

Abstract
Maintaining a pool of functional mitochondria requires degradation of damaged ones within the cell. PINK1 is critical in this quality-control process: loss of mitochondrial membrane potential causes PINK1 to accumulate on the mitochondrial surface, triggering mitophagy. However, little is known about how PINK1 is regulated. Recently, we showed that PINK1 content is kept low in healthy mitochondria by continuous ubiquitination and proteasomal degradation of its mature form via a mechanism inconsistent with the proposed N-end rule process. Using both human female and monkey cell lines, we now demonstrate that once generated within the mitochondria, 52 kDa PINK1 adopts a mitochondrial topology most consistent with it being at the mitochondrial–endoplasmic reticulum (ER) interface. From this particular submitochondrial location, PINK1 interacts with components of the ER-associated degradation pathway, such as the E3 ligases gp78 and HRD1, which cooperate to catalyze PINK1 ubiquitination. The valosin-containing protein and its cofactor, UFD1, then target ubiquitinated PINK1 for proteasomal degradation. Our data show that PINK1 in healthy mitochondria is negatively regulated via an interplay between mitochondria and ER, and shed light on how this mitochondrial protein gains access to the proteasome.
SIGNIFICANCE STATEMENT Regulation of mitochondrial content of PINK1, a contributor to mitophagy, is an important area of research. Recently, we found that PINK1 content is kept low in healthy mitochondria by continuous ubiquitination and proteasomal degradation. We now extend and refine this novel finding by showing that PINK1 localizes at the mitochondrial–endoplasmic reticulum (ER) interface, from where it interacts with the ER-associated degradation machinery, which catalyzes its ubiquitination and transfer to the proteasome. Thus, these data show that PINK1 in healthy mitochondria is negatively regulated via a mitochondria and ER interplay, and how this mitochondrial protein gains access to the proteasome.
Keywords: ERAD, mitochondria, Parkinson's disease, PINK1, proteasome, ubiquitination
Introduction
Parkinson's disease (PD) is an adult-onset neurodegenerative disorder that is characterized pathologically by the loss of ventral midbrain dopaminergic neurons and by the presence of intracytoplasmic proteinaceous inclusions, or Lewy bodies, in the few neurons that are spared (Dauer and Przedborski, 2003). Although PD presents primarily as a sporadic condition, mutations in PINK1, which encodes a mitochondrial kinase, have been associated with an early onset form of the disease, inherited as an autosomal-recessive trait (Valente et al., 2004).
Most of our current knowledge about the function of PINK1 and how PINK1 mutations can cause neuronal death are derived from studies in invertebrate and cell-culture models (Valente et al., 2004; Clark et al., 2006; Park et al., 2006; Narendra et al., 2010; Vives-Bauza et al., 2010; Thomas et al., 2014). One conclusion from these studies is that PINK1 cooperates with the E3 ubiquitin ligase Parkin to promote the turnover of mitochondria by autophagy (also called mitophagy). Upon a profound loss of mitochondrial membrane potential (ΔΨm), it is believed that PINK1 accumulates at the surface of mitochondria, where it phosphorylates ubiquitin (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014) thereby promoting the recruitment of cytosolic Parkin to the organelle (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010). Thereafter, Parkin-decorated mitochondria migrate toward the perinuclear area of the cell, where they are eliminated by the autophagy/lysosomal pathway (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010). This process of mitophagy involves the autophagy receptors optineurin and NDP52 (Lazarou et al., 2015) and the autophagy adaptor TBK1 (Heo et al., 2015).
In mammalian cells deficient in PINK1, the lowering of ΔΨm fails to trigger Parkin-mediated mitochondrial recruitment and mitophagy (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010). Conversely, Parkin translocation and mitophagy are observed in cells overexpressing wild-type (WT) PINK1, but not its kinase-dead mutant counterpart, even when ΔΨm is preserved (Vives-Bauza et al., 2010). These findings indicate that the fine-tuning of the mitochondrial content of PINK1 is not only essential to drive Parkin-dependent mitophagy, but also to allow the autophagic machinery to discriminate between damaged (low ΔΨm) and healthy (normal ΔΨm) mitochondria, a process that is fundamental to preserving a pool of functional mitochondria within cells. The PARL-processed (“mature”) 52 kDa form of PINK1, located in part on the mitochondrial outer membrane (MOM), retains most of the known functional properties of its full-length 63 kDa counterpart. We, and others, have shown that PINK1 is expressed constitutively, and its level is kept low in healthy mitochondria by the preferential polyubiquitination and ensuing proteasomal degradation of its 52 kDa form (Lin and Kang, 2008; Takatori et al., 2008; Zhou et al., 2008; Jin et al., 2010; Liu et al., 2017). However, our recent analyses of the PINK1 degradation process indicate that the bulk of ubiquitinated (Ub)-PINK1 is mitochondrially anchored and not cytosolic and that the N-terminal phenylalanine (F104) of PINK1, which was proposed to form an N-degron motif, is, according to our study (Liu et al., 2017), not facing the cytosol but rather located inside the MOM. These data suggest that the low PINK1 steady-state level in healthy mitochondrial is primarily regulated by a conventional polyubiquitin/proteasome process and not by an N-end rule mechanism, as previously proposed (Yamano and Youle, 2013). However, how mitochondrial PINK1 becomes ubiquitinated and then is turned over to the cytosolic proteasome remains poorly understood.
Here we show that, in mammalian cells, degradation of 52 kDa PINK1 by the proteasome relies on the AAA+ ATPase segregase valosin-containing protein (VCP) and on components of the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway, including the E3 ligases gp78 and HRD1. The ERAD-assisted degradation of 52 kDa PINK1 correlates with the accumulation of mature PINK1 in the vicinity of the ER. Our study emphasizes the key role of 52 kDa PINK1 at all points of this protein metabolic pathway, including ubiquitination, involvement of the ER, and proteasomal degradation. Moreover, by studying PINK1 processing and turnover, we have uncovered a new functional link between mitochondria and ER, as well as a role for ERAD in regulating mitochondrial protein content.
Materials and Methods
Cell culture, transfection, and plasmids.
Human female embryonic kidney epithelial HEK293T, human female cervical epithelial HeLa, monkey kidney fibroblast COS-7 (unknown sex), and human female bone marrow epithelial SH-SY5Y cell lines were from ATCC and maintained in medium suggested by the provider. HA-Ubiquitin and GFP-Sec61β plasmids were from Addgene. 293T Hrd1 CRISPR cells were generous gifts from Yihong Ye (National Institutes of Health). Transient transfections were performed as previously described (Vives-Bauza et al., 2010; Becker et al., 2012). Epitope-tagged PINK1 and PINK1 mutants or deletion constructs were generated by PCR-based DNA mutagenesis. Ufd2A-Flag was a gift from David Sidransky (Johns Hopkins University) (Chatterjee et al., 2008). The backbone vector used to construct C-terminal Venus-tagged PINK1 (Vc-PINK1) was from Kjungjin Kim (Korea Brain Research Institute) (Lee et al., 2010). The APEX backbone was from Addgene, and the IMS-APEX2 plasmid was a generous gift from Alice Ting (MIT).
Antibodies and reagent used in the study.
The following antibodies were used: PINK1 #BC100-494 (Novus); PINK1 #P0075, Flag M2, and HA #H3663 (Sigma-Aldrich); Myc, PDI, Calnexin, and Hrd1 (Abcam); Sec61β (Millipore); Myc 9E10, Ufd1 and Smac (Santa Cruz Biotechnology); VCP, ATP5A (Invitrogen); Cyto C, Tim23, Tom20, and GM130 (BD); KDEL (Calbiochem). Gp78 antibody was gift from Yihong Ye (NIH). The following chemicals were used: MitoTracker (Invitrogen); MG132 (Calbiochem).
Subcellular fractionation, protease K protection assay, and alkaline extraction.
Crude mitochondria, purified mitochondria, and ER from cultured cells were fractioned as described previously (Area-Gomez, 2014; Guardia-Laguarta et al., 2014). The alkaline extraction was performed as described previously (Zhou et al., 2008).
Immunoblotting, immunoprecipitation, and in vivo ubiquitination.
Immunoblottting (IB) and immunoprecipitation (IP) assays were performed as previously described (Zhou et al., 2008; Vives-Bauza et al., 2010). Coimmunoprecipitation (co-IP) protocols for PINK1-gp78 interaction were adapted from Zhang et al. (2015). In vivo ubiquitination assay was performed in stringent buffer containing 1% SDS before proceeding to IP as described previously (Vives-Bauza et al., 2010). APEX technology was adapted from previously published studies (Rhee et al., 2013; Hung et al., 2014).
Gene silencing.
Lentiviral vectors expressing Sec61β, VCP, Ufd1 shRNA were from Sigma-Aldrich. Gp78 shRNA plasmid was a gift from Yihong Ye (NIH). siRNA used in the study were from Dharmacon or Qiagen. Protocols used for gene silencing were recommended by the providers. All shRNA experiments in the study were performed with multiple controls for off-target effects including empty control vector (Sigma-Aldrich, Mission pLKO.1-puro SHC001, which contain no shRNA insert), non-targeting shRNA control (Sigma-Aldrich, Mission pLKO.1-puro SHC002, which does not target any human or mouse genes). For each knock-down experiment, at least two independent Sigma-Aldrich Mission-specific shRNAs to a single target gene with validated high efficiency (>70%) were tested in both HEK239T and HeLa cells. Any significant phenotype/change that resulted from a specific shRNA was also confirmed by alternative approach using independent siRNA (Dharmacon/Qiagen) at final concentration of 15 nm.
Immunofluorescence and imaging.
Immunofluorescence procedures were performed as described previously (Guardia-Laguarta et al., 2014). The Manders colocalization coefficient M1 (Manders et al., 1993) was calculated using the Fiji distribution (Schindelin et al., 2012) of ImageJ (Schneider et al., 2012). PINK1 and organelle images were manually threshold by a skilled observer CG-L. The background-subtracted integrated intensity was calculated for each label and for the overlapping areas. The colocalization coefficient was calculated as the percentage of PINK1 integrated density overlapping with the indicated organelle. For super-resolution structured illumination microscopy (SIM), cells were imaged on a Nikon N-SIM, based on an Eclipse Ti inverted microscope using an SR Apo-TIRF 100×/1.49 oil-immersion objective and an Andor iXon 3 EMCCD camera.
Mass spectrometry analysis.
PINK1 purified protein complexes were resolved using SDS-PAGE and protein bands were excised, digested with trypsin and purified using Poros 50 R2 (Applied Biosystems) reversed-phased beads, then purified peptide mixtures were subjected to nano-liquid chromatography-mass spectrometry (LC-MS/MS) analysis as described previously (Beverly et al., 2012).
Experimental design and statistical analysis.
All experiments were performed in 1–3 technical replicates and in at least three biological replicates, unless stated otherwise. Data are expressed as mean ± SEM. Samples were compared by two-tailed Student's t test or one- or two-way ANOVA followed by a Holm–Sidak's multiple-comparison test. The null hypothesis was rejected at the 0.05 level. All analyses were performed with SigmaPlot for Windows v12.0 (Systech Software).
Results
Ub-PINK1 interacts with valosin-containing protein
The degradation of the mature form of PINK1 by the ubiquitin/proteasome pathway has recently been clarified (Liu et al., 2017). However, the mechanism by which Ub-PINK1 gains access to the cytosolically-located proteasome remains uncertain. To identify PINK1-interacting partners that might provide clues as to how it is extracted from mitochondria and then targeted to the proteasome, we immunoprecipitated PINK1 from HeLa cells, separated the resulting material by SDS-PAGE gels, excised Coomassie brilliant blue-stained bands, and subjected them to LC-MS/MS (Fig. 1A). We identified ∼480 candidate interaction partners (Fig. 1-1), including VCP (Fig. 1A, Band 2), as well as several VCP cofactors, including ubiquitin conjugation factor E4B (UBE4B or UFD2A; Fig. 1A, Band 1) and nuclear protein localization protein 4 homolog (NPL4; Fig. 1A, Band 3). We were particularly interested in these candidates because VCP, which is an AAA+ ATPase, is known to facilitate the degradation of proteins anchored in intracellular membranes (Meyer and Weihl, 2014) and was found, upon mutation, to phenocopy the mitochondrial defect caused by PINK1 mutations in flies (Kim et al., 2013). In the PINK1 proteomics analysis, the IgG lane was not processed. To exclude spurious interactions between the selected putative partners and PINK1, we performed a series of co-IP validation experiments in HEK293T cells stably transfected with WT PINK1 and treated with the proteasome inhibitor MG132 to enrich the cell content in Ub-PINK1. As shown in Figure 1B, VCP was successfully pulled-down upon IP PINK1 with a previously validated antibody (Zhou et al., 2008). The reciprocal IP also showed that 52 kDa PINK1 was the PINK1 species that bound preferentially to VCP. Despite low levels of endogenous PINK1 in cells with healthy mitochondria (Zhou et al., 2008; Liu et al., 2017), some endogenous 52 kDa PINK1 was pulled-down upon IP VCP even in the absence of MG132 (Fig. 1C). Furthermore, co-IP experiments using HEK293T cells expressing the ubiquitination-defective mutant PINK1K137R (Liu et al., 2017) revealed lower, but not abolished, VCP/PINK1 interaction compared with HEK293T cells expressing WT PINK1 (Fig. 1D). Collectively, these experiments support a bona fide physical interaction between VCP and PINK1, even when the latter is not highly ubiquitinated. We also validated the interaction between PINK1 and UFD2A (Fig. 1E).
Figure 1.

Ub-PINK1 interacts with VCP. A, Endogenous PINK1 interacting proteins revealed by Coomassie blue-stained SDS-PAGE gel. Endogenous PINK1 and its associated complexes were enriched by IP with anti-PINK1 antibody in HeLa cells. Anti-HA antibody and preimmune IgG (Ctr IgG) were used as controls. Bands labeled 1, 2, and 3 were identified as Ufd2A, VCP, and Npl4, respectively. Individual LC-MS/MS data can be found in Figure 1-1. B, PINK1 and VCP interaction in HEK293T cells cotransfected with WT PINK1 and VCP evidenced by co-IP using either an anti-PINK1 or anti-VCP antibody. *Denotes IgG heavy chain. C, Endogenous PINK1 and VCP interaction in SH-SY5Y cells evidenced by co-IP using anti-VCP-antibody. D, As in B, except that cells were transfected either with WT PINK1 or mutant PINK1K137R. *Denotes IgG heavy chain. E, PINK1 and Ufd2A interaction in HEK293T cells stably-expressing WT PINK1 and transfected with Ufd2A-flag evidenced by IP using an anti-PINK1 antibody. F, Ub-PINK1 IB in WT PINK1 stably-transfected HEK293T cells. Endogenous VCP was efficiently knocked down by VCP shRNAs. G, Ub-PINK1 IB in WT PINK1 stably-transfected HEK293T cells. Endogenous Ufd2A was efficiently knocked down by Ufd2A shRNAs. H, Ub-PINK1 IB in PINK1 stably-transfected HEK293T cells. Endogenous Ufd1 was efficiently knocked down by Ufd1 shRNAs. Data are representative of three independent experiments performed under identical conditions.
Related to Figure 1A. PINK1 interacted proteins identified by LC-MS/MS. The protein ID and peptide ID setting is 95% and above, with a cut-off of minimum 2 peptides that need to match and identify a protein. Download Figure 1-1, XLS file (112KB, xls)
To confirm the functional significance of the interactions of Ub-PINK1 with UFD2A and with VCP, we silenced each of these two factors in HeLa cells stably expressing PINK1, using shRNA lentiviral vectors. HeLA cells infected with a VCP shRNA showed a 65 ± 2% reduction in VCP protein expression as compared with HeLa cells infected with a scrambled shRNA control (Student's t test: t(5) = 22.17, p = 3.4E−5). Consistent with a role of VCP in PINK1 degradation, we found that upon knockdown of VCP using our validated shRNA, we observed an increase in the amount of the 52 kDa form, as well as that of Ub-PINK1 species, in cells both treated and untreated with MG132 compared with control cells (Fig. 1F). As for UFD2A, HeLA cells infected with a UFD2A shRNA showed a 82 ± 9% reduction in UFD2A protein expression as compared with HeLa cells infected with scrambled shRNA control (Student's t test: t(4) = 10.96, p = 4.0E−4). This marked silencing in HeLa cells was associated with a moderate increase in both 52 kDa PINK1 and Ub-PINK1 (Fig. 1G).
UFD1 encodes a key adaptor protein that contributes to the VCP-specific activity in the removal of ER proteins (Ye et al., 2001) and, interestingly, of damaged mitochondria as well (Kim et al., 2013). Thus, to further support the role of VCP in PINK1 degradation process, we targeted UFD1 with a shRNA that significantly reduced its protein expression as compared with HeLa cells infected with scrambled shRNA control (78 ± 8%, Student's t test: t(4) = 10.64, p = 4.4E−4). Strikingly, we found that silencing UFD1 phenocopied the effects of VCP knockdown, in that it markedly increased 52 kDa PINK1 and Ub-PINK1 (Fig. 1H).
Thus, our data indicate that conditions in which the functions of VCP, UFD1 and, to a lesser extent, UFD2A are reduced, promote 52 kDa PINK1 stability, suggesting that all of these three factors operate in the molecular pathway between Ub-PINK1 and its proteasomal degradation.
52 kDa PINK1 interacts with the ERAD machinery
The VCP machinery collaborates with the ERAD pathway to remove and degrade selected integral membrane proteins (Brodsky, 2012). Thus, in light of the above, we then asked whether PINK1 interacts with ER-resident components of the ERAD pathway, namely, the factors ERLIN-1 and -2, Sec61β (that is also associated with the VCP retrotranslocation complex), and the E3 ubiquitin ligase gp78 (gene AMFR; Spiro, 2004; Scott and Schekman, 2008; Wang et al., 2008; Christianson and Ye, 2014; Habeck et al., 2015). We found no evidence of interactions between PINK1 and ERLIN-1 and -2 (data not shown). In contrast, we found that PINK1 interacts with endogenous Sec61β in both MG132-treated and non-treated HEK293T cells stably expressing PINK1 (Fig. 2A). This interaction was also detected in cells expressing endogenous PINK1 and endogenous Sec61β (Fig. 2B). Moreover, in a similar experiment, we found that gp78 was successfully pulled down by overexpressed PINK1 IP in both MG132-treated and non-treated HEK293T cells; the reciprocal IP also indicated that 52 kDa PINK1 binds to gp78 (Fig. 2C). Our results indicate that PINK1 interacts directly or indirectly with specific members of the ERAD machinery, raising the possibility that PINK1 degradation involves the ERAD pathway.
Figure 2.

52 kDa PINK1 interacts with the ERAD machinery. A, PINK1 and Sec61β interaction in WT PINK1 stably-expressing HEK293T cells transfected with Sec61β evidenced by IP using either an anti-PINK1 or anti-Sec61β antibody. *Denotes IgG heavy chain. B, Endogenous PINK1 and Sec61β interaction in SH-SY5Y cells evidenced by IP using an anti-Sec61β antibody. C, WT PINK1 and endogenous gp78 interaction in stably PINK1 expressing HEK293T cells evidenced by co-IP using either an anti-PINK1 or anti-gp78 antibody. D, Ub-PINK1 IB in PINK1 stably-transfected HEK293T cells. Endogenous Sec61β was efficiently knocked down by Sec61β shRNAs. E, Ub-PINK1 IB in PINK1 stably-transfected HEK293T cells. Endogenous gp78 was efficiently knocked down by gp78 shRNA validated by IB. F, Ub-PINK1 IB in Hrd1 deficient HEK293T cells generated by CRISPR that were transfected with WT PINK1. Experiment shown in F was repeated once independently with similar results while those shown in A–E are representative of 3–4 independent experiments performed under identical conditions.
PINK1 polyubiquitination and degradation rely on ERAD machinery and E3 ligases
To determine the contribution of Sec61β to PINK1 stability, cells stably overexpressing PINK1 were infected with a lentiviral shRNA to knockdown Sec61β. This Sec61β shRNA caused a 89 ± 6% (Student's t test: t(4) = 17.3, p = 6.6E−5) reduction in Sec61β protein expression and a consistent increase in 52 kDa PINK1 and Ub-PINK1 in whole-cell extracts (Figs. 2D). Thus, Sec61β interacts with and contributes to PINK1 stability.
Furthermore, given the physical interaction between PINK1 and gp78 (Fig. 2C), we also asked whether this E3 ubiquitin ligase catalyzes PINK1 polyubiquitination. Silencing gp78 (68 ± 5%; Student's t test: t(5) = 9.4, p = 2.3E−4) in PINK1 and HA-Ub cotransfected HEK293T cells reduced the signal for Ub-PINK1 in associate ion with a moderate increase of 52 KDa PINK1, but in an unusual pattern (Fig. 2E): the reduction in the Ub-PINK1 was greatest in the region corresponding to the higher molecular mass species (Fig. 2E), suggesting that PINK1 polyubiquitination, and in particular that of the lower molecular mass species, may not be mediated solely by gp78. Another ER-related E3 ligase, HRD1, can form a transient interaction and cooperate with gp78 to catalyze polyubiquitination of both luminal and membrane ERAD substrates (Bernardi et al., 2010; Ishikura et al., 2010; Zhang et al., 2015). Accordingly, to examine whether HRD1 is also involved in PINK1 ubiquitination, we performed an in vivo ubiquitination assay in HRD1-deficient HEK293T cells (Zhang et al., 2015). Strikingly, the lack of HRD1 did abrogate the IB signal for all Ub-PINK1 species (Fig. 2F). These results suggest that PINK1 polyubiquitination arises from the dual action of HRD1 and gp78, where the former initiates the formation of the polyubiquitin chain, whereas the latter elongates it, an interpretation consistent with our previous results that PINK1 polyubiquitination is made of heterotypic chains (Liu et al., 2017).
PINK1 colocalizes with both mitochondria and the ER
In light of the proposed link of PINK1 with ERAD, we revisited PINK1's subcellular distribution. Strikingly, confocal microscopy revealed that PINK1 immunoreactivity in HeLa cells transfected stably with WT PINK1 colocalized with the ER marker Sec61β [M1 colocalization coefficients (CCs) = 0.593; Fig. 3A] as well as with the mitochondrial markers MitoTracker Green FM and cytochrome c (M1 CCs = 0.531 and 0.685, respectively). In contrast, PINK1 did not colocalize with the Golgi marker GM130 (M1 CC = 0.045; Fig. 3A). Using super-resolution SIM imaging (Gustafsson et al., 2008) we also observed that PINK1 colocalized with both Mito-DsRed and Sec61β-GFP in COS-7 cells cotransfected with PINK1 and Sec61β-GFP or Mito-DsRed (Fig. 3B).
Figure 3.

PINK1 is localized in both the mitochondria and the ER. A, Colocalization between PINK1 and organelle markers. Cells were transiently transfected with PINK1 (red) and costained with cytochrome c (cyt c) or MitoTracker (mitochondria), Sec61β-GFP (ER), and GM130 (Golgi; green). Representative images were shown (left) and the quantification (right) was done by ImageJ software. Scale bar, 10 μm. **p < 0.001 difference from GM130, but p > 0.05 cyto C versus MitoTracker versus Sec61β (one-way ANOVA, F(3,8) = 48.13, p < 0.001, followed by a Holm-Sidak's post hoc test). B, COS-7 cells (ATCC) were transfected with PINK1 expression vectors and ER marker Sec61β-GFP or mitochondrial marker Mito-DsRed. Images were acquired in 3D-SIM mode using excitation at 488 and 561 nm and standard filter sets for green and red emission. Image z-stacks were collected with a z interval of 200 nm. SIM image reconstruction, channel alignment, and 3D reconstruction were performed using NIS-Elements AR and Fiji (Schindelin et al., 2012). SIM reconstructed images were threshold, using consistent thresholds for each channel. A binarized mask was created for each channel. Representative images of ER or mitochondria (left) PINK1 (center), and a composition of the PINK1 mask (yellow) on top of the ER/mitochondria mask (red) are shown. Unmasked overlays are shown in the first panel. Scale bar, 2 μm. C, PINK1 IB of purified subcellular fractions from HeLa cells (n = 3). Endogenous PINK1 in was enriched by MG132 treatment for 6 h, and then proceeded for IP using anti-PINK1 antibody. Note that 52 kDa PINK1 migrates slightly above the heavy chain of IgG (*).
Because these microscopy methods do not allow one to conclude whether the observed colocalization is linked to PINK1 immunofluorescence generated by the full-length or cleaved form, or both, two additional methods were used. First, we used gradient-centrifugation to generate enriched subcellular fractions to detect endogenous 52 kDa PINK1. With this method endogenous mature 52 kDa PINK1 was unambiguously detected in the ER-enriched fractions of HEK293T cells treated with MG132 (Fig. 3C). Second, we performed a subcellular biotinylation assay using an intermembrane space-specific targeted ascorbate peroxidase (IMS-APEX2; Rhee et al., 2013; Hung et al., 2014) to investigate the presence of PINK1 in specific subcellular compartments. Upon addition of biotin to cells cotransfected with PINK1 and IMS-APEX2, biotin-labeled 52 kDa PINK1 was recovered from the both the ER and mitochondrial fractions following sequential pull-down with anti-PINK1 and streptavidin magnetic beads (Fig. 4A,B). The IMS-resident protein SMAC was also biotinylated (Fig. 4C). In contrast, none of the ER-resident proteins tested, such as BIP, Sec61β, or calreticulin, were biotinylated by the IMS-APEX2-catalyzed reaction (Fig. 4C). These results are consistent with 52 kDa PINK1 residing in or close proximity of both mitochondria and the ER, such as the mitochondrial–ER interface, and thus in keeping with the finding of Gelmetti et al. (2017).
Figure 4.

Ub-PINK1 is found in both the ER and mitochondria. A, HeLa cells were cotransfected with PINK1 and IMS-APEX2, purified mitochondrial (mito) and ER fractions were used in the sequential pull-down. This experiment was repeated once independently with similar results. B, Mito-DsRed and Sec61β were used as mitochondrial and ER outer membrane markers, respectively. IMS-APEX2 specifically colocalized with mitochondrial marker Mito-DsRed in COS-7 cells. Scale bar, 10 μm. C, 293T cells were cotransfected with PINK1 and IMS-APEX2, labeled with biotin-phenol and processed for IP. Biotin-labeled protein was detected in elutes that was pulled-down by streptavidin magnetic beads using the total cell extract (Input). BIP, Sec61β, and calreticulin were used as ER markers; ATP5A, Smac, and Tom20 were used as mitochondrial markers; PKC and GAPDH were used as cytosolic markers. This experiment was repeated once independently with similar results. D, Ub-PINK1 IP followed by IB analysis of purified subcellular fractionation isolated from cotransfected PINK1 and HA-Ub HeLa cell exposed to MG132 (n = 3). E, F, Alkaline extraction (0.1 m Na2CO3, pH 11) of purified mitochondrial (E) or ER fractions (F) from the same cells as in Figure 3D followed by IP and IB analysis (n = 3). P, Particulate; S, supernatant. Tom70, Smac, and calnexin are markers of mitochondrial outer membrane, mitochondrial interspace and ER membrane integral proteins, respectively. G, Left, The Venus signal (yellow) was only observed in HeLa cells cotransfected with Vn-Ub and PINK1-Vc constructs. Middle, Ub-PINK1 signal (yellow) in HeLa cells coexpressing Vn-Ub and PINK1-Vc and immunostained with antibody against Tom20, Sec61β, KDEL, or GM130 (red). Scale bar, 10 μm. Right, Quantification of Ub-PINK1 and organelle marker colocalization index. *p < 0.005, **p < 0.001 difference from GM130 (one-way ANOVA, F(3,8) = 22.83, p < 0.001, followed by a Holm-Sidak's post hoc test). H, Top, PINK1 IB of purified subcellular fractions of HeLa cells expressing PINK1K137R. Bottom, PINK1 52 kDa:63 kDa ratio relative to WT PINK1 expressing cells. **p < 0.001, ***p < 0.0001 difference from WT controls (two-way ANOVA, interaction mutation × subcellular fraction: F(3,16) = 58.35, p < 0.001, followed by a Holm–Sidak's post hoc test). I, SIM imaging analysis was performed similar to that in Figure 3B, except that cells were transfected with PINK1K137R. Scale bar, 100 μm.
Ub-PINK1 is found in both the ER and mitochondrial fractions
The observed partitioning of 52 kDa PINK1 between the mitochondrial and the ER enriched fractions raises the question as to whether PINK1 is differentially ubiquitinated in these two fractions. We therefore performed an in vivo ubiquitination assay using purified subcellular fractions isolated from MG132-treated HEK293T cells. We found that Ub-PINK1 was associated with both mitochondrial and ER fractions (Fig. 4D) and, using alkaline extraction, that Ub-PINK1 was poorly extractable from both subcellular fractions (Fig. 4E,F). To confirm these observations, we performed an in vivo ubiquitin-mediated fluorescence complementation assay (Fang and Kerppola, 2004) using one plasmid that encoded for ubiquitin fused to the N-terminal half of the fluorescent protein Venus (Vn-Ub), and another that encoded for PINK1 fused to the remaining C-terminal half of Venus (Pink1-Vc). As shown in Figure 4G, Venus fluorescent signal was only observed in HeLa cells successfully transfected with both plasmids (left). In those cells, the yellow fluorescence colocalized with both the ER markers Sec61β and KDEL immunofluorescence (M1 CCs = 0.847 and 0.628, respectively) and mitochondrial marker TOM20 (M1 CCs = 0.381), but not with the Golgi matrix protein GM130 (M1 CCs = 0.065; Fig. 4G, right). Our findings thus provide compelling evidence that Ub-PINK1 is associated with both the ER and the mitochondria-enriched fractions. Furthermore, we showed, by IB and SIM, that the Ub-inefficient PINK1K137R mutant was also found in the mitochondrial (Fig. 4H,I) and in the ER fractions, indicating that ubiquitination is not required for 52 kDa PINK1 dual subcellular association.
Ubiquitination-defective PD-linked PINK1 mutations accumulate in the ER fraction
We note that K137, which is required for ubiquitination (Liu et al., 2017), is located in a 130-aa “linker” region (aa 104–233) connecting the transmembrane domain and the kinase domain. To confirm the importance of the PINK1 linker region and the ER to PINK1 degradation, we screened a series of nine pathogenic PD mutations (Bonifati, 2005; Song et al., 2013) that reside in the linker region in the vicinity of K137. Of the nine such PINK1 mutations examined, we found that six were associated with little or no ubiquitination of PINK1, even after treatment with MG132 (Fig. 5A). Remarkably, of these six ubiquitination-defective mutants, five were selected for further studies and were disproportionally enriched in the ER-enriched fractions in the absence of MG132, as revealed by the ratio of 52:63 kDa PINK1 (Fig. 5B). In contrast, we did not detect ubiquitination defects or ER enrichment in two representative kinase-domain mutants, PINK1G309D and PINK1G386A (Valente et al., 2004; Okatsu et al., 2012; data not shown). These results suggest that the PINK1 ER-assisted degradation and kinase activity may be two independent determinants of PINK1 pathogenicity in PD.
Figure 5.

Ubiquitination level and subcellular fraction in PD pathogenic PINK1 mutants. A, Ub-PINK1 IB of HEK293T cells cotransfected with HA-Ub and PINK1 pathogenic mutants exposed to MG132 for 6 h. This experiment was repeated once independently with similar results. B, Top, PINK1 IB of purified subcellular fractions of cells that express PINK1 WT and pathogenic mutants. Bottom, PINK1 52 kDa:63 kDa ratio relative to WT PINK1 expressing cells in each subcellular fraction. *p < 0.01, **p < 0.001 difference from WT controls (two-way ANOVA, interaction mutation × subcellular fraction: F(21,64) = 42.85, p < 0.001, followed by a Holm-Sidak's post hoc test).
Discussion
We show here that 52 kDa PINK1 can be found in both the mitochondria and the ER-enriched fractions, that it interacts with the ERAD machinery, and that its degradation by the proteasome is controlled by the ERAD E3 ubiquitin ligases HRD1 and gp78 and by the ERAD-associated proteins VCP, UFD1, and UFD2A. Thus, these data begin to reveal a novel picture of PINK1 metabolic processing that revolves around possible mitochondria–ER interface and involvement of the ERAD machinery.
Our discoveries of PINK1 linkage to heterotypic polyubiquitin chains (Liu et al., 2017) and interaction with the components of the ERAD pathway have enabled us to obtain further insights into PINK1 degradation process. We note that some E3 ligases (e.g., UBE3C; You and Pickart, 2001) can generate both K29 and K48 linkages, whereas in other cases, the formation of heterotypic K29/K48 polyubiquitin chains requires at least two E3 enzymes (Koegl et al., 1999; Saeki et al., 2004). In keeping with the latter scenario, we found that silencing the ERAD-associated E3 ligase gp78 decreased mainly the higher-order polyubiquitin species (Fig. 2E), whereas silencing the E3 ligase HRD1 abolished essentially all PINK1 ubiquitination, suggesting that HRD1 initiates PINK1 ubiquitination and that gp78 extends it. This is reminiscent of the proteasomal degradation of two other ERAD substrates, the cytokine autocrine motility factor (Wang et al., 2014) and the cystic fibrosis transmembrane conductance regulator (Morito et al., 2008), where, following the initiation of ubiquitination by one E3 ligase (i.e., TRIM25 and RMA1, respectively; Morito et al., 2008), gp78 then recognizes the ubiquitin chains already conjugated to the substrates and operates as an ubiquitin chain elongase (i.e., E4 ubiquitin factor) to catalyze further polyubiquitination.
The role of the ubiquitin-proteasome system in the degradation of mitochondrial proteins, and especially MOM-localized proteins, is increasingly recognized (Karbowski and Youle, 2011). Even though the knowledge of the direct involvement for PINK1 in mitochondrial–ER crosstalk is limited (Hattori et al., 2017), a recent study showed that endogenous PINK1 and BECN1 were both found to re-localize at mitochondria-associated membranes, where they enhance ER-mitochondria contact sites and the formation autophagosome precursors (Gelmetti et al., 2017). However, how integral MOM proteins are removed from the membrane and targeted to the cytosolic proteasome remains uncertain. Mounting evidence indicates that VCP, which is important for the extraction of proteins from the ER and from other organelle membranes, is also instrumental in the removal and ensuing proteasomal degradation of MOM proteins, such as mitofusin (Kim et al., 2013; Kimura et al., 2013; Zhang et al., 2017). The specific action and subcellular recruitment of the multifunctional protein VCP is governed via its interaction with a variety of adaptors. For example, it has been shown in yeast that for VCP to be recruited to the mitochondria and to contribute to the degradation of MOM proteins, specific VCP adaptors, such as Vms1 (Heo et al., 2010), are necessary. Although our data demonstrate that VCP is involved in PINK1 turnover, the knockdown of ANKZF1 (the human homolog of yeast Vms1; data not shown) or of mitochondria-associated E3 ligases (e.g., Parkin, Mulan, and March V) did not affect PINK1 ubiquitination/degradation (Liu et al., 2017). Conversely, the physical interaction between PINK1 and ER proteins, including Sec61β, and components of the ERAD pathway, such as HRD1, gp78, UFD1, and UFD2A, suggests that the VCP-dependent degradation of PINK1 is associated with the ER, not with the mitochondria. Relevant to the proposed PINK1 association with the ER proteostasis machinery, it is also worth mentioning that Weihofen et al. (2008) showed that PINK1 can be detected in the ER-rich microsome fractions of cell homogenates.
At this time, there is no evidence for PINK1 cotranslation in either mitochondria or ER nor for PINK1 import in the ER. Although protein movement between the mitochondria and the ER have been reported, especially for proteins belonging to the Bcl-2 family (Saita et al., 2013), it is more likely that our results reflect the localization of PINK1 at the contact sites, which refers to zones of close contact between mitochondrial and ER membranes (Rowland and Voeltz, 2012). Consistent with this interpretation are the observations made by both Silvestri et al. (2005) and ourselves (Zhou et al., 2008) that PINK1 is found in both the soluble and particulate fractions of mitoplast preparations and is poorly extractable by detergents, features often linked to proteins residing at the mitochondrial/ER contact side.
Although the focus of this study is on PINK1 degradation, it is worthy to note that Figures 3 and 4 reveal some 52 kDa PINK1 in the cytosol, a subcellular localization that we (Fedorowicz et al., 2014) and others (Haque et al., 2008; Lin and Kang, 2008; Weihofen et al., 2008; Sha et al., 2010; Murata et al., 2011) have previously mentioned. In light of a growing body of literature on the topic, we believe that cytosolic PINK1 is not only rapidly and mainly targeted for proteasomal degradation, but that it may also play a significant role in modulating homeostasis processes such as Parkin subcellular location (Fedorowicz et al., 2014), dendritic complexity (Dagda et al., 2014), vesicular release of dopamine, calcium-activated potassium channels, long-term potentiation (Kitada et al., 2007; Shan et al., 2009), mitochondrial trafficking (Liu et al., 2012; Matenia et al., 2012) and even possibly, Parkin-PARIS regulated PGC-1α expression (Lee et al., 2017).
In Drosophila, it seems that PINK1 follows two metabolic routes, one in which a fraction of PINK1 is degraded by the protease Lon inside of the mitochondria and a second that involves the proteasome (Thomas et al., 2014). Although the intra-mitochondrial/Lon route has not been found in vertebrate cells (Jin et al., 2010; Greene et al., 2012), the role of the cytosolic/proteasome route in PINK1 degradation is well established, and our study and that those already in the literature lead us to propose the following PINK1 metabolic cycle for vertebrate organisms (Fig. 6). Upon translation of PINK1, the N-terminal part of the full-length 63 kDa PINK1 is imported into mitochondria where it is cleaved by the combination of MPP and PARL proteases, producing 52 kDa PINK1. This shorter form of PINK1 is subjected to two critical events. First, it adopts a distinct conformation, and engages in interactions with bilayer membranes, distinct from those of the 63 kDa form, thereby, promoting exposure of the lysine residues of the transmembrane-kinase domain linker region, such as K137, which are critical for ubiquitin linkage to PINK1. Second, PINK1 localizes to the contact sites where it associates with the ER, at which point 52 kDa PINK1 interacts with the ERAD machinery, including HRD1, gp78, and VCP, enabling its ubiquitination and transfer to the proteasome for degradation. Whereas conventional ERAD substrates are degraded when misfolded, PINK1, akin to the non-canonical BCL-2 factor BOK (Llambi et al., 2016), is degraded continuously in healthy cells by a gp78/VCP-dependent process. Furthermore, like mitofusin (Zhang et al., 2017), PINK1 interacts with VCP even when poorly ubiquitinated (Fig. 1D) suggesting that PINK1, like mitofusin, may be degraded in a VCP-dependent manner even under unstressed conditions. However, if VCP contributes to PINK1 degradation, as we suggest herein, and PINK1 deletion is associated with gross mitochondrial alterations in flies (Clark et al., 2006; Park et al., 2006; Kim et al., 2013; Zhang et al., 2017), why does VCP overexpression rescue rather than exacerbate PINK1-null mutant-associated mitochondrial damage (Kim et al., 2013; Zhang et al., 2017)? The explanation likely resides in the study of Zhang et al. (2017), which provides compelling evidence that the observed mitochondrial phenotype associated with PINK1-null mutant is actually due to an upregulation of mitofusin, and not to a direct effect of the loss of PINK1 function. Additionally, VCP regulation of mitofusin expression does not require PINK1. Thus, the novel molecular pathway outlined here allows for PINK1 metabolic stability and activity to be tightly regulated in a timely and spatially restricted manner by mechanisms that share some similarities with, but that are independent of mitofusin turnover, and ensures that PINK1 serves its function as a mitochondrial quality control factor.
Figure 6.
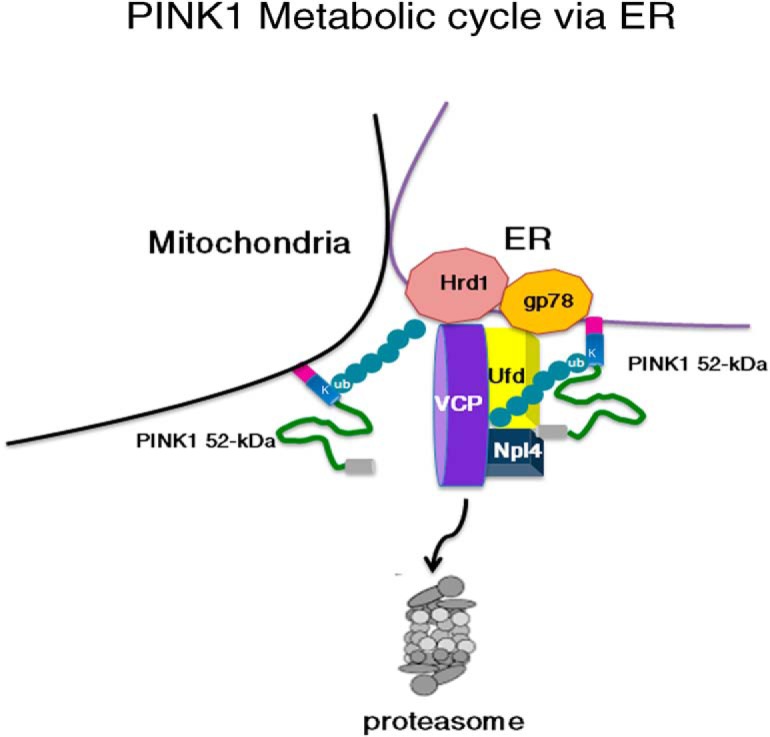
A model for PINK1 metabolic cycle via ER.
Footnotes
This work was supported by the American Parkinson Disease Association and the Leir Foundation to C.G.-L., by research grants from The Research Council of Norway and travel grants from The Fulbright Foundation and The Research Council of Norway to K.H.L., by the Herbert Irving Comprehensive Cancer Center and NIH Grant P30 CA013696 to T.C.S., by NIH Grants R01CA166413 and R01GM113013, and William H. and Alice Goodwin and the Commonwealth Foundation for Cancer Research of the MSKCC Experimental Therapeutics Center to X.J., and by Department of Defense W81XWH-08-1-0465, R21NS111176, R21AG064596, R01NS107442, and PDF-RCE-1401 to S.P. Structured illumination microscopy and image analysis were performed in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center, supported by NIH Grant P30 CA013696. We thank Yihong Ye for critical comments on the paper and providing reagents for assays on Hrd1 and gp78, Laura Munteanu for assistance on image collection and analysis, Zhezheng Li and Kevin Velasco for technical support on cell culture and plasmid constructions, Eric A. Schon for insightful comments and editing, and Estela Area-Gomez, Vernice Jackson-Lewis, Andrew Koff, and Pengbo Zhou for helpful discussions and suggestions.
The authors declare no competing financial interests.
References
- Area-Gomez E. (2014) Assessing the function of mitochondria-associated ER membranes. Methods Enzymol 547:181–197. 10.1016/B978-0-12-801415-8.00011-4 [DOI] [PubMed] [Google Scholar]
- Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W (2012) Pink1 kinase and its membrane potential (δψ)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem 287:22969–22987. 10.1074/jbc.M112.365700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B (2010) The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell 21:140–151. 10.1091/mbc.e09-07-0586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, Varmus H (2012) Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc Natl Acad Sci U S A 109:E119–E126. 10.1073/pnas.1119167109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V. (2005) Genetics of Parkinson's disease. Minerva Med 96:175–186. [PubMed] [Google Scholar]
- Brodsky JL. (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151:1163–1167. 10.1016/j.cell.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Upadhyay S, Chang X, Nagpal JK, Trink B, Sidransky D (2008) U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 7:1231–1237. 10.4161/cc.7.9.5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 21:325–335. 10.1038/nsmb.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166. 10.1038/nature04779 [DOI] [PubMed] [Google Scholar]
- Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem 128:864–877. 10.1111/jnc.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Fang D, Kerppola TK (2004) Ubiquitin-mediated fluorescence complementation reveals that jun ubiquitinated by Itch/AIP4 is localized to lysosomes. Proc Natl Acad Sci U S A 101:14782–14787. 10.1073/pnas.0404445101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorowicz MA, de Vries-Schneider RL, Rüb C, Becker D, Huang Y, Zhou C, Alessi Wolken DM, Voos W, Liu Y, Przedborski S (2014) Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep 15:86–93. 10.1002/embr.201337294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131. 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM, Valente EM (2017) PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13:654–669. 10.1080/15548627.2016.1277309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and parkin recruitment. EMBO Rep 13:378–385. 10.1038/embor.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, Voos W, Schon EA, Przedborski S (2014) α-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34:249–259. 10.1523/JNEUROSCI.2507-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW (2008) Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J 94:4957–4970. 10.1529/biophysj.107.120345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck G, Ebner FA, Shimada-Kreft H, Kreft SG (2015) The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J Cell Biol 209:261–273. 10.1083/jcb.201408088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS (2008) Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A 105:1716–1721. 10.1073/pnas.0705363105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori N, Arano T, Hatano T, Mori A, Imai Y (2017) Mitochondrial-associated membranes in Parkinson's disease. Adv Exp Med Biol 997:157–169. 10.1007/978-981-10-4567-7_12 [DOI] [PubMed] [Google Scholar]
- Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP, Ashrafi K, Glickman MH, Rutter J (2010) A stress-responsive system for mitochondrial protein degradation. Mol Cell 40:465–480. 10.1016/j.molcel.2010.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20. 10.1016/j.molcel.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, Ting AY (2014) Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 55:332–341. 10.1016/j.molcel.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikura S, Weissman AM, Bonifacino JS (2010) Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem 285:23916–23924. 10.1074/jbc.M110.127936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942. 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153. 10.1083/jcb.201402104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23:476–482. 10.1016/j.ceb.2011.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460:127–139. 10.1042/BJ20140334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78:65–80. 10.1016/j.neuron.2013.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Fukushi J, Hori S, Matsuda N, Okatsu K, Kakiyama Y, Kawawaki J, Kakizuka A, Tanaka K (2013) Different dynamic movements of wild-type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin-mediated mitochondrial quality control system. Genes Cells 18:1131–1143. 10.1111/gtc.12103 [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A 104:11441–11446. 10.1073/pnas.0702717104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96:635–644. 10.1016/S0092-8674(00)80574-7 [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate Parkin. Nature 510:162–166. 10.1038/nature13392 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Stevens DA, Kang SU, Jiang H, Lee YI, Ko HS, Scarffe LA, Umanah GE, Kang H, Ham S, Kam TI, Allen K, Brahmachari S, Kim JW, Neifert S, Yun SP, Fiesel FC, Springer W, Dawson VL, Shin JH, et al. (2017) PINK1 primes parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell reports 18:918–932. 10.1016/j.celrep.2016.12.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee J, Kwon I, Nakajima Y, Ohmiya Y, Son GH, Lee KH, Kim K (2010) Coactivation of the CLOCK-BMAL1 complex by CBP mediates resetting of the circadian clock. J Cell Sci 123:3547–3557. 10.1242/jcs.070300 [DOI] [PubMed] [Google Scholar]
- Lin W, Kang UJ (2008) Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 106:464–474. 10.1111/j.1471-4159.2008.05398.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, Takahashi R, Hattori N, Imai Y, Lu B (2012) Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet 8:e1002537. 10.1371/journal.pgen.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Guardia-Laguarta C, Yin J, Erdjument-Bromage H, Martin B, James M, Jiang X, Przedborski S (2017) The ubiquitination of PINK1 is restricted to its mature 52-kDa form. Cell Rep 20:30–39. 10.1016/j.celrep.2017.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S, Moldoveanu T, Chen T, Green DR (2016) BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165:421–433. 10.1016/j.cell.2016.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA (1993) Measurement of colocalization of objects in dual-colour confocal images. J Microsc 169:375–382. 10.1111/j.1365-2818.1993.tb03313.x [DOI] [PubMed] [Google Scholar]
- Matenia D, Hempp C, Timm T, Eikhof A, Mandelkow EM (2012) Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at thr-313, a mutation site in Parkinson disease: effects on mitochondrial transport. J Biol Chem 287:8174–8186. 10.1074/jbc.M111.262287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221. 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127:3877–3883. 10.1242/jcs.093831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata K (2008) Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell 19:1328–1336. 10.1091/mbc.e07-06-0601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Sakaguchi M, Jin Y, Sakaguchi Y, Futami J, Yamada H, Kataoka K, Huh NH (2011) A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem 286:7182–7189. 10.1074/jbc.M110.179390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. 10.1371/journal.pbio.1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, Shiba-Fukushima K, Sato S, Shimizu H, Fukunaga Y, Taniguchi H, Komatsu M, Hattori N, Mihara K, Tanaka K, Matsuda N (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3:1016. 10.1038/ncomms2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by Parkin. Nature 441:1157–1161. 10.1038/nature04788 [DOI] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339:1328–1331. 10.1126/science.1230593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AA, Voeltz GK (2012) Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13:607–625. 10.1038/nrm3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki Y, Tayama Y, Toh-e A, Yokosawa H (2004) Definitive evidence for Ufd2-catalyzed elongation of the ubiquitin chain through Lys48 linkage. Biochem Biophys Res Commun 320:840–845. 10.1016/j.bbrc.2004.05.216 [DOI] [PubMed] [Google Scholar]
- Saita S, Shirane M, Nakayama KI (2013) Selective escape of proteins from the mitochondria during mitophagy. Nat Commun 4:1410. 10.1038/ncomms2400 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Schekman R (2008) Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol 181:1095–1105. 10.1083/jcb.200804053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha D, Chin LS, Li L (2010) Phosphorylation of Parkin by Parkinson disease-linked kinase PINK1 activates Parkin E3 ligase function and NF-κB signaling. Hum Mol Genet 19:352–363. 10.1093/hmg/ddp501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Liu B, Li L, Chang N, Li L, Wang H, Wang D, Feng H, Cheung C, Liao M, Cui T, Sugita S, Wan Q (2009) Regulation of PINK1 by NR2B-containing NMDA receptors in ischemic neuronal injury. J Neurochem 111:1149–1160. 10.1111/j.1471-4159.2009.06398.x [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14:3477–3492. 10.1093/hmg/ddi377 [DOI] [PubMed] [Google Scholar]
- Song S, Jang S, Park J, Bang S, Choi S, Kwon KY, Zhuang X, Kim E, Chung J (2013) Characterization of PINK1 (PTEN-induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. J Biol Chem 288:5660–5672. 10.1074/jbc.M112.430801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro RG. (2004) Role of N-linked polymannose oligosaccharides in targeting glycoproteins for endoplasmic reticulum-associated degradation. Cell Mol Life Sci 61:1025–1041. 10.1007/s00018-004-4037-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatori S, Ito G, Iwatsubo T (2008) Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett 430:13–17. 10.1016/j.neulet.2007.10.019 [DOI] [PubMed] [Google Scholar]
- Thomas RE, Andrews LA, Burman JL, Lin WY, Pallanck LJ (2014) PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet 10:e1004279. 10.1371/journal.pgen.1004279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304:1158–1160. 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107:378–383. 10.1073/pnas.0911187107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Heath-Engel H, Zhang D, Nguyen N, Thomas DY, Hanrahan JW, Shore GC (2008) BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 133:1080–1092. 10.1016/j.cell.2008.04.042 [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha SW, Zhang T, Kho DH, Raz A, Xie Y (2014) Polyubiquitylation of AMF requires cooperation between the gp78 and TRIM25 ubiquitin ligases. Oncotarget 5:2044–2051. 10.18632/oncotarget.1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ (2008) Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet 17:602–616. 10.1093/hmg/ddm334 [DOI] [PubMed] [Google Scholar]
- Yamano K, Youle RJ (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9:1758–1769. 10.4161/auto.24633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414:652–656. 10.1038/414652a [DOI] [PubMed] [Google Scholar]
- You J, Pickart CM (2001) A HECT domain E3 enzyme assembles novel polyubiquitin chains. J Biol Chem 276:19871–19878. 10.1074/jbc.M100034200 [DOI] [PubMed] [Google Scholar]
- Zhang T, Xu Y, Liu Y, Ye Y (2015) gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol Biol Cell 26:4438–4450. 10.1091/mbc.E15-06-0354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Mishra P, Hay BA, Chan D, Guo M (2017) Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. eLife 6:e17834. 10.7554/eLife.17834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S (2008) The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A 105:12022–12027. 10.1073/pnas.0802814105 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 1A. PINK1 interacted proteins identified by LC-MS/MS. The protein ID and peptide ID setting is 95% and above, with a cut-off of minimum 2 peptides that need to match and identify a protein. Download Figure 1-1, XLS file (112KB, xls)