in the chamber. Subsequently, animals either underwent a cocaine challenge session, where both cocaine-treated and saline-treated mice received a 15 mg/kg IP injection of cocaine or were anesthetized and perfused for preparation of acute slices. Behavior was recorded via an overhead camera linked to Ethovision software (Noldus), which quantified locomotor activity during each session. Locomotor sensitization data presented were acquired from pairs of animals used only for behavior, and represent distance traveled (m) during the 20-minute post-injection session. For electrophysiology experiments, D1-tdTomato mice were injected with optogenetic constructs and given at least 10 days to recover prior to initiating behavioral sessions, as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Electrophysiology and two-photon imaging data were acquired using custom software in MATLAB (MathWorks). All electrophysiology, anatomical, and behavioral data were collected from a minimum of 3 animals from different litters. Electrophysiology data analysis was performed in Igor Pro (Wavemetrics). EPSC amplitudes were averaged from a 1 ms window around the peak current for AMPAR EPSCs, and 50 ms after stimulation for NMDAR EPSCs. Average traces in figures depict mean ± SEM of all recorded pairs. Statistical significance of differences between two groups was evaluated using the Wilcoxon Signed-Rank test for paired data or the Mann-Whitney test for unpaired data, and with repeated-measure 2-way ANOVA followed by Sidak’s multiple comparison test for drug effects with p values <0.05 considered significant. Image analysis was conducted using NIH ImageJ. Statistical tests and graph generation were performed using Prism 7 (GraphPad).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-RFP | Rockland Antibodies and Assays | Rockland Cat# 600-401-379; RRID:AB_2209751 |
| Goat Anti-Rabbit 594 | Abcam | Abcam Cat# ab150080; RRID:AB_2650602 |
| Bacterial and Virus Strains | ||
| AAV1-hSYn-hChR2-eYFP | Penn Vector Core | AV-1-26973P |
| AAV1 -hSYn-eYFP | Penn Vector Core | AV-1-PV1696 |
| AAV1-EF1a-DIO-eYFP | Penn Vector Core | AV-1-27056 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cholera toxin subunit B-488 | Thermo Fisher | Cat# C22841 |
| Cholera toxin subunit B-647 | Thermo Fisher | Cat# C34778 |
| Gabazine | Tocris | Cat# 1262 |
| D-Serine | Tocris | Cat# 0226 |
| Prolong Gold with DAPI | Invitrogen | Cat# P36931 |
| VectaShield with DAPI | Vector Labs | RRID: AB_2336790 |
| Cocaine Hydrochloride | Sigma-Aldrich | Cat# C5776 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Lab | RRID: IMSR_JAX:000664 |
| Mouse: D1-tdTomato | Jackson Lab | RRID:IMSR_JAX:016204 |
| Mouse: D1-Cre | Jackson Lab | RRID:MMRRC_030989-UCD |
| Software and Algorithms | ||
| Igor Pro 6 | WaveMetrics | RRID: SCR_000325 |
| MATLAB | MathWorks | RRID: SCR_001622 |
| Prism 7.0 | GraphPad Software | RRID: SCR_002798 |
| Volume Integration and Alignment System (VIAS) | Icahn School of Medicine at Mount Sinai | RRID:SCR_013800 |
| Neuron Studio Software | Icahn School of Medicine at Mount Sinai | RRID:SCR 013798 |
Highlights.
NAc medial shell has multiple D1 receptor-expressing (D1+) medium spiny neurons (MSNs)
Distinct D1+ MSNs project to ventral tegmental area (VTA) and ventral pallidum (VP)
Hippocampus (vHPC) and amygdala (BLA) differentially target D1+VTA and D1+VP MSNs
Cocaine selectively alters vHPC inputs at D1+VTA MSNs and BLA inputs at D1+VP MSNs
ACKNOWLEDGMENTS
We thank the Carter lab and Nic Tritsch for helpful discussions and comments on the manuscript. This work was supported by a CIHR postdoctoral fellowship (C.B.), NIH grant T32 NS086750 (L.M.M.), and NIH R01 DA038138 (A.G.C.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.074.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ade KK, Wan Y, Chen M, Gloss B, and Calakos N (2011). An improved BAC transgenic fluorescent reporter line for sensitive and specific identification of striatonigral medium spiny neurons. Front. Syst. Neurosci 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo C-O, Park SI, Marcinkiewcz CM, Crowley NA, et al. (2015). Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggi F, Ishikawa A, Fields HL, and Nicola SM (2008). Basolateral amygdala neurons facilitate reward-seeking behavior by exciting nucleus accumbens neurons. Neuron 59, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, and Alvarez VA (2013). Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat. Neurosci 16, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JCY, House DRC, Yvon C, de Roo M, Tan KR, and Lüscher C (2013). Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341, 1521–1525. [DOI] [PubMed] [Google Scholar]
- Britt JP, and Bonci A (2013). Optogenetic interrogations of the neural circuits underlying addiction. Curr. Opin. Neurobiol 23, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, and Bonci A (2012). Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine SB, Humby T, Robbins TW, and Everitt BJ (2001). Behavioral effects of psychomotor stimulants in rats with dorsal or ventral subiculum lesions: locomotion, cocaine self-administration, and prepulse inhibition of startle. Behav. Neurosci 115, 880–894. [DOI] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Peña CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, et al. (2016). In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc. Natl. Acad. Sci. U S A 113, 2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, and Sabatini BL (2004). State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44, 483–493. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, and Carter AG (2010). GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron 66, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Pascoli VJ, and Lüscher C (2015). Addiction therapy. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science 347, 659–664. [DOI] [PubMed] [Google Scholar]
- Creed M, Ntamati NR, Chandra R, Lobo MK, and Lüscher C (2016). Convergence of reinforcing and anhedonic cocaine effects in the ventral pallidum. Neuron 92, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu D, Laplant Q, Grossman YS, Dias C, Janssen WG, Russo SJ, Morrison JH, and Nestler EJ (2012). Subregional, dendritic compartment, and spine subtype specificity in cocaine regulation of dendritic spines in the nucleus accumbens. J. Neurosci 32, 6957–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faget L, Zell V, Souter E, McPherson A, Ressler R, Gutierrez-Reed N, Yoo JH, Dulcis D, and Hnasko TS (2018). Opponent control of behavioral reinforcement by inhibitory and excitatory projections from the ventral pallidum. Nat. Commun 9, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr., and Sibley DR (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432. [DOI] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, and Surmeier DJ (2008). Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci 28, 10814–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GD, Prasad AA, Jean-Richard-Dit-Bressel P, Yau JOY, Millan EZ, Liu Y, Campbell EJ, Lim J, Marchant NJ, Power JM, et al. (2018). Distinct accumbens shell output pathways promote versus prevent relapse to alcohol seeking. Neuron 98, 512–520.e6. [DOI] [PubMed] [Google Scholar]
- Goto Y, and Grace AA (2008). Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 31, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Rothwell PE, and Malenka RC (2012). Integrating synaptic plasticity and striatal circuit function in addiction. Curr. Opin. Neurobiol 22, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Robison AJ, Neve RL, Nestler EJ, and Malenka RC (2013). DFosB differentially modulates nucleus accumbens direct and indirect pathway function. Proc. Natl. Acad. Sci. U S A 110, 1923–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, and Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci 29, 565–598. [DOI] [PubMed] [Google Scholar]
- Kim J, Park B-H, Lee JH, Park SK, and Kim J-H (2011). Cell type-specific alterations in the nucleus accumbens by repeated exposures to cocaine. Biol. Psychiatry 69, 1026–1034. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PRL, Kay K, Thwin MT, Deisseroth K, and Kreitzer AC (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci 15, 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, and Malenka RC (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647. [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, and Kalivas PW (2015). Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat. Neurosci 18, 1230–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-W, Kim Y, Kim AM, Helmin K, Nairn AC, and Greengard P (2006). Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc. Natl. Acad. Sci. U S A 103, 3399–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, Ma Y-Y, Huang YH, Wang X, Otaka M, Ishikawa M, Neumann PA, Graziane NM, Brown TE, Suska A, et al. (2013). Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat. Neurosci 16, 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Weitz AJ, Bernal-Casas D, Duffy BA, Choy M, Kravitz AV, Kreitzer AC, and Lee JH (2016). Activation of direct and indirect pathway medium spiny neurons drives distinct brain-wide responses. Neuron 91, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, and Nestler EJ (2011). The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front. Neuroanat 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C (2016). The emergence of a circuit model for addiction. Annu. Rev. Neurosci 39, 257–276. [DOI] [PubMed] [Google Scholar]
- Lüscher C, and Malenka RC (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Little JP, Cassel JM, and Carter AG (2012). Subcellular connectivity underlies pathway-specific signaling in the nucleus accumbens. Nat. Neurosci 15, 1624–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Cassel JM, and Carter AG (2014). Cocaine exposure reorganizes cell type- and input-specific connectivity in the nucleus accumbens. Nat. Neurosci 17, 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, Wilson SP, Deisseroth K, Woodward JJ, and Aston-Jones G (2014). Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat. Neurosci 17, 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Covington HE 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, et al. (2010). Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogenson GJ, Swanson LW, and Wu M (1983). Neural projections from nucleus accumbens to globus pallidus, substantia innominata, and lateral preoptic-lateral hypothalamic area: an anatomical and electrophysiological investigation in the rat. J. Neurosci 3, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor EC, Kremer Y, Lefort S, Harada M, Pascoli V, Rohner C, and Lüscher C (2015). Accumbal D1R neurons projecting to lateral hypothalamus authorize feeding. Neuron 88, 553–564. [DOI] [PubMed] [Google Scholar]
- Pardo-Garcia TR, Garcia-Keller C, Penaloza T, Richie CT, Pickel J, Hope BT, Harvey BK, Kalivas PW, and Heinsbroek JA (2019). Ventral pallidum is the primary target for accumbens D1 projections driving cocaine seeking. J. Neurosci 39, 2041–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, and Lüscher C (2014). Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509, 459–464. [DOI] [PubMed] [Google Scholar]
- Phillipson OT, and Griffiths AC (1985). The topographic order of inputs to the nucleus accumbens in the rat. Neuroscience 16, 275–296. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Gorny G, Mitton E, and Kolb B (2001). Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse 39, 257–266. [DOI] [PubMed] [Google Scholar]
- Scheyer AF, Loweth JA, Christian DT, Uejima J, Rabei R, Le T, Dolubizno H, Stefanik MT, Murray CH, Sakas C, and Wolf ME (2016). AMPA receptor plasticity in accumbens core contributes to incubation of methamphetamine craving. Biol. Psychiatry 80, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudder SL, Baimel C, Macdonald EE, and Carter AG (2018). Hippocampal-evoked feedforward inhibition in the nucleus accumbens. J. Neurosci 38, 9091–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, and Grace AA (2010). Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology 35, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C-J, Zheng D, Li K-X, Yang J-M, Pan H-Q, Yu X-D, Fu J-Y, Zhu Y, Sun Q-X, Tang M-Y, et al. (2019). Cannabinoid CB1 receptors in the amygdalar cholecystokinin glutamatergic afferents to nucleus accumbens modulate depressive-like behavior. Nat. Med 25, 337–349. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, and Bolam JP (1998). Commentary: microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86, 353–387. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, Tye KM, Kempadoo KA, Zhang F, Deisseroth K, and Bonci A (2011). Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature 475, 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, and Malenka RC (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat. Neurosci 4, 1217–1223. [DOI] [PubMed] [Google Scholar]
- Tooley J, Marconi L, Alipio JB, Matikainen-Ankney B, Georgiou P, Kravitz AV, and Creed MC (2018). Glutamatergic ventral pallidal neurons modulate activity of the habenula - tegmental circuitry and constrain reward seeking. Biol. Psychiatry 83, 1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, de Jong JW, Tak Y, Peck J, Bateup HS, and Lammel S (2018). Nucleus accumbens subnuclei regulate motivated behavior via direct inhibition and disinhibition of VTA dopamine subpopulations. Neuron 97, 434–449.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.