

Abstract
Clinical and experimental data indicate striatal cholinergic dysfunction in dystonia, a movement disorder typically resulting in twisted postures via abnormal muscle contraction. Three forms of isolated human dystonia result from mutations in the TOR1A (DYT1), THAP1 (DYT6), and GNAL (DYT25) genes. Experimental models carrying these mutations facilitate identification of possible shared cellular mechanisms. Recently, we reported elevated extracellular striatal acetylcholine by in vivo microdialysis and paradoxical excitation of cholinergic interneurons (ChIs) by dopamine D2 receptor (D2R) agonism using ex vivo slice electrophysiology in Dyt1ΔGAG/+ mice. The paradoxical excitation was caused by overactive muscarinic receptors (mAChRs), leading to a switch in D2R coupling from canonical Gi/o to noncanonical β-arrestin signaling. We sought to determine whether these mechanisms in Dyt1ΔGAG/+ mice are shared with Thap1C54Y/+ knock-in and Gnal+/− knock-out dystonia models and to determine the impact of sex. We found Thap1C54Y/+ mice of both sexes have elevated extracellular striatal acetylcholine and D2R-induced paradoxical ChI excitation, which was reversed by mAChR inhibition. Elevated extracellular acetylcholine was absent in male and female Gnal+/− mice, but the paradoxical D2R-mediated ChI excitation was retained and only reversed by inhibition of adenosine A2ARs. The Gi/o-preferring D2R agonist failed to increase ChI excitability, suggesting a possible switch in coupling of D2Rs to β-arrestin, as seen previously in a DYT1 model. These data show that, whereas elevated extracellular acetylcholine levels are not always detected across these genetic models of human dystonia, the D2R-mediated paradoxical excitation of ChIs is shared and is caused by altered function of distinct G-protein-coupled receptors.
SIGNIFICANCE STATEMENT Dystonia is a common and often disabling movement disorder. The usual medical treatment of dystonia is pharmacotherapy with nonselective antagonists of muscarinic acetylcholine receptors, which have many undesirable side effects. Development of new therapeutics is a top priority for dystonia research. The current findings, considered in context with our previous investigations, establish a role for cholinergic dysfunction across three mouse models of human genetic dystonia: DYT1, DYT6, and DYT25. The commonality of cholinergic dysfunction in these models arising from diverse molecular etiologies points the way to new approaches for cholinergic modulation that may be broadly applicable in dystonia.
Keywords: acetylcholine, cholinergic interneuron, dystonia, G-protein-coupled receptors, mice, striatum
Introduction
Dystonia is a common movement disorder affecting at least half a million individuals in the United States. Current treatments are generally insufficient to address symptoms, which cause substantial pain, disability, and even death (Albanese et al., 2019). Although the majority of cases are sporadic, recent work has uncovered a number of genetically determined forms; >20 dystonia-causing mutations have been established to date (Lohmann and Klein, 2017). However, even in these genetic forms in which the proximate cause is known, the neural mechanism(s) are still for the most part unknown (Breakefield et al., 2008; Balint et al., 2018). These limitations have made it difficult to develop targeted treatments for dystonia.
The prototypical form of genetic isolated dystonia is Oppenheim's or DYT1 dystonia, characterized by early onset and incomplete penetrance (Albanese et al., 2013). The discovery of mutations in the gene TOR1A, encoding TorsinA, as the cause of DYT1 dystonia has yielded many of the insights that we have into the pathophysiology of dystonia. In genetic mouse models, behavioral phenotypes are typically mild, but functional and neurochemical alterations are profound (Dang et al., 2005; Sharma et al., 2005; Zhao et al., 2008; Song et al., 2012; Ip et al., 2016; Richter et al., 2017). Specifically, striatal dopamine release is dysregulated (Balcioglu et al., 2007; Song et al., 2012) and cholinergic function is abnormal (Pisani et al., 2006; Scarduzio et al., 2017). Furthermore, corticostriatal synapses onto medium spiny neurons (MSNs) in these mice have heightened long-term potentiation (LTP) and an inability to undergo long-term depression (LTD) in response to high-frequency stimulation, elements of striatal plasticity thought to be necessary for the expression of normal movement (Pisani et al., 2006). We have previously found that Dyt1ΔGAG/+ mice, replicating the genetic mutation in human DYT1, have elevated striatal acetylcholine release. This hypercholinergic state and associated excessive activation of muscarinic acetylcholine receptors (mAChRs) causes a switch in dopamine D2 receptor (D2R) coupling in striatal cholinergic interneurons (ChIs) from the canonical inhibitory Gi/o pathway to the noncanonical excitatory β-arrestin pathway (Scarduzio et al., 2017). This switch underlies the well documented D2R-mediated paradoxical excitation of ChIs (Pisani et al., 2006; Sciamanna et al., 2012; Eskow Jaunarajs et al., 2015). Interestingly, blocking striatal cholinergic function through inhibition of mAChRs reverses the D2R-mediated paradoxical excitation and the loss of LTD at corticostriatal synapses onto MSNs, likely through restoring normal Gi/o signaling (Martella et al., 2009; Dang et al., 2012; Maltese et al., 2014; Scarduzio et al., 2017).
Recently, additional genetic mouse models of dystonia have become available. DYT6 is caused by heterozygous mutations in the gene for the transcription factor THAP1 (Bressman et al., 2009; Djarmati et al., 2009; Fuchs et al., 2009; Ruiz et al., 2015; Yellajoshyula et al., 2017) and DYT25 is caused by mutations of the gene for Gαolf (GNAL), a G-protein signaling molecule downstream of striatal dopamine D1 rceptors (D1Rs) and adenosine A2A receptors (A2ARs) (Bressman et al., 1994; Belluscio et al., 1998; Fuchs et al., 2013; Pelosi et al., 2017). In patients, both DYT6 and DYT25 share some features with DYT1: they are isolated dystonias, they exhibit incomplete penetrance, and none of them respond very well to currently available therapies. In contrast to DYT1 models, little is known about the pathophysiology of DYT6 and DYT25 models, especially with regard to function of the cholinergic system. Additionally, the majority of investigations have been limited to males, though sex has been shown to be a key variable in the manifestation of many types of dystonia (ESDE Collaborative Group, 1999; Defazio et al., 2003; Butler et al., 2015). Thus, the goals of the current set of experiments were to determine whether similar changes in cholinergic function observed in DYT1 models exist in two additional genetic models of isolated dystonia (DYT6 and DYT25) and their sex dependency. Elucidation of common pathophysiology involved in genetic causes of isolated dystonias may reveal common mechanisms, potentially bridging the gap between gene mutation, physiology, and behavior.
Materials and Methods
Animal Models
Mice.
Male and female heterozygous Thap1 KI (Thap1C54Y/+; Ruiz et al., 2015) and Gnal heterozygous KO (Gnal+/−; Belluscio et al., 1998; Pelosi et al., 2017) mice were maintained congenically through backcross with C57BL/6J mice from The Jackson Laboratory. Mice were housed in a 12 h light/dark cycle. Food and water were provided ad libitum. Mice were evaluated between 3 and 6 months of age. All experimental protocols were approved by the Institutional Animal Care and Use Committee at University of Alabama at Birmingham. Tail DNA was genotyped for each strain: Thap1C54Y/+ (primer A, GCGTATAATTCAGGCTGTCAG; primer B, GCATTCACCCAAAGCCAATGC) and Gnal+/− (primer A, TGATCTTCCGCGGTCTTGCT; primer B, CATTCCTCCCACTCATGATC; primer C, TTCAGTATCAGAGAGCGGAG). PCR products were developed using a 2% agarose gel.
In vivo microdialysis.
Male and female Thap1C54Y/+ and Gnal+/− mice and their control littermates were anesthetized with isoflurane (1–3%) and placed in a stereotaxic apparatus. Unilateral microdialysis cannulae (CMA Microdialysis) were implanted vertically above the striatum (anterior +0.6; lateral +1.9; ventral −1.8 mm from bregma, according to the coordinates of Paxinos and Franklin (2004). Two anchor screws (Plastics One) were placed behind the cannula and then fixed to the skull with dental cement (Lang Dental). Buprenorphine (0.03 mg/kg, i.p.) was injected for pain relief. Three to 4 d after surgery, the guide cannulae were removed under light sedation with isoflurane and microdialysis probes were inserted (CMA7/2 mm, CMA Microdialysis). Awake, behaving mice were habituated to the microdialysis environment for 2–3 h while the probes were perfused with artificial CSF (aCSF) containing the following: 127.6 mm NaCl, 4.02 mm KCl, 750 μm NaH2PO4, 2.1 mm Na2HPO4, 2.00 mm MgCl2, 1.71 mm CaCl2, pH 7.4, at a constant rate of 2 μl/min for the duration of the experiment. After the conclusion of the experiment, mice were killed and the brains were removed and postfixed in 4% paraformaldehyde in 0.1 m PBS for at least 5 d. Brains were then sectioned using a sliding microtome and relevant sections were cresyl violet stained for verification of probe placement. Two animals were removed from analyses due to improper probe placement.
To determine the basal levels of acetylcholine, three dialysate samples were collected every 20 min for 1 h after habituation. After baseline, 10, 100, and 500 nm neostigmine bromide (Sigma-Aldrich) were added to aCSF and samples were collected every 20 min for 1 h per dose in an escalating dose design for a total of 4 h of sample collection (1 h baseline + 3 h neostigmine dose–response). All samples were analyzed for acetylcholine using HPLC-ED (HTEC-500 with Autosampler INSIGHT, Amuza) with an enzyme reactor (AC-ENZYM II 1.0 × 4.0 mm, Amuza) and a platinum electrode (WE-PT; +450 mV vs Ag/AgCl, Amuza). The mobile phase consisted of a 50 mm dibasic potassium phosphate buffer, pH 8.5, with 300 mg/L sodium decanesulfonate and 50 mg/L Na2EDTA-2H2O and was delivered at a rate of 150 μl/min. Microdialysate sample chromatographs were analyzed based on established concentration curves for acetylcholine (1–100 nm) using Envision 6.0 Chromatography Software (Amuza) software. The limit of detection was ∼10 pm.
DYT1 mouse models also exhibit dampened amphetamine-induced striatal dopamine release, with varying effects on basal dopamine levels (Balcioglu et al., 2007; Song et al., 2012). Therefore, to determine basal levels of dopamine in Thap1C54Y/+ and Gnal+/− mouse lines, three dialysate samples were collected every 20 min for 1 h after habituation while infusing aCSF + 0.25 mm ascorbic acid (Sigma-Aldrich) to reduce oxidation. After baseline, amphetamine (2.5 mg/kg, i.p.) was administered and samples collected for an additional 2 h for a total of 3 h of sample collection (1 h baseline + 2 h after amphetamine injection). All samples were analyzed for dopamine using HPLC-ED as for acetylcholine with some alterations: graphite working electrode (WE-3G, +400 mV vs Ag/AgCl, Amuza) and a C18 column (4.6 × 30 mm, PP-ODS2, Amuza). Mobile phase included 0.1 m phosphate buffer, pH 5.4, with 1.5% methanol, 500 mg/L sodium decanesulfonate and 50 mg/L Na2EDTA-2H2O delivered at a rate of 500 μl/min. Microdialysate sample chromatographs were analyzed based on established concentration curves for dopamine (0.1–100 nm) as for acetylcholine analyses. The limit of detection was ∼100 pm.
Electrophysiology.
Male and female Thap1C54Y/+ and Gnal+/− mice and their control littermates of 3–6 months of age were deeply anesthetized with isoflurane and perfused transcardially with ice-cold aCSF, bubbled with 95% O2-5% CO2, and containing the following (in mm): 2.5 KCl, 126 NaCl, 26 NaHCO3, 1.25 Na2HPO4, 2 CaCl2, 2 MgSO4 and 10 glucose. The brain was removed, blocked in the sagittal plane, and sectioned at a thickness of 280 μm in ice-cold aCSF. Slices were then submerged in room temperature aCSF bubbled with 95% O2–5% CO2, and stored at room temperature for at least 1 h before recording.
The slices were transferred to a recording chamber mounted on an Olympus BX50WI upright, fixed-stage microscope and perfused (2 ml/min) with oxygenated aCSF at 30°C. A 40×/0.9 numerical aperture water-immersion objective was used to examine the slice using standard infrared differential interference contrast video microscopy. Electrophysiological recordings were obtained with an Axopatch 200B amplifier (Molecular Devices), using borosilicate glass pipette pulled on a P97 puller (Sutter Instruments). Patch pipette resistance was typically 3–4 MΩ when filled with aCSF.
ChIs were identified by their distinct morphology (i.e., scarce, evenly distributed, very large neurons) and by the presence of spontaneous firing in the range of 0.5–6 Hz. ChI spontaneous activity was recorded in the loose, cell-attached configuration (seal resistance: 50 MΩ→200 MΩ), voltage-clamp mode, at a command potential at which the amplifier current, Iamp, is at 0 pA. Signals were digitized at 100 kHz and logged onto a personal computer with the Clampex 10.5 software (Molecular Devices). For the time courses, event frequency was averaged at 2 min intervals and transformed to the average of baseline (first 8 min) to yield the normalized event frequency, an indicator of how frequency changes over time. These values were multiplied by 100 when expressed as percentage of baseline.
Data analysis and statistics.
Data were analyzed using pClampfit 10.5 (Molecular Devices) and Prism 6.0e (GraphPad 6.0 for Mac OSX). Numerical data are presented as means ± SEM; n values represent independent observations. For acetylcholine, a sex effect was observed in Thap1C54Y/+ and Gnal+/− lines. In addition, we considered basal and neostigmine-induced accumulation of acetylcholine separately since the increase in acetylcholine due to neostigmine was so large that it made determining basal effects unrealistic. Therefore, mice were analyzed by sex and genotype using a two-way ANOVA to determine their effects on basal acetylcholine levels in the striatum. Acetylcholine accumulations determined by neostigmine dose–response time courses were also sex-dependent. Therefore, each sex was considered by independent repeated-measures two-way ANOVAs, which considered genotype and time as independent variables. Post hoc tests were completed using Tukey's or Sidak's multiple-comparisons tests. For dopamine in vivo microdialysis, we first determined that there was no effect of sex for dopamine efflux using two-way ANOVA. Therefore, male and female Thap1C54Y/+ and Gnal+/− mice were collated and analyzed together. As for acetylcholine, basal dopamine efflux was examined separately from amphetamine-induced efflux using independent two-sample t tests. Amphetamine-induced dopamine in Thap1C54Y/+ and Gnal+/− lines was analyzed by time and genotype using two-way repeated-measures ANOVAs.
For electrophysiological experiments, the statistical evaluation of the drug effects was performed using Wilcoxon signed-rank test (matched pairs) to compare firing rates before and after quinpirole (last baseline time point and last quinpirole time point). Mixed-design, repeated-measures two-way ANOVA was used to compare genotypes over time within each sex independently. To directly compare genotype and sex, we averaged the last four time points of quinpirole for each genotype of each sex and run a two-way ANOVA test. When a significant interaction was determined, Sidak's post hoc comparison tests were completed where appropriate. Significance was considered at p < 0.05 and was adjusted for multiple comparisons.
Results
Thap1C54Y/+ mice display a striatal hypercholinergic state and dopamine D2R-induced paradoxical excitation of striatal cholinergic interneurons
Thap1C54Y/+ mice are a heterozygous knock-in model with a mutation in Thap1 that is known to be a cause of human isolated dystonia (Bressman et al., 2009; Djarmati et al., 2009; Fuchs et al., 2009; Blanchard et al., 2011; Lohmann et al., 2012; Ruiz et al., 2015; Yellajoshyula et al., 2017). In the Thap1C54Y/+ line, basal extracellular striatal acetylcholine levels varied by sex and genotype (Fig. 1A, sex: F(1,14) = 13.93, p < 0.01; genotype: F(1,14) = 17.23, p < 0.001), with a significant interaction (F(1,14) = 6.818, p < 0.05). Specifically, basal acetylcholine levels in male Thap1C54Y/+ mice were significantly higher than male Thap1+/+ controls (p < 0.0001) and female Thap1C54Y/+ mice (p < 0.001). Interestingly, there was no significant difference in basal extracellular acetylcholine levels between female Thap1C54Y/+ and their Thap1+/+ female littermate controls (n.s.). However, female Thap1C54Y/+ mice did exhibit elevated acetylcholine accumulation upon neostigmine infusion at the highest dose (Fig. 1B, time: F(11,66) = 244.6, p < 0.0001; genotype: F(1,6) = 3.466, n.s.; interaction: F(11,66) = 3.274, p < 0.01; 500 nm B and C samples, both p < 0.001), as did male Thap1C54Y/+ mice (Fig. 1C, time: F(11,121) = 20.97, p < 0.0001; genotype: F(1,11) = 4.608, p = 0.055; interaction: F(11,121) = 3.831, p < 0.0001; 500 nm B and C samples, both p < 0.01).
Figure 1.

Cholinergic efflux and D2R-induced paradoxical excitation in male and female Thap1C54Y/+ mice. A, Male, but not female, Thap1C54Y/+ mice displayed elevated basal striatal acetylcholine. Interestingly, both male (B) and female (C) Thap1C54Y/+ mice had higher neostigmine-induced release at the 500 nm dose, compared with littermate Thap1+/+ controls (n = 4–8/group for A–C). D, Time courses and trace samples of ChI spontaneous firing rate of female Thap1+/+ mice (black) showed no change in normalized firing rate in the presence of quinpirole (Quin) over time, while female Thap1C54Y/+ mice (red) displayed paradoxical excitation in response to Quin application (*p < 0.05, 2-way ANOVA). D1, *p < 0.05, d versus c. Likewise, time courses and trace samples of male Thap1+/+ mice (E) (black) showed no change in striatal ChI firing with Quin, while male Thap1C54Y/+ mice (blue) showed a significant increase in firing (*p < 0.05, 2-way ANOVA). E1, *p < 0.05, d versus c. These results are collated in F, with Thap1C54Y/+ mice showing significantly elevated quinpirole-induced firing compared with Thap1+/+ regardless of sex. The average of the last four time points of quinpirole are compared with the average of baseline time points in percentage. G, Quin-induced paradoxical excitation of ChI firing in male and female Thap1C54Y/+ (black) is compared with the effect of Quin in the presence of mAChR antagonist, scopolamine (Scop). Time course and trace samples (green) show a block of expression of the paradoxical excitation in male and female Thap1C54Y/+ ChIs in the presence of Scop. G1, n.s., d versus c; *p < 0.05, b versus a.
Since we previously showed in Dyt1ΔGAG/+ mice that the D2R-induced paradoxical excitation of striatal ChIs is dependent upon overactivation of mAChRs due to a striatal hypercholinergic state (Scarduzio et al., 2017), we would also expect that Thap1C54Y/+ mice would display mAChR-dependent D2R-induced paradoxical excitation since they share this same hypercholinergic state (Fig. 1A–C). Interestingly, basal firing rates of ChIs were no different based on genotype (p = 0.67). However, quinpirole (10 μm) produced a significant increase of ChI firing rate in female Thap1C54Y/+ (Fig. 1D, red, baseline (c): 3.8 ± 1.3 Hz, 30 min in quinpirole (d): 6.4 ± 1 Hz, n = 7, 1D1, d vs c, p < 0.05), whereas little change in firing was seen in littermate controls (Fig. 1D, black, baseline (a): 3.3 ± 0.7 Hz, 30 min in quinpirole (b): 3 ± 0.9 Hz, n = 8, 1D1, b vs a, n.s.). Indeed, upon quinpirole treatment female Thap1C54Y/+ mice showed significantly elevated ChI firing compared with littermate controls at several time points (Fig. 1D, 2-way ANOVA, time: F(19.247) = 6.396, p < 0.0001; genotype: F(1,13) = 6.424, p = 0.025; interaction: F(19,247) = 5.189, p < 0.0001; at 26–28 and 32–40 min, all p < 0.05). Likewise, in males, quinpirole also induced paradoxical excitation in Thap1C54Y/+ (Fig. 1E, blue, baseline (c): 1.3 ± 0.3 Hz, 30 min in quinpirole (d): 2.3 ± 0.3 Hz, n = 11, Fig. 1E1, d vs c, p < 0.05), while no change was seen in littermate controls (Fig. 1E, black, baseline (a): 1.8 ± 0.4 Hz, 30 min in quinpirole (b): 2.1 ± 0.4 Hz, n = 9, Fig. 1E1, b vs a, n.s.). Direct comparison of male genotypes confirmed that Thap1C54Y/+ mice had significantly higher ChI firing after quinpirole application on slices compared with Thap1+/+ mice (Fig. 1E, 2-way ANOVA, time: F(19,342) = 5.523, p < 0.0001; genotype: F(1,18) = 3.075, p = 0.09; interaction: F(19,342) = 3.009, p < 0.0001; at 34–36 min, all p < 0.05). These changes were sex-independent, with mutants displaying a higher percentage of baseline firing of ChIs in response to quinpirole (Fig. 1F; sex: F(1,31) = 0.509, p > 0.05; genotype: F(1,31) = 9.372, p = 0.004; interaction: F(1,31) = 0.0004, n.s.). As predicted, we also found that after incubating striatal slices in the broad-spectrum mAChR antagonist, scopolamine (10 μm, >1 h), the D2R agonist-induced paradoxical excitation was blocked in Thap1C54Y/+ mice of both sex (Fig. 1G, green, baseline in >1 h scopolamine (c): 0.7 ± 0.2 Hz, 30 min in scopolamine + quinpirole (d): 0.6 ± 0.3 Hz, n = 7, Fig. 1G1, d vs c n.s.). The scopolamine effect was compared with the effect of quinpirole alone in Thap1C54Y/+ mice of both sex combined (Fig. 1G, black, baseline (a): 2.2 ± 0.5 Hz, 30 min in quinpirole (b): 3.9 ± 0.6 Hz n = 18, 1G1 b vs a p < 0.05; 2-way ANOVA, time: F(19,418) = 2.802, p < 0.0001; pretreatment: F(1,22) = 8.567, p = 0.008; interaction: F(19,418) = 4.844, p < 0.0001; at 28–40 min, all p < 0.05). These data support a causal role for mAChRs in mediating the paradoxical excitation of ChIs in Thap1C54Y/+ mice, likely through a mechanism similar to that we described previously in Dyt1ΔGAG/+ mice involving the interaction of mAChRs with D2Rs and the recruitment of β-arrestin downstream of D2R activation (Scarduzio et al., 2017).
Gnal+/− mice display a striatal hypocholinergic state and mAChR-independent dopamine D2R-induced paradoxical excitation of striatal cholinergic interneurons
Gnal+/− mice have a heterozygous deletion of the gene encoding Gαolf, modeling human DYT25 dystonia (Bressman et al., 1994; Belluscio et al., 1998; Fuchs et al., 2013; Pelosi et al., 2017). Basal striatal acetylcholine levels in the Gnal+/− line were also dependent upon sex and genotype (Fig. 2A, sex: F(1,17) = 8.442, p < 0.01; genotype: F(1,17) = 10.37, p < 0.01), with a significant interaction (F(1,17) = 5.257, p < 0.05). Unlike DYT1 and DYT6 mouse models, this effect was driven exclusively by a decrease in basal extracellular acetylcholine levels in male Gnal+/−, compared with male Gnal+/+ mice (p < 0.01). There was no difference in basal acetylcholine in female Gnal+/− compared with female littermate controls (n.s.), though Gnal+/+ females had lower basal acetylcholine levels than male Gnal+/+ mice (p < 0.01). Neostigmine-induced acetylcholine accumulation was reduced in male Gnal+/− mice compared with male littermate controls (Fig. 2C, time: F(11,99) = 29.14, p < 0.0001; genotype: F(1,9) = 3.344, n.s.; interaction: F(11,99) = 3.110, p < 0.01), most prominently at the highest neostigmine doses (500 nm B and C samples, both p < 0.01); in females, neostigmine-induced acetylcholine release was similar in Gnal+/− mice and littermate controls (Fig. 2B, time: F(11,121) = 51.63, p < 0.0001; genotype: F(1,11) = 0.880, n.s., interaction: F(11,121) = 0.937, n.s.).
Figure 2.

Cholinergic efflux and D2R-induced paradoxical excitation in male and female Gnal+/− mice. A–C, Gnal+/− male mice had lower levels of acetylcholine at baseline (A) and upon neostigmine-induced elevation (C) compared with littermate Gnal+/+ controls. These changes were not as pronounced in female Gnal+/− mice and did not reach significance (B; n = 3–8/group for A–C). D, Time courses and trace samples of ChIs firing of female Gnal+/+ (black) showed no change in firing rate in the presence of Quin, while female Gnal+/− mice (red) displayed profound paradoxical excitation in response to Quin application (*p < 0.05, 2-way ANOVA). D1, *p < 0.05, d versus c. E, Likewise, time courses and trace samples of ChIs firing of male Gnal+/+ (black) showed no change in striatal ChI firing rate with Quin, whereas male Gnal+/− (blue) showed a significant increase in firing (*p < 0.05, 2-way ANOVA). E1, *p < 0.05, d versus c. These results are collated in F, with Gnal+/− mice expressing significantly elevated quinpirole-induced firing compared with Gnal+/+ regardless of sex. The average of the last four time points of quinpirole are compared with the average of baseline time points in percentage. G, Quin-induced paradoxical excitation of ChI firing in male and female Gnal+/− (black) is compared with the effect of Quin in the presence of mAChR antagonist, scopolamine (Scop). Time course and trace samples (green) show that mAChR antagonism failed to block Quin-induced paradoxical excitation in male and female Gnal+/− mice, and interestingly, actually enhanced ChI normalized firing rates (*p < 0.05, 2-way ANOVA). G1, *p < 0.05, d versus c and b versus a.
In contrast to Thap1C54Y/+, Gnal+/− mice lack a striatal hypercholinergic state and, at least in males, exhibit significantly reduced striatal acetylcholine levels (Fig. 2A–C). Thus, we would not expect quinpirole to induce mAChR-sensitive striatal ChI paradoxical excitation in Gnal+/− mice of either sex. Similarly to the DYT6 line, there was no difference in basal ChI firing rate in Gnal+/− compared with littermate controls (p = 0.12). Surprisingly, a very prominent D2R-mediated paradoxical excitation was observed in the Gnal+/− line. In female Gnal+/− slices, quinpirole markedly enhanced ChI spontaneous firing rates (Fig. 2D, red, baseline (c): 1 ± 0.3 Hz, 30 min in quinpirole (d): 3.5 ± 0.9 Hz, n = 9, Fig. 2D1, d vs c, p < 0.05), whereas it had no effect in littermate controls (Fig. 2D, black, baseline (a): 2.1 ± 0.5 Hz, 30 min in quinpirole (b): 2.5 ± 0.4 Hz, n = 8, 2D1, b vs a, n.s.). Female Gnal+/− ChIs displayed significantly elevated firing at several time points after quinpirole application when compared with Gnal+/+ ChIs (Fig. 2D, 2-way ANOVA, time: F(19,285) = 6.702, p < 0.0001; genotype: F(1,15) = 5.215, p < 0.04; interaction: F(19,285) = 4.845, p < 0.0001; at 30–36 and 40 min, all p < 0.05). In males, quinpirole also enhanced ChI spontaneous firing rates in slices from Gnal+/− mice (Fig. 2E, blue, baseline (c): 0.6 ± 0.2 Hz, 30 min in quinpirole (d): 2.9 ± 0.7 Hz, n = 8, Fig. 2E1, d vs c, p < 0.05), whereas it had no effect in littermate controls (Fig. 2E, black, baseline (a): 1.3 ± 0.6 Hz, 30 min in quinpirole (b): 1.5 ± 0.6 Hz, n = 7, Fig. 2E1, b vs a, n.s.). Upon quinpirole application, slices from male Gnal+/− mice displayed significantly elevated ChI firing compared with Gnal+/+ at 36 and 38 min time points (Fig. 2E, 2-way ANOVA, time: F(19,209) = 4.426, p < 0.0001; genotype: F(1,12) = 3.396, p = 0.09.; interaction: F(19,209) = 2.783, p < 0.001; at 36–38 min, all p < 0.05). Direct comparisons of males to females in both genotypes revealed these changes were sex-independent, with mutants displaying a higher percentage of baseline firing of ChIs in response to quinpirole (Fig. 2F; sex: F(1,25) = 1.583, n.s.; genotype: F(1,25) = 15.13, p < 0.001; interaction: F(1,25) = 0.885, n.s.). Antagonizing mAChRs with scopolamine did not block the excitatory effect of quinpirole on Gnal+/− ChIs of either sex (Fig. 2G, green; baseline in scopolamine (c): 0.4 ± 0.2 Hz,; 30 min in scopolamine + quinpirole (d): 6.3 ± 1.4 Hz, n = 6, Fig. 2G1, d vs c, p < 0.05). In fact, with scopolamine pretreatment quinpirole significantly elevated ChI firing rate compared with quinpirole alone in Gnal+/− mice (Fig. 2G; drug: F(1,20) = 4.852, p < 0.05; time: F(19,380) = 8.942, p < 0.0001; interaction: F(19,380) = 4.089, p < 0.0001; at 30, 34, and 40 min, all p < 0.05). These data suggest an altogether distinct mechanism underlying D2R-induced paradoxical excitation in Gnal+/− mice versus that in Dyt1ΔGAG/+ and Thap1C54Y/+ mice.
Dopamine D2R-induced paradoxical excitation of striatal ChIs is driven by an adenosine A2AR-dependent mechanism in Gnal+/− mice, but not Thap1C54Y/+ mice
One potential contributor to paradoxical excitation in Gnal+/− mice is defective interaction of D2R and A2ARs, which traditionally signal using Gαolf (the protein encoded by Gnal), and are known to complex together forming D2R-A2AR heteromers. Importantly, A2AR agonist-induced activation of these heteromers can favor the binding of β-arrestin to the D2L protomer (Borroto-Escuela et al., 2011; see also, Huang et al., 2013; Ferré et al., 2016; Sahlholm et al., 2018). Thus, given the known D2R–A2AR interaction and the role of β-arrestin in this interaction (Fig. 3C), we tested whether altered A2AR function in Gnal+/− mice could underlie the D2R-induced paradoxical excitation of ChIs. We incubated slices from Gnal+/− mice in the potent and selective A2AR antagonist, ZM241385. Interestingly, A2AR blockade with ZM241385 (>1 h, 500 nm) prevented the quinpirole-induced increase of ChI firing rates (Fig. 3A, cyan, baseline in 500 nm ZM241385 (c): 0.8 ± 0.2 Hz, 20 min in ZM241385 + quinpirole (d): 0.49 ± 0.2 Hz, n = 6, Fig. 3A1, d vs c, p = 0.56, ns; Figure 3A, 2-way ANOVA, time: F(13,390) = 4.571, p < 0.0001; drug: F(1,30) = 6.587, p = 0.016; interaction: F(13,390) = 4.865, p < 0.0001; at 22–28 min, all p < 0.05).
Figure 3.

Involvement of A2ARs and β-arrestin in the D2R-induced paradoxical excitation of ChI in male and female Gnal+/− mice. A, Quin-induced excitation of ChI firing in male and female Gnal+/− (black) is compared with the effect of Quin in the presence of the A2AR antagonist ZM241385. Time course and trace samples (cyan) show that A2AR antagonism blocked Quin-induced paradoxical excitation in Gnal+/− mice, likely indicating a role for the Gαolf-coupled A2AR in this effect. A1, n.s., d versus c; *p < 0.05, b versus a. B, In male and female Gnal+/−, D2-induced ChI firing excitation (black) is compared with the effect of a G-protein-biased D2R agonist, MLS1547. Time course and trace samples (orange) showed an expected reduction in ChI firing after application of MLS1547 to slices. B1, *p < 0.05 significant reduction, d versus c; *p < 0.05 significant potentiation, b versus a. C, Schematic representation of A2A/D2 heteroreceptor on striatal ChI membrane. Activation of the allosteric A2A–D2R interaction induces a conformational state of the D2R promoter, which moves away from Gi/o signaling and instead favors binding of β-arrestin2 and β-arrestin2-mediated signaling. D, Male and female Thap1C54Y/+ quin-induced excitation of ChI firing (black) is compared with the effect of Quin in the presence of the A2AR antagonist, ZM241385. Time course and trace samples (cyan) show that ZM421385 failed to block Quin-induced paradoxical excitation in Thap1C54Y/+ mice. D1, *p < 0.05 d, versus c and b versus a.
These results suggest the involvement of an alternative signaling pathway, likely the β-arrestin pathway that we have previously implicated in the paradoxical excitation seen in Dyt1ΔGAG/+ mice (Scarduzio et al., 2017). If this is the case, a Gi/o-preferring D2R agonist should be ineffective at inducing the paradoxical excitation in striatal ChIs in Gnal+/− mice. We used a G-protein-biased D2R agonist, MLS1547 (10 μm, 20 min), which we have previously shown is able to normalize ChI function in male Dyt1ΔGAG/+ instead of quinpirole (Scarduzio et al., 2017). In slices from male and female Gnal+/− mice, MLS1547 induced a depression of spontaneous firing rate, rather than excitation (Fig. 3B, orange; baseline in DMSO (c): 0.2 ± 0.07 Hz, 20 min in MLS1547 (d): 0.05 ± 0.02 Hz, n = 6, Fig. 3B1, d vs c, p < 0.05; Figure 3B, 2-way ANOVA, time: F(17,442) = 2.103, p < 0.01; drug: F(1,26) = 3.928, p = 0.058; interaction: F(17,442) = 2.974, p < 0.0001; at 32–36 min, all p < 0.05). This findings indicate that activation of the Gi/o pathway does not mediate the D2R-induced paradoxical excitation in ChIs as expected and, instead, suggests that Gnal haploinsufficiency (like Dyt1 and Thap1 mutation) drives D2R signaling toward a β-arrestin signaling strategy, albeit through a separate A2AR-dependent mechanism.
This mechanism appears to be specific to the Gnal+/− mice, since quinpirole-induced paradoxical excitation in Thap1C54Y/+ mice was unaffected by A2AR antagonism (Fig. 3D, cyan, baseline in ZM241385 (c): 1 ± 0.2 Hz, 20 min in ZM241385 + quinpirole (d): 1.8 ± 0.3 Hz, n = 11, Fig. 3D1, d vs c, p < 0.05. Indeed, the paradoxical excitation induced in Thap1C54Y/+ mice by quinpirole was not different with or without ZM241385 (Fig. 3D, 2-way ANOVA, time: F(19,513) = 11.64, p < 0.0001; drug: F(1,27) = 1.094, p = 0.30; interaction: F(19,513) = 0.7988, p = 0.71). Given their similarities to the DYT1 mouse model, we would predict that the Thap1C54Y/+ mutation exerts a switch to D2R noncanonical signaling on ChIs via a muscarinic-dependent mechanism (Scarduzio et al., 2017), whereas the switch in Gnal+/− mice is exerted by interaction between D2R and A2AR pathways (Fig. 3C).
Amphetamine-induced striatal dopamine release is dampened in Thap1C54Y/+, but not Gnal+/− mice
Numerous studies have reported reduced amphetamine-induced dopamine efflux in DYT1 mouse models (Balcioglu et al., 2007; Hewett et al., 2010; Song et al., 2012). We investigated the effect of systemic amphetamine treatment on dopamine release in male and female Thap1C54Y/+ and Gnal+/− mice, to determine whether dopamine function was also altered in these dystonia models. In these two models, sex did not statistically alter any measure of dopamine efflux and was ignored as an independent variable (Thap1C54Y/+: F(1,10) = 1.915, n.s.; Gnal+/−: F(1,15) = 1.364, n.s.). Likewise, basal levels of striatal extracellular dopamine were unaffected by genotype in both the Thap1C54Y/+ (Thap1+/+: 2.063 ± 0.5392; Thap1C54Y/+: 1.770 ± 0.3572; t8 = 0.50, n.s.) and the Gnal+/− line (Gnal+/+: 0.3999 ± 0.1221; Gnal+/−: 0.6022 ± 0.1826; t19 = 0.74, n.s.). Amphetamine-stimulated dopamine release was perturbed in Thap1C54Y/+ mice (Fig. 4A, time: F(8,96) = 23.18, p < 0.0001; genotype: F(1,12) = 2.884, n.s.; interaction: F(8,96) = 2.157, p < 0.05), with significantly reduced levels at peak time points in Thap1C54Y/+ compared with littermate controls (100 min; p < 0.0001). In contrast, dopamine efflux after amphetamine administration in Gnal+/− mice was indistinguishable from littermate controls (Fig. 4B, time: F(8,152) = 24.28, p < 0.0001; genotype: F(1,19) = 0.134, n.s.; interaction: F(8,152) = 0.194, n.s.).
Figure 4.
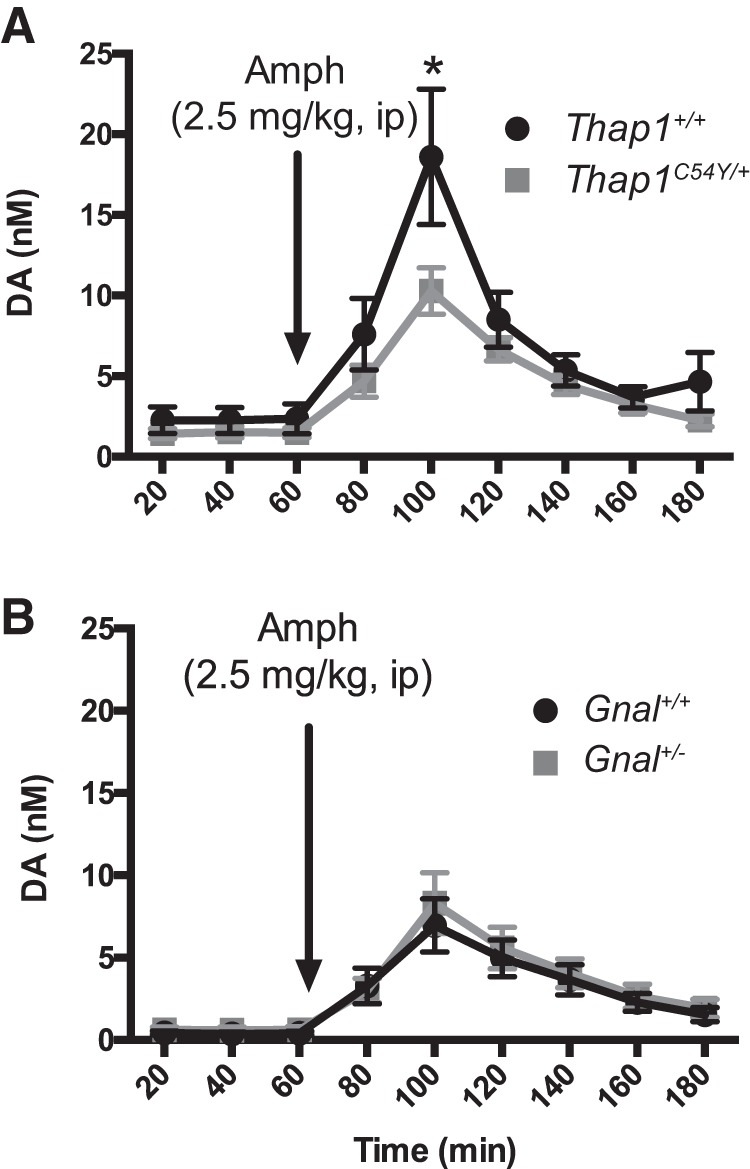
Amphetamine-stimulated (2.5 mg/kg, i.p.) striatal dopamine efflux in Thap1C54Y/+ and Gnal+/− mice determined by in vivo microdialysis. A, Thap1C54Y/+ mice had blunted amphetamine-stimulated striatal dopamine compared with Thap1+/+ littermate controls (n = 4–5/group). B, Both Gnal+/− and Gnal+/+ littermate controls had similarly enhanced dopamine efflux in response to amphetamine administration (n = 7–14/group). *p < 0.05 versus littermate controls.
Discussion
The studies described here using Thap1C54Y/+ and Gnal+/− mouse models, considered in light of our earlier work with Dyt1ΔGAG/+ mice (Scarduzio et al., 2017), led to three primary observations. First, Dyt1ΔGAG/+ and Thap1C54Y/+ mice share a core physiology characterized by elevated extracellular striatal acetylcholine and prominent D2R-induced paradoxical excitation of striatal ChIs which can be reversed by mAChR inhibition. Second, the Gnal+/− mouse model has a fundamentally different striatal physiology. Although D2R-induced paradoxical excitation of ChIs is present in these mice, extracellular striatal acetylcholine is reduced and paradoxical excitation is not affected by mAChR blockade. Finally, striatal neurochemistry in Thap1C54Y/+ and Gnal+/− mouse models was sex-dependent, with males in general having more dramatic abnormalities than females, although the functional consequences of these changes were similar.
Cholinergic function has long been considered a central mechanism in the manifestations of dystonia, due to the ability of cholinergic agonists to induce dystonia (Shafrir et al., 1986) and the efficacy of mAChR antagonists for the treatment of dystonia (Burke and Fahn, 1983; Burke et al., 1986). We recently found that extracellular striatal acetylcholine levels are many times higher in male Dyt1ΔGAG/+ mice, compared with controls (Scarduzio et al., 2017). Here, we examined the Thap1C54Y/+ mouse model, and found similar elevations in both basal and neostigmine-induced accumulation of striatal acetylcholine (Fig. 1A–C). In our previous publication, we also suggested that in Dyt1ΔGAG/+ mice the hypercholinergic striatal state drives the paradoxical excitation of ChIs in response to D2R activation by switching signaling from a canonical (Gi/o) to a noncanonical (β-arrestin) signaling pathway (Scarduzio et al., 2017). The present data suggest that this is also the case in the Thap1C54Y/+ mouse model: mAChR antagonism was capable of reversing D2R-mediated paradoxical excitation (Fig. 1G). Other evidence suggests that TorsinA and THAP1 proteins may have important interactions: THAP1 is a transcription factor linked to regulation of TOR1A transcription (Gavarini et al., 2010; Kaiser et al., 2010; Cheng et al., 2014). Indeed, C54Y and similar missense mutations (i.e., F81L) in THAP1 are found in the DNA-binding domain (Bressman et al., 2009; Djarmati et al., 2009; Fuchs et al., 2009; Ruiz et al., 2015; Zakirova et al., 2018). It is possible that this relationship accounts for the similar phenotypic effects of mutations in both genes and should be a target for future investigation.
In contrast, we found that male Gnal+/− mice had lower levels of extracellular basal and neostigmine-induced accumulation of striatal acetylcholine, while females have relatively normal levels, as measured by microdialysis (Fig. 2A–C); nevertheless, both sexes displayed D2R-induced ChI paradoxical excitation (Fig. 2D–F). This effect was not blocked by mAChR antagonism (Fig. 2G), demonstrating that in these animals a hypercholinergic state is not the cause of the aberrant response to D2R activation and suggesting an alternative mechanism. In support of this notion, we observed that a Gi/o-biased D2R agonist was unable to mimic the effect of quinpirole, suggesting that the excitatory response in Gnal+/− mice is not mediated by the D2R–Gi/o pathway (Fig. 3B).
The GNAL gene encodes for Gαolf, a G-protein known to be involved in striatal D1R, as well as A2AR signaling. A2ARs are well known for their role in ChIs and have been indicated in the pathogenesis of movement disorders (Tozzi et al., 2011; Peterson et al., 2012). Previous work has demonstrated the formation of A2AR–D2R heteromers (Fuxe et al., 2005) and A2AR agonist-induced activation of this complex can favor the binding of β-arrestin2 to the D2L protomer (Borroto-Escuela et al., 2011; Huang et al., 2013; Ferré et al., 2016; Sahlholm et al., 2018). Indeed, in Gnal+/− slices incubated in the A2AR antagonist ZM241385, the D2R-induced paradoxical excitation of striatal ChIs was completely blocked (Fig. 3A). Though further studies will be necessary to validate the potential formation of D2R-A2AR heteromers in DYT25 models, we propose that the reduction of Gαolf levels in the Gnal+/− mouse may induce an upregulation of A2AR expression, leading to abnormal coupling with D2Rs and facilitating the switch to β-arrestin signaling downstream of dopamine receptor activation (Fig. 3C). Classically, β-arrestins block GPCR signaling by binding phosphorylated receptors promoting endocytosis. More roles for β-arrestin have been discovered recently, including acting as a scaffold for the Raf-MEK-ERK signaling cascade, both dependently and independently of GPCRs (Gurevich and Gurevich, 2019). This cascade is involved in development of pathological synaptic plasticity in hyperkinetic disorders (Peterson et al., 2010; Cerovic et al., 2015; Picconi et al., 2018). β-arrestins are thought to bind with more affinity to the same conformational “pocket” as the G-protein in many GPCRs and would reduce Gi/o signaling impact on neuronal output. Indeed, in a DYT1 mouse model, there is a decline in the ability of striatal D2Rs to activate Gi/o receptors (Napolitano et al., 2010). β-arrestin signaling may underlie the development of ChI paradoxical excitation by reducing Gi/o activity in ChIs through occupation of its conformational “pocket” on D2Rs and/or by acting to promote Raf-MEK-ERK signaling's effects on synaptic plasticity.
Finally, we found that amphetamine-stimulated dopamine is blunted in Thap1C54Y/+, but not Gnal+/− mice (Fig. 4). Blunted amphetamine-induced dopamine has been a common phenotype observed in a number of DYT1 animal models (Balcioglu et al., 2007; Sciamanna et al., 2012; Song et al., 2012). In transgenic human mutant TOR1A (hMT1) mice, reduced amphetamine-induced dopamine release may be related to deficits in the dopamine transporter (DAT; Hewett et al., 2010), a primary site of action for amphetamine. The overlapping molecular pathways and amphetamine response phenotype for both TorsinA and THAP1, but not Gαolf, likely further indicates a common underlying mechanism in DYT1 and DYT6 mouse models. Because GNAL encodes for a postsynaptic protein (Gαolf), this may account for the lack of a direct influence on amphetamine-induced dopamine release in this DYT25 mouse model (Fig. 4B).
The disparity in extracellular acetylcholine between males and females found here is interesting. In particular, Thap1C54Y/+ females displayed muted hypercholinergic striatal states when studied with microdialysis, with abnormalities only becoming clear after unmasking with neostigmine (Fig. 1A–B). However, paradoxical excitation at the cellular level in the response to D2R activation was sex-independent (Fig. 1D–E). Likewise, Gnal+/− females also did not display statistically relevant changes in basal acetylcholine, whereas male mice had ∼50% less striatal acetylcholine efflux (Fig. 2A–C). Electrophysiologically, ChIs responded similarly in both males and females in response to quinpirole, though the response in males was more profound (Fig. 2D–F). These data collectively suggest that although changes in interstitial acetylcholine may not be sufficient for detection by in vivo microdialysis, female mutant mice likely still have a physiologically relevant change in acetylcholine. Many features of striatal acetylcholine function are known to be sex dependent and may explain our current results: higher choline acetlytransferase in females would decrease available acetylcholine in the extracellular space and higher affinity of cholinergic receptors in females, particularly autoreceptors, would make them less likely to exhibit detectable hypercholinergic phenotypes (Miller, 1983; Becker and Beer, 1986). An additional potential contributor is sex-dependent sensitivity to Ras homolog enriched in striatum (Rhes), which has been suggested as a key player in the development of paradoxical excitation in striatal ChIs (Sciamanna et al., 2015). Female mice have been shown to display enhanced sensitivity to Rhes KO on cAMP/PKA activation (Ghiglieri et al., 2015). Thus, female mutant mice may display a less pronounced biochemical hypercholinergic state, but are more attuned to changes in neurotransmitter concentration in terms of receptor signaling. Although this is only one of many possibilities, it highlights the need for adequate investigation of sex-dependent phenotypes in dystonia models. Indeed, significant sex disparity exists in human dystonia, at least in sporadic forms, where women have a threefold higher incidence compared with men (ESDE Collaborative Group, 1999; Williams et al., 2017), though males have a significantly earlier age of onset (ESDE Collaborative Group, 1999; Defazio et al., 2003; Butler et al., 2015).
Considering the current data in context, we can conclude that multiple genetic mouse models of dystonia–though arising from diverse genetic abnormalities and differing in striatal dopamine and cholinergic efflux–have concordant changes in the response of ChIs to dopaminergic input (Fig. 5). This provides credence to the idea that ChI paradoxical excitation is a common endophenotype of dystonias (Eskow Jaunarajs et al., 2015), though its precise role in the manifestation of dystonic movements remains unknown. Further, we have observed that one of the key factors in the pathophysiology in these genetic mouse models of dystonia is altered signaling downstream of D2Rs, with a switch from canonical Gi/o receptor signaling to noncanonical β-arrestin signaling pathways. In genetic DYT1 and DYT6 mouse models, this appears to be driven by abnormal interactions of D2R and mAChRs, while in DYT25 it is the result of abnormal D2R-A2AR interaction. These common features raise the possibility that treatments which alter striatal cholinergic interneuron function and signaling pathways downstream of D2Rs may be broadly applicable in many forms of dystonia.
Figure 5.

Common and distinct phenotypes in three genetic mouse models of isolated dystonia. Compared with previously published phenotypes observed in DYT1 mouse models (Pisani et al., 2006; Song et al., 2012; Scarduzio et al., 2017), DYT6 and DYT25 also share the phenomenon of D2R-induced paradoxical excitation of striatal ChIs. Animals from both DYT1 and DYT6 mouse models exhibited elevated acetylcholine release and impaired amphetamine-induced dopamine release. Furthermore, mAChR antagonism was able to completely block ChI paradoxical excitation in response to D2R agonism. In contrast, ChI paradoxical excitation in a DYT25 mouse model was unaffected by mAChR antagonism, though A2AR antagonism was sufficient to block D2R-induced paradoxical excitation. Neurochemically, these mice had normal responsivity to amphetamine in terms of dopamine release and there was a reduction in extracellular acetylcholine in the striatum.
Footnotes
This work was supported by the University of Alabama at Birmingham Nell Johnson Dystonia Research Acceleration Fund (D.G.S.) and the National Institutes of Health (Grant P01NS087997 to D.G.S. and Grant R01NS081282 to M.E.E.). We thank Jeffrey Conn for the sharing of his DYT25 mouse colony and Josh Randolph for his technical assistance.
The authors declare no competing financial interests.
References
- Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK (2013) Phenomenology and classification of dystonia: a consensus update. Mov Disord 28:863–873. 10.1002/mds.25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese A, Di Giovanni M, Lalli S (2019) Dystonia: diagnosis and management. Eur J Neurol 26:5–17. 10.1111/ene.13762 [DOI] [PubMed] [Google Scholar]
- Balcioglu A, Kim MO, Sharma N, Cha JH, Breakefield XO, Standaert DG (2007) Dopamine release is impaired in a mouse model of DYT1 dystonia. J Neurochem 102:783–788. 10.1111/j.1471-4159.2007.04590.x [DOI] [PubMed] [Google Scholar]
- Balint B, Mencacci NE, Valente EM, Pisani A, Rothwell J, Jankovic J, Vidailhet M, Bhatia KP (2018) Dystonia. Nat Rev Dis Primers 4:25. 10.1038/s41572-018-0023-6 [DOI] [PubMed] [Google Scholar]
- Becker JB, Beer ME (1986) The influence of estrogen on nigrostriatal dopamine activity: behavioral and neurochemical evidence for both pre- and postsynaptic components. Behav Brain Res 19:27–33. 10.1016/0166-4328(86)90044-6 [DOI] [PubMed] [Google Scholar]
- Belluscio L, Gold GH, Nemes A, Axel R (1998) Mice deficient in G(olf) are anosmic. Neuron 20:69–81. 10.1016/S0896-6273(00)80435-3 [DOI] [PubMed] [Google Scholar]
- Blanchard A, Roubertie A, Simonetta-Moreau M, Ea V, Coquart C, Frederic MY, Gallouedec G, Adenis JP, Benatru I, Borg M, Burbaud P, Calvas P, Cif L, Damier P, Destee A, Faivre L, Guyant-Marechal L, Janik P, Janoura S, Kreisler A, et al. (2011) Singular DYT6 phenotypes in association with new THAP1 frameshift mutations. Mov Disord 26:1775–1777. 10.1002/mds.23641 [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Romero-Fernandez W, Tarakanov AO, Ciruela F, Agnati LF, Fuxe K (2011) On the existence of a possible A2A-D2-beta-Arrestin2 complex: A2A agonist modulation of D2 agonist-induced beta-arrestin2 recruitment. J Mol Biol 406:687–699. 10.1016/j.jmb.2011.01.022 [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG (2008) The pathophysiological basis of dystonias. Nat Rev Neurosci 9:222–234. 10.1038/nrn2337 [DOI] [PubMed] [Google Scholar]
- Bressman SB, de Leon D, Kramer PL, Ozelius LJ, Brin MF, Greene PE, Fahn S, Breakefield XO, Risch NJ (1994) Dystonia in ashkenazi jews: clinical characterization of a founder mutation. Ann Neurol 36:771–777. 10.1002/ana.410360514 [DOI] [PubMed] [Google Scholar]
- Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R (2009) Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol 8:441–446. 10.1016/S1474-4422(09)70081-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Fahn S (1983) Double-blind evaluation of trihexyphenidyl in dystonia. Adv Neurol 37:189–192. [PubMed] [Google Scholar]
- Burke RE, Fahn S, Marsden CD (1986) Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl. Neurology 36:160–164. 10.1212/WNL.36.2.160 [DOI] [PubMed] [Google Scholar]
- Butler JS, Beiser IM, Williams L, McGovern E, Molloy F, Lynch T, Healy DG, Moore H, Walsh R, Reilly RB, O'Riordan S, Walsh C, Hutchinson M (2015) Age-related sexual dimorphism in temporal discrimination and in adult-onset dystonia suggests GABAergic mechanisms. Front Neurol 6:258. 10.3389/fneur.2015.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerovic M, Bagetta V, Pendolino V, Ghiglieri V, Fasano S, Morella I, Hardingham N, Heuer A, Papale A, Marchisella F, Giampà C, Calabresi P, Picconi B, Brambilla R (2015) Derangement of ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and extracellular signal-regulated kinase (ERK) dependent striatal plasticity in L-DOPA-induced dyskinesia. Biol Psychiatry 77:106–115. 10.1016/j.biopsych.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Cheng FB, Feng JC, Ma LY, Miao J, Ott T, Wan XH, Grundmann K (2014) Combined occurrence of a novel TOR1A and a THAP1 mutation in primary dystonia. Mov Disord 29:1079–1083. 10.1002/mds.25921 [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y (2005) Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol 196:452–463. 10.1016/j.expneurol.2005.08.025 [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Cheetham CC, Lu J, Vo V, Lovinger DM, Li Y (2012) An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 DeltaGAG knock-in mice. Behav Brain Res 226:465–472. 10.1016/j.bbr.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defazio G, Abbruzzese G, Girlanda P, Vacca L, Currà A, Marchese R, Martino D, Masi G, Majorana G, Mazzella L, Livrea P, Berardelli A (2003) Does sex influence age at onset in cranial-cervical and upper limb dystonia? J Neurol Neurosurg Psychiatry 74:265–267. 10.1136/jnnp.74.2.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djarmati A, Schneider SA, Lohmann K, Winkler S, Pawlack H, Hagenah J, Brüggemann N, Zittel S, Fuchs T, Raković A, Schmidt A, Jabusch HC, Wilcox R, Kostić VS, Siebner H, Altenmüller E, Münchau A, Ozelius LJ, Klein C (2009) Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study. Lancet Neurol 8:447–452. 10.1016/S1474-4422(09)70083-3 [DOI] [PubMed] [Google Scholar]
- ESDE Collaborative Group (1999) Sex-related influences on the frequency and age of onset of primary dystonia. epidemiologic study of dystonia in europe (ESDE) collaborative group. Neurology 53:1871–1873. 10.1212/WNL.53.8.1871 [DOI] [PubMed] [Google Scholar]
- Eskow Jaunarajs KL, Bonsi P, Chesselet MF, Standaert DG, Pisani A (2015) Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol 127–128:91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Bonaventura J, Tomasi D, Navarro G, Moreno E, Cortés A, Lluís C, Casadó V, Volkow ND (2016) Allosteric mechanisms within the adenosine A2A-dopamine D2 receptor heterotetramer. Neuropharmacology 104:154–160. 10.1016/j.neuropharm.2015.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ (2009) Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet 41:286–288. 10.1038/ng.304 [DOI] [PubMed] [Google Scholar]
- Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, Lang AE, Liang TW, Trosch RM, White S, Ainehsazan E, Hervé D, Sharma N, Ehrlich ME, Martemyanov KA, Bressman SB, Ozelius LJ (2013) Mutations in GNAL cause primary torsion dystonia. Nat Genet 45:88–92. 10.1038/ng.2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe K, Ferré S, Canals M, Torvinen M, Terasmaa A, Marcellino D, Goldberg SR, Staines W, Jacobsen KX, Lluis C, Woods AS, Agnati LF, Franco R (2005) Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J Mol Neurosci 26:209–220. 10.1385/JMN:26:2-3:209 [DOI] [PubMed] [Google Scholar]
- Gavarini S, Cayrol C, Fuchs T, Lyons N, Ehrlich ME, Girard JP, Ozelius LJ (2010) Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann Neurol 68:549–553. 10.1002/ana.22138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglieri V, Napolitano F, Pelosi B, Schepisi C, Migliarini S, Di Maio A, Pendolino V, Mancini M, Sciamanna G, Vitucci D, Maddaloni G, Giampà C, Errico F, Nisticò R, Pasqualetti M, Picconi B, Usiello A (2015) Rhes influences striatal cAMP/PKA-dependent signaling and synaptic plasticity in a gender-sensitive fashion. Sci Rep 5:10933. 10.1038/srep10933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV (2019) GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol 10:125. 10.3389/fphar.2019.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewett J, Johanson P, Sharma N, Standaert D, Balcioglu A (2010) Function of dopamine transporter is compromised in DYT1 transgenic animal model in vivo. J Neurochem 113:228–235. 10.1111/j.1471-4159.2010.06590.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JJ, Yen CT, Liu TL, Tsao HW, Hsu JW, Tsai ML (2013) Effects of dopamine D2 agonist quinpirole on neuronal activity of anterior cingulate cortex and striatum in rats. Psychopharmacology (Berl) 227:459–466. 10.1007/s00213-013-2965-4 [DOI] [PubMed] [Google Scholar]
- Ip CW, Isaias IU, Kusche-Tekin BB, Klein D, Groh J, O'Leary A, Knorr S, Higuchi T, Koprich JB, Brotchie JM, Toyka KV, Reif A, Volkmann J (2016) Tor1a+/- mice develop dystonia-like movements via a striatal dopaminergic dysregulation triggered by peripheral nerve injury. Acta Neuropathol Commun 4:108. 10.1186/s40478-016-0375-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser FJ, Osmanoric A, Rakovic A, Erogullari A, Uflacker N, Braunholz D, Lohnau T, Orolicki S, Albrecht M, Gillessen-Kaesbach G, Klein C, Lohmann K (2010) The dystonia gene DYT1 is repressed by the transcription factor THAP1 (DYT6). Ann Neurol 68:554–559. 10.1002/ana.22157 [DOI] [PubMed] [Google Scholar]
- Lohmann K, Uflacker N, Erogullari A, Lohnau T, Winkler S, Dendorfer A, Schneider SA, Osmanovic A, Svetel M, Ferbert A, Zittel S, Kühn AA, Schmidt A, Altenmüller E, Münchau A, Kamm C, Wittstock M, Kupsch A, Moro E, Volkmann J et al. (2012) Identification and functional analysis of novel THAP1 mutations. Eur J Hum Genet 20:171–175. 10.1038/ejhg.2011.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann K, Klein C (2017) Update on the genetics of dystonia. Curr Neurol Neurosci Rep 17:26. 10.1007/s11910-017-0735-0 [DOI] [PubMed] [Google Scholar]
- Maltese M, Martella G, Madeo G, Fagiolo I, Tassone A, Ponterio G, Sciamanna G, Burbaud P, Conn PJ, Bonsi P, Pisani A (2014) Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors. Mov Disord 29:1655–1665. 10.1002/mds.26009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A (2009) Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain 132:2336–2349. 10.1093/brain/awp194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC. (1983) Sex differences in dopaminergic and cholinergic activity and function in the nigro-striatal system of the rat. Psychoneuroendocrinology 8:225–236. 10.1016/0306-4530(83)90059-8 [DOI] [PubMed] [Google Scholar]
- Napolitano F, Pasqualetti M, Usiello A, Santini E, Pacini G, Sciamanna G, Errico F, Tassone A, Di Dato V, Martella G, Cuomo D, Fisone G, Bernardi G, Mandolesi G, Mercuri NB, Standaert DG, Pisani A (2010) Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiol Dis 38:434–445. 10.1016/j.nbd.2010.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2004) The Mouse Brain in Stereotaxic Coordinates, Ed 2 London, UK: Gulf Professional Publishing. [Google Scholar]
- Pelosi A, Menardy F, Popa D, Girault JA, Hervé D (2017) Heterozygous gnal mice are a novel animal model with which to study dystonia pathophysiology. J Neurosci 37:6253–6267. 10.1523/JNEUROSCI.1529-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DA, Sejnowski TJ, Poizner H (2010) Convergent evidence for abnormal striatal synaptic plasticity in dystonia. Neurobiol Dis 37:558–573. 10.1016/j.nbd.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JD, Goldberg JA, Surmeier DJ (2012) Adenosine A2a receptor antagonists attenuate striatal adaptations following dopamine depletion. Neurobiol Dis 45:409–416. 10.1016/j.nbd.2011.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, De Leonibus E, Calabresi P (2018) Synaptic plasticity and levodopa-induced dyskinesia: electrophysiological and structural abnormalities. J Neural Transm 125:1263–1271. 10.1007/s00702-018-1864-6 [DOI] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG (2006) Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis 24:318–325. 10.1016/j.nbd.2006.07.006 [DOI] [PubMed] [Google Scholar]
- Richter F, Gerstenberger J, Bauer A, Liang CC, Richter A (2017) Sensorimotor tests unmask a phenotype in the DYT1 knock-in mouse model of dystonia. Behav Brain Res 317:536–541. 10.1016/j.bbr.2016.10.028 [DOI] [PubMed] [Google Scholar]
- Ruiz M, Perez-Garcia G, Ortiz-Virumbrales M, Méneret A, Morant A, Kottwitz J, Fuchs T, Bonet J, Gonzalez-Alegre P, Hof PR, Ozelius LJ, Ehrlich ME (2015) Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum Mol Genet 24:7159–7170. 10.1093/hmg/ddv384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlholm K, Gómez-Soler M, Valle-León M, López-Cano M, Taura JJ, Ciruela F, Fernández-Dueñas V (2018) Antipsychotic-like efficacy of dopamine D2 receptor-biased ligands is dependent on adenosine A2A receptor expression. Mol Neurobiol 55:4952–4958. 10.1007/s12035-017-0696-y [DOI] [PubMed] [Google Scholar]
- Scarduzio M, Zimmerman CN, Jaunarajs KL, Wang Q, Standaert DG, McMahon LL (2017) Strength of cholinergic tone dictates the polarity of dopamine D2 receptor modulation of striatal cholinergic interneuron excitability in DYT1 dystonia. Exp Neurol 295:162–175. 10.1016/j.expneurol.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Tassone A, Mandolesi G, Puglisi F, Ponterio G, Martella G, Madeo G, Bernardi G, Standaert DG, Bonsi P, Pisani A (2012) Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J Neurosci 32:11991–12004. 10.1523/JNEUROSCI.0041-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Napolitano F, Pelosi B, Bonsi P, Vitucci D, Nuzzo T, Punzo D, Ghiglieri V, Ponterio G, Pasqualetti M, Pisani A, Usiello A (2015) Rhes regulates dopamine D2 receptor transmission in striatal cholinergic interneurons. Neurobiol Dis 78:146–161. 10.1016/j.nbd.2015.03.021 [DOI] [PubMed] [Google Scholar]
- Shafrir Y, Levy Y, Beharab A, Nitzam M, Steinherz R (1986) Acute dystonic reaction to bethanechol–a direct acetylcholine receptor agonist. Dev Med Child Neurol 28:646–648. [DOI] [PubMed] [Google Scholar]
- Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, Breakefield XO (2005) Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci 25:5351–5355. 10.1523/JNEUROSCI.0855-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CH, Fan X, Exeter CJ, Hess EJ, Jinnah HA (2012) Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis 48:66–78. 10.1016/j.nbd.2012.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, Ghiglieri V, Giampà C, Fusco FR, Picconi B, Calabresi P (2011) The distinct role of medium spiny neurons and cholinergic interneurons in the D(2)/A(2)A receptor interaction in the striatum: implications for Parkinson's disease. J Neurosci 31:1850–1862. 10.1523/JNEUROSCI.4082-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L, McGovern E, Kimmich O, Molloy A, Beiser I, Butler JS, Molloy F, Logan P, Healy DG, Lynch T, Walsh R, Cassidy L, Moriarty P, Moore H, McSwiney T, Walsh C, O'Riordan S, Hutchinson M (2017) Epidemiological, clinical and genetic aspects of adult onset isolated focal dystonia in ireland. Eur J Neurol 24:73–81. 10.1111/ene.13133 [DOI] [PubMed] [Google Scholar]
- Yellajoshyula D, Liang CC, Pappas SS, Penati S, Yang A, Mecano R, Kumaran R, Jou S, Cookson MR, Dauer WT (2017) The DYT6 dystonia protein THAP1 regulates myelination within the oligodendrocyte lineage. Dev Cell 42:52–67.e4. 10.1016/j.devcel.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakirova Z, Fanutza T, Bonet J, Readhead B, Zhang W, Yi Z, Beauvais G, Zwaka TP, Ozelius LJ, Blitzer RD, Gonzalez-Alegre P, Ehrlich ME (2018) Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet 14:e1007169. 10.1371/journal.pgen.1007169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, DeCuypere M, LeDoux MS (2008) Abnormal motor function and dopamine neurotransmission in DYT1 DeltaGAG transgenic mice. Exp Neurol 210:719–730. 10.1016/j.expneurol.2007.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]