

Abstract
Aim:
Imbalances in cytochrome P450 (CYP)-dependent eicosanoid formation may play a central role in ischemic acute kidney injury (AKI). We reported previously that inhibition of 20-hydroxyeicosatetraenoic acid (20-HETE) action ameliorated ischemia/reperfusion (I/R)-induced AKI in rats. Now we tested the hypothesis that enhancement of epoxyeicosatrienoic acid (EET) actions may counteract the detrimental effects of 20-HETE and prevent the initiation of AKI.
Methods:
Male Lewis rats underwent right nephrectomy and ischemia was induced by 45 min clamping of the left renal pedicle followed by up to 48 h of reperfusion. Circulating CYP-eicosanoid profiles where compared in patients who underwent cardiac surgery with (n=21) and without (n=38) developing postoperative AKI.
Results:
Ischemia induced an about 8-fold increase of renal 20-HETE levels, whereas free EETs were not accumulated. To compensate for this imbalance, a synthetic 14,15-EET analog was administered by intrarenal infusion before ischemia. The EET analog improved renal reoxygenation as monitored by in vivo parametric MRI during the initial 2 h reperfusion phase. The EET analog improved PI3K- as well as mTORC2-dependent rephosphorylation of Akt, induced inactivation of GSK-3β, reduced the development of tubular apoptosis and attenuated inflammatory cell infiltration. The EET analog also significantly alleviated the I/R-induced drop in creatinine clearance. Patients developing postoperative AKI featured increased preoperative 20-HETE and 8,9-EET levels.
Conclusions:
Pharmacological interventions targeting the CYP-eicosanoid pathway could offer promising new options for AKI prevention. Individual differences in CYP-eicosanoid formation may contribute to the risk of developing AKI in clinical settings.
Keywords: Acute kidney injury, CYP-eicosanoids, Inflammation, Reoxygenation, Signalling
INTRODUCTION
Ischemia-reperfusion (I/R) is one of the major causes of acute kidney injury (AKI)1. Ischemic AKI occurs frequently after cardiovascular surgery and kidney transplantation. Beyond acute and chronic impairment of kidney function, AKI also greatly contributes to patients’ short- and long-term morbidity and mortality2–4.
Early events during the initiation phase of ischemic AKI include ATP depletion and Ca2+-overload followed by activation of phospholipases A2 (PLA2)5. PLA2 activation plays a critical role in I/R injury of the heart and brain6–8, however, its contribution to AKI initiation remains to be defined. PLA2 activation results in the generation of potentially toxic lyso-phospholipids as well as accumulation of free arachidonic acid (AA) that in turn may trigger disturbances in eicosanoid formation. We found that 20-hydroxyeicosatetraenoic acid (20-HETE), a cytochrome P450 (CYP)-dependent AA metabolite, is excessively released during ischemia and that inhibiting the formation or action of 20-HETE ameliorated I/R-induced AKI in rat9. Others confirmed a detrimental role of 20-HETE when using a 1-kidney model, but not in a bilateral ischemic model10,11.
In addition to 20-HETE, epoxyeicosatrienoic acids (EETs) are produced as a second class of CYP-eicosanoids throughout the renal vascular and tubular system12–14. Unlike 20-HETE, whose formation is catalyzed by CYP4A and CYP4F enzymes, EETs are produced by CYP2C and CYP2J isoforms15,16. EETs oppose the vasoconstrictor, pro-inflammatory and pro-apoptotic properties of 20-HETE14,17,18. EETs mediate vasodilator responses and represent the major endothelium-derived hyperpolarizing factor in renal arterioles19,20. EETs repress pro-inflammatory activation of endothelial cells by inhibiting cytokine-induced NF-κB activation and VCAM-1 expression21. EET-mediated prevention of hypoxia/reoxygenation-induced apoptosis and cell death has been demonstrated in endothelial cells and cardiomyocytes22–24. Deficiency in renal EET formation was observed in various rodent models of hypertension and target organ damage25–28. Induction or transgenic overexpression of EET-generating CYP enzymes attenuated renal injury in rat and mouse models of angiotensin II-induced hypertension29,30. Genetic ablation of the soluble epoxide hydrolase (sEH), an enzyme mediating the degradation of EETs to dihydroxyeicosatrienoic acids (DHETs), reduced renal injury in diabetic mice31. SEH-inhibitors and most recently also metabolically robust synthetic EET analogs are under development that may provide novel therapeutic options for the treatment of cardiovascular and renal disease32–36.
In the present study, we tested the hypothesis that synthetic EET analogs may protect the kidney against I/R-induced AKI. Experiments were performed in uninephrectomized Lewis rats, a model that partially mimics the situation after renal transplantation9. The synthetic EET analog was designed to share the functional features of the naturally occurring 11,12- and 14,15-EETs32.The animals were pretreated with the EET analog via renal artery infusion directly before inducing ischemia. Initiation of pro-apoptotic pathways and renal reoxygenation were analyzed in the early reperfusion phase. Effects on the subsequent development of renal inflammation and functional deterioration were determined two days after reperfusion.
Finally, the experimental results obtained in the rat model prompted us to speculate that individual differences in 20-HETE and EET formation may be linked to the risk of developing AKI in clinical settings. To start addressing this question, we took advantage of plasma samples collected during a recently concluded clinical trial37 and compared the circulating CYP-eicosanoid profiles in patients who underwent open heart surgery with and without developing postoperative AKI.
RESULTS
Ischemia-induced imbalance between renal free 20-HETE and EET levels
The formation of free CYP-eicosanoids in the kidney was analyzed in uninephrectomized rats under the same conditions as otherwise used to induce I/R injury. The left kidneys were pretreated with vehicle or the synthetic EET analog and harvested either immediately at the end of the 45 min period of warm ischemia (0 h reperfusion group) or after a subsequent 2 h reperfusion phase. The contralateral kidneys obtained during uninephrectomy before I/R served as native control. As shown in Fig. 1, the EET analog was present in the pretreated kidneys during the ischemic period in a concentration of about 1,500 ng per gram wet weight. Subsequent reperfusion resulted in a rapid washout lowering the intrarenal concentration of the EET analog to less than 5 ng/g after 2 h.
Figure 1: Determination of intrarenal EET analog levels.
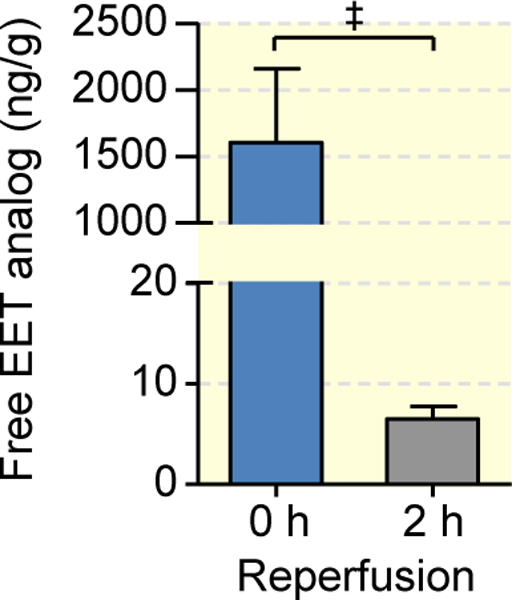
The EET analog or its vehicle was infused into the left kidney 5 min before inducing ischemia. The kidneys were harvested either immediately after 45 min of warm ischemia (0 h reperfusion) or 2 h after reperfusion. Data are given as mean ± SEM (n = 6–8 per group). Statistically significant differences were observed as indicated: ‡(p<0.001).
In the native kidneys, about 96 % of total 20-HETE, 98 % of total EETs and 99 % of total DHETs (dihydroxyeicosatrienoic acids, the products of sEH-mediated EET hydrolysis) became only detectable after alkaline hydrolysis indicating that the metabolites were predominantly esterified into membrane phospholipids under control conditions (Figs. 2).
Figure 2: Ischemia induces the release of 20-HETE, but not of EETs in the rat kidney.
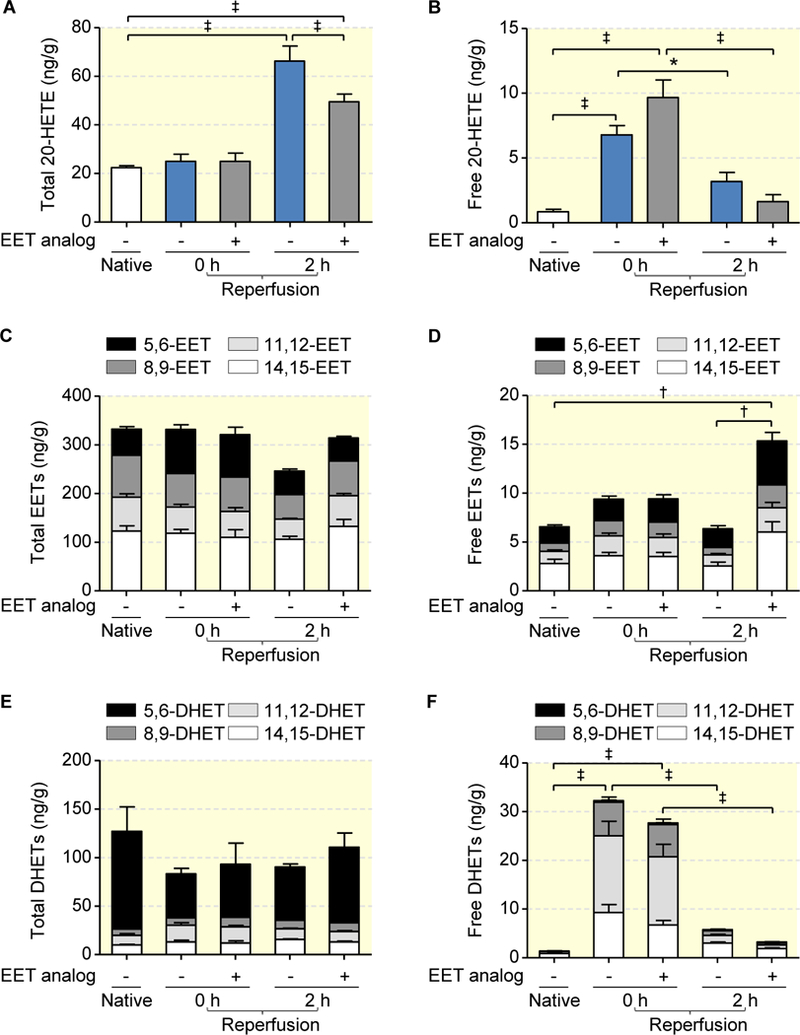
Total amounts of 20-HETE (a), EETs (c) and DHETs (e) were determined after alkaline hydrolysis of the kidney samples and represent the sum of membrane-stored (esterified into phospholipids) and free metabolites. Free metabolites (b, d and f) were extracted by treating the kidney samples with methanol without prior hydrolysis. Data are given as mean ± SEM (n = 5 per group). Statistically significant differences were observed as indicated: *(p<0.05), †(p<0.01), and ‡(p<0.001) vs. native.
Ischemia for 45 min increased free 20-HETE levels 8-fold in the vehicle group and 11-fold in the EET analog pretreated kidneys (Fig. 2b). In contrast, free renal EET levels were not significantly higher in both groups after ischemia compared with native controls (Fig. 2d). Free DHET levels increased almost 23- and 20-fold upon ischemia, but were not significantly different comparing vehicle- and EET analog-pretreated kidneys (Fig. 2f). As a net effect, the relative abundance of free 20-HETE and free EETs was shifted from about 0.1:1 in native kidneys to 0.7:1 (vehicle group) and 1:1 (EET analog pretreated kidneys) during 45 min of warm ischemia. The presence of the EET analog (compare Fig. 1) had no significant effect on the ischemia-induced changes in the endogenous CYP-eicosanoid profile compared to the vehicle control.
2h after reperfusion, the renal levels of free 20-HETE and DHETs strongly declined in both experimental groups compared to the ischemic period (Figs. 2b and 2f). The free EET-levels remained largely unchanged in the vehicle group, but were significantly increased by a factor of about 2.4-fold in the kidneys pretreated with the EET analog (Fig. 2d). Considering the total levels of renal CYP-eicosanoids, we observed reperfusion-induced significant elevations in esterified 20-HETE that were most pronounced in the vehicle group (Fig. 2a). There were no changes in the total EET and DHET levels (Figs. 2c and 2e).
EET analog alleviates I/R-induced deterioration of renal function and tubular epithelial cell death
The EET analog (for structural formula see Fig. 3a) was administered 5 minutes prior to induction of ischemia via the renal artery. This single application largely preserved renal function (Figs. 3b–3d). Animals pretreated with the EET analog featured significantly lower serum creatinine (0.61±0.06 mg/dl vs. vehicle 2.41±0.23 vs. sham 0.31±0.01 mg/dl, p<0.001; Fig. 3b) and serum urea levels (73.67±7.65 mg/dl vs. vehicle 287.80±34.81, p<0.001 vs. sham 37.20±1.02 mg/dl; Fig. 3c), and showed largely maintained creatinine clearance (1.03±0.18 ml/min vs. vehicle 0.26±0.02, p<0.01; vs. sham 1.37±0.04 ml/min; Fig. 3d) 48h after reperfusion.
Figure 3: EET analog alleviates I/R-induced acute renal failure.
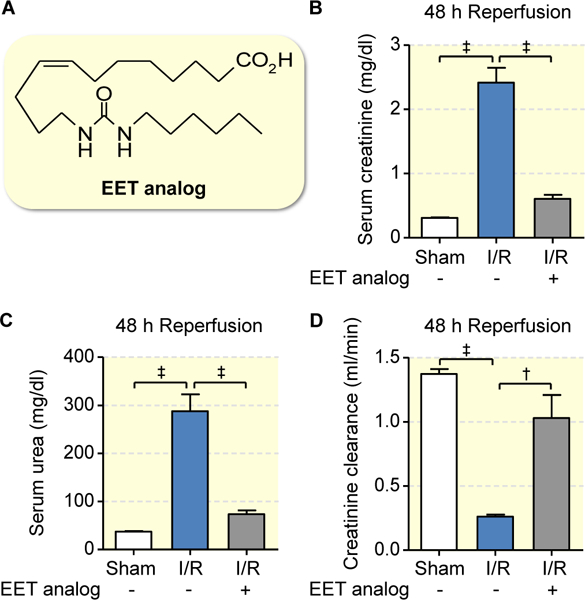
Structural formula of the EET analog (a) used in the present study. Serum creatinine (b), urea (c) and creatinine clearance (d) were determined 48 hours after reperfusion: Compared to vehicle, EET analog alleviated development of I/R induced renal function impairment. Data are given as mean ± SEM (n = 5–6 per group). Statistically significant differences were observed as indicated: †(p<0.01) and ‡(p<0.001).
Pretreatment with the EET analog also had a potent anti-apoptotic effect as shown by representative images of TUNEL staining in the outer medulla (Figs. 4a and 4b) and reflected by a significantly lower number of apoptotic cells obtained by semi-quantitative morphometric evaluation (0.26±0.02 vs. vehicle 4.10±0.24 vs. sham 0.03±0.01 % per field of view (FOV), p<0.001; Fig. 4c) 48 hours after reperfusion. Renal tubular damage was quantified using the acute tubular necrosis (ATN) score38. Severe damage occurred in the vehicle group as indicated by the presence of flattened tubular epithelium, exfoliated tubular epithelial cells, widened tubular lumina, hyaline cast formation, and necrotic tubules. These features were markedly reduced upon pretreatment with the EET analog (ATN score in the outer medulla: 0.70±0.11 vs. vehicle 2.50±0.18, p<0.001; Fig 4d). Dense infiltration with ED1-positive monocytes/macrophages was observed in the vehicle group 48 h post-ischemia indicating I/R-induced inflammation in the damaged areas of outer medulla. The EET analog significantly repressed inflammatory cell infiltration as shown by morphometric quantification (11.5±2.2 vs. vehicle 53.4±1.5 cells per FOV, p<0.001; Fig. 4e).
Figure 4: EET analog alleviates I/R-induced renal epithelial cell death and intrarenal inflammation.
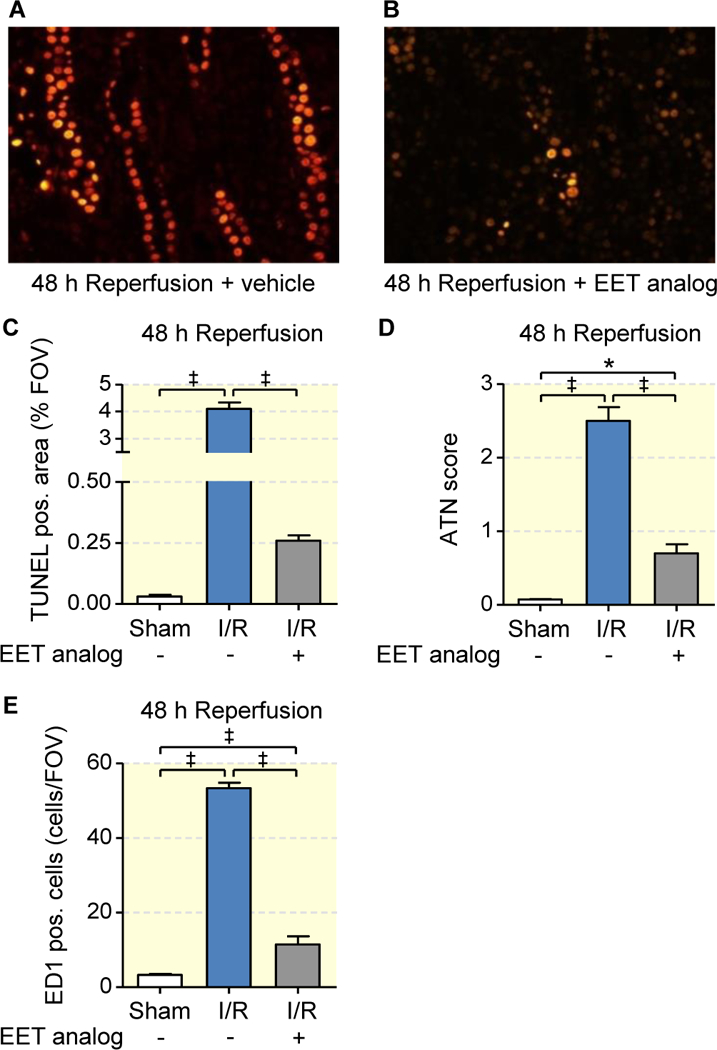
Representative images of outer medullary sections stained by TUNEL assay (a, b) with corresponding quantitative evaluation (c), and semiquantitative acute tubular necrosis score (ATN) (d) show that EET analog treatment significantly attenuates I/R induced abundant epithelial apoptosis and necrosis 48 hours after I/R as observed upon vehicle treatment. Quantification of monocyte/macrophage cell infiltration (ED1-positive cells/FOV) in the outer medulla (e) shows minimal ED1-positive cell infiltration upon EET analog treatment in comparison to vehicle at 48 hours after I/R injury. Data are given as mean ± SEM (n = 4–6 per group). Statistically significant difference were observed as indicated: *(p<0.05) and ‡(p<0.001).
EET analog improves renal reoxygenation
In an additional set of experiments, we applied a hydraulic occluder to enable remote induction of renal I/R in a 9.4 Tesla small animal MR-scanner. With this setup, we performed high spatial resolution monitoring of blood oxygenation by means of parametric magnetic resonance imaging (MRI; mapping of the relaxation time T2*) during the initial I/R phase as described previously39. In the outer medulla, T2* remained below baseline level after reperfusion during the entire observation period of 2 hours post reperfusion (Fig. 5a). In contrast, cortical T2* exceeded baseline levels in the initial phase after reperfusion (Fig. 5b). Outer medullary T2* levels were higher after pretreatment with the EET analog versus vehicle, but were not restored to pre-ischemia level. Cortical T2* levels remained significantly elevated in the EET analog group, whereas in vehicle treated animals T2* slowly returned to baseline. Color-coded maps showing T2* ratio of end-reperfusion/baseline indicate markedly improved reoxygenation 2 hours post reperfusion in renal cortex and outer medulla after treatment with the EET agonist (Fig. 5d) as compared to vehicle control (Fig. 5c).
Figure 5: EET analog improves intrarenal oxygenation measured by high temporal resolution parametric MRI during the initial I/R phase.
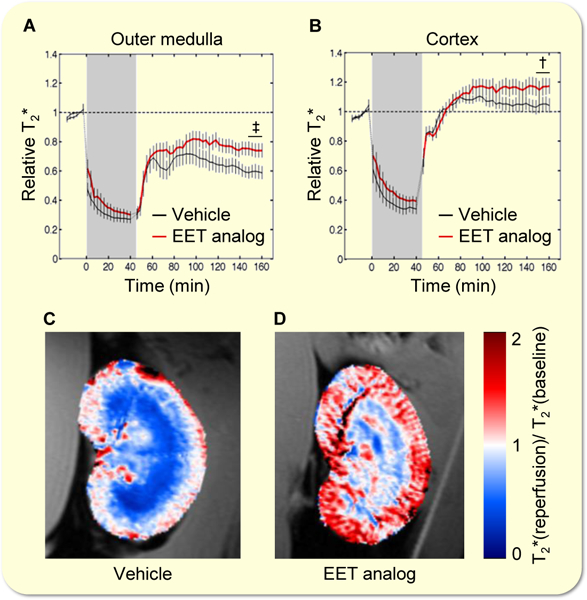
Kinetics of T2* changes (a, b) and color-coded T2* ratio maps (reperfusion/baseline) of two exemplary kidneys superimposed to the anatomical MR image (c, d) show lower extent of hypoxia and better reoxygenation after EET analog treatment. Data are given as mean ± SEM (n = 7–10 per group). Statistically significant differences (2-tailed t-test) were observed as indicated: †(p<0.01) and ‡(p<0.001) vs. vehicle.
EET analog contributes to induction of early pro-survival signaling
We next tested the hypothesis that the EET analog may initiate activation of key cell survival pathways and, thereby, might restrain pro-apoptotic programming in the early I/R phase (Figs. 6 and 7). To this end, kidneys were subjected to 45 min of ischemia and harvested immediately after ischemia (0h reperfusion) or after 2h of reperfusion. Consistent with the initiation of cell protective pathways, the rephosphorylation of Akt at both Thr-308 (Fig. 6a) and Ser-473 (Fig. 6b) was significantly increased from 0 to 2 hours of reperfusion in the EET analog treated kidneys, but not in the vehicle group. In native kidneys, GSK-3β kinase was largely inactivated by phosphorylation at Ser-9 (Fig. 7a). Dephosphorylation essential for GSK-3β activation subsequently leading to apoptosis and inflammation was induced by ischemia. The data obtained 2 h after reperfusion showed that GSK-3β dephosphorylation remained unchanged in the vehicle control, whereas the EET analog mediated strong rephosphorylation up to the level of native kidneys (Fig. 7a). The EET analog also reduced the occurrence of apoptotic tubular epithelial cells in the early reperfusion phase, as revealed by TUNEL staining of kidney sections harvested 2 h after reperfusion (Fig. 7b).
Figure 6: EET analog facilitates the restoration of Akt phosphorylation in the early reperfusion phase.
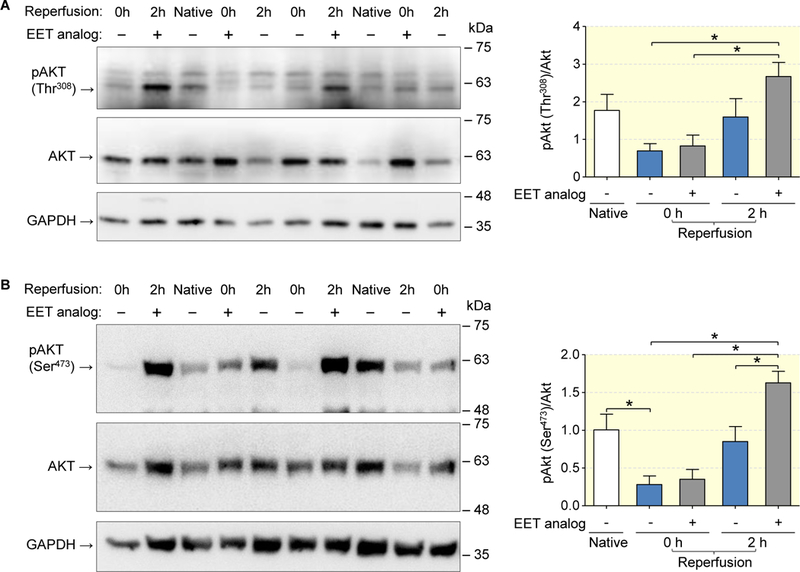
Akt phosphorylation at both Thr-308 (a) and Ser-473 (b) was increased in the EET analog compared to the vehicle group 2 hours after reperfusion. Representative Western blots are shown on the left panel. Data are given as mean ± SEM (n = 6–8 per group). Statistically significant differences were observed as indicated: *(p<0.05).
Figure 7: EET analog facilitates the restoration of GSK-3ß phosphorylation and acts anti-apoptotic.
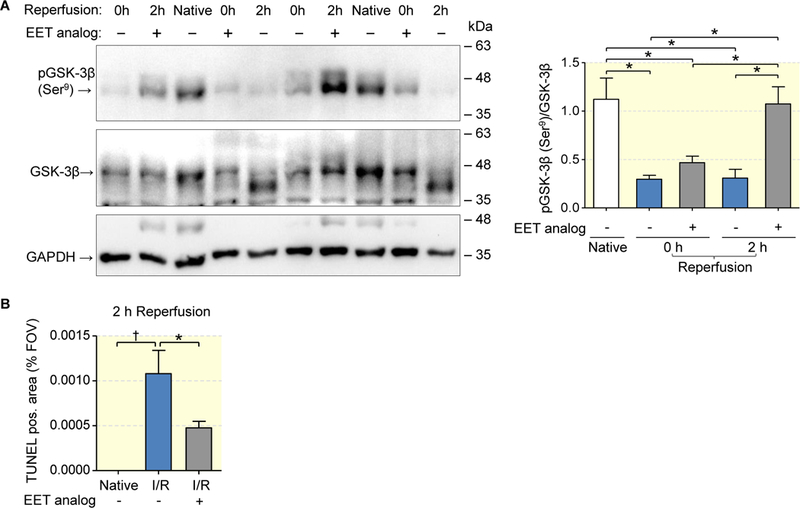
Lower magnitude of dephosphorylation after ischemia and successful rephosphorylation of GSK-3β 2 hours after reperfusion in the EET analog group compared to maintained dephosphorylation of GSK-3β indicative of pro-apoptotic kinase activity in vehicle group (a). Quantitative evaluation of outer medullary sections stained by TUNEL assay confirming lower apoptosis rate in EET analog treated group (b). Data are given as mean ± SEM (n = 6–8 per group in a; n= 4–6 in b). Statistically significant differences were observed as indicated: *(p<0.05); †(p<0.01).
Circulating CYP-eicosanoids in patients who do or do not develop AKI after cardiac surgery
We analyzed a subset of plasma samples originating from a clinical trial on the utility of sodium bicarbonate to prevent AKI in patients following open heart surgery37. This subset was derived from the non-bicarbonate treated control arm of the study at the German Heart Center (n=81) and included all available patients with postoperative AKI (AKI-group, n=21) as well as age- and gender-matched patients without postoperative AKI (control group; n=39); Table 1. The circulating CYP-eicosanoid profile was determined in plasma samples collected directly before starting the surgical intervention in order to evaluate the potential predictive value of 20-HETE and EET levels for the subsequent development of AKI. The basal 20-HETE levels were significantly higher in the AKI compared to the control group (1.14±0.13 vs. 0.82±0.08 ng/ml; p=0.028); Fig 8a. On average, patients developing postoperative AKI also showed nearly 50 % higher 8,9-EET levels than controls (4.60±0.45 vs. 3.19 ng/ml; p=0.015); Fig. 8b. No significant differences were observed comparing the levels of other regioisomeric EETs in AKI and control patients (Fig. 8c). Moreover, we did not detect significantly increased DHET/EET- or DiHOME/EpoME-ratios in the AKI compared to the control group indicating that differences in sEH activities were not involved in predisposing to AKI (Figs. 8d and 8e).
Table 1:
Characteristics of patients who do or do not develop AKI after cardiac surgery
| AcuteKidneyInjury | Control | p-value | |
|---|---|---|---|
| n (total) | 21 | 39 | |
| Gender (f=female, m=male) | f = 4 (19%), m = 17 (81%) | f = 7 (18%), m = 32 (82%) | n.s. (0.931) |
| Age (yr) | 70 (52–90) | 69 (50–84) | n.s. (0.683) |
| Body weight (kg) | 83 ± 14 | 82 ± 19 | n.s. (0.918) |
| BMI | 27 ± 4 | 27 ± 5 | n.s. (0.642) |
| eGFR (pre-OP) (ml/min per 1.73m2) | 63 ± 20 | 68 ± 16 | n.s. (0.355) |
| PreexistingComorbidities | |||
| NYHA Class III-IV or EF<35% | Yes = 4 (19%), No = 17 (81%) | Yes = 10 (26%), No = 29 (74%) | n.s. (0.607) |
| Peripheral Vascular Disease | Yes = 4 (19%), No = 17 (81%) | Yes = 8 (21%), No = 31 (79%) | n.s. (0.931) |
| AtrialFibrillation | Yes = 7 (33%), No = 14 (67%) | Yes = 14 (36%), No = 25 (64%) | n.s. (0.897) |
| COPD | Yes = 4 (19%), No = 17 (81%) | Yes = 4 (10%), No = 35 (90%) | n.s. (0.328) |
| Smoker | Yes = 3 (14%), No = 18 (86%) | Yes = 4 (10%), No = 35 (90%) | n.s. (0.625) |
| Diabetes mell. on med. | Yes = 11 (52%, No = 10 (48%) | Yes = 28 (72%), No = 11 (28%) | n.s. (0.179) |
| Insulin-dep. Diabetes mell. | Yes = 0 (0%), No = 21 (100%) | Yes = 2 (5%), No = 37 (95%) | n.s. |
| Art. Hypertension | Yes = 15 (71%), No = 6 (29%) | Yes = 32 (82%), No = 7 (18%) | n.s. (0.458) |
| Pulmonary Hypertension | Yes = 5 (24%), No = 16 (76%) | Yes = 7 (18%), No = 32 (82%) | n.s. (0.564) |
| Hypercholesterinemia | Yes = 15 (71%),No = 6 (29%) | Yes = 25 (64%),No = 14 (36%) | n.s. (0.494) |
| Medication on admission | |||
| ACE-Inhibitors/ ARB | Yes = 13 (62%), No = 8 (38%) | Yes = 20 (51%), No = 19 (49%) | n.s. (0.384) |
| Ca-Channel-blocker | Yes = 8 (38%), No = 13 (62%) | Yes = 13 (33%), No = 26 (67%) | n.s. (0.668) |
| Betablocker | Yes = 19 (91%), No = 2 (9%) | Yes = 29 (74%), No = 10 (26%) | n.s. (0.107) |
| Diuretics (pre-OP) | Yes = 14 (67%), No = 7 (33%) | Yes = 27 (69%), No = 12 (31%) | n.s. (0.949) |
| Statins | Yes = 12 (57%), No = 9 (43%) | Yes = 23 (59%), No = 16 (41%) | n.s. (0.979) |
| Type of Surgery /duration | |||
| Duration of Surgery | 132 ± 55 | 122 ± 33 | n.s. (0.358) |
| CABG | 5 | 11 | n.s. (0.760) |
| Valveimplantation | 10 | 17 | n.s. (0.708) |
| Combined (CABG + Valve) | 6 | 8 | n.s. (0.458) |
| RedoSurgery | 5 | 10 | n.s. (0.920) |
| Medicationduringsurgery | |||
| Red Blood Cell Transfusion Mean tranfsuion Volume (ml) |
no 6, yes 15 1008 (250–5920) |
no 18, yes 29 452 (0–3830) |
p = 0.042 |
| Cristalloids (ml/0–24h) | 6077 ± 2067 | 8427 ± 1972 | p = 0.001 |
| Plasmaexpander (ml/0–24h) | 286 ± 435 | 325 ± 446 | n.s. (0.743) |
| Furosemide (mg/0–24h) | 82 ± 106 | 22 ± 23 | n.s. (0.920) |
| Drains (ml/0–24h) | 1006 ± 1126 | 609 ± 430 | p = 0.052 |
| Anyinotropic (surgeryday) | Yes = 14 (67%), No = 7 (33%) | Yes = 25 (64%),No = 14 (36%) | n.s. (0.752) |
| Any inotropic (day 1 post OP) | Yes = 12 (57%), No = 9 (43%) | Yes = 18 (46%), No = 21 (54%) | n.s. (0.376) |
| Hemoglobin (g/dl) | |||
| Presurgery | 12 ± 2 | 13 ± 1 | n.s. (0.274) |
| 0h Post surgery | 11 ± 2 | 11 ± 2 | n.s. (0.605) |
| 24h Post surgery | 10 ± 1 | 11 ± 1 | p = 0.006 |
| 48h Post surgery | 11 ± 2 | 10 ± 3 | n.s. (0.556) |
Figure 8: Circulating CYP-eicosanoids in patients who do or do not develop AKI after cardiac surgery.
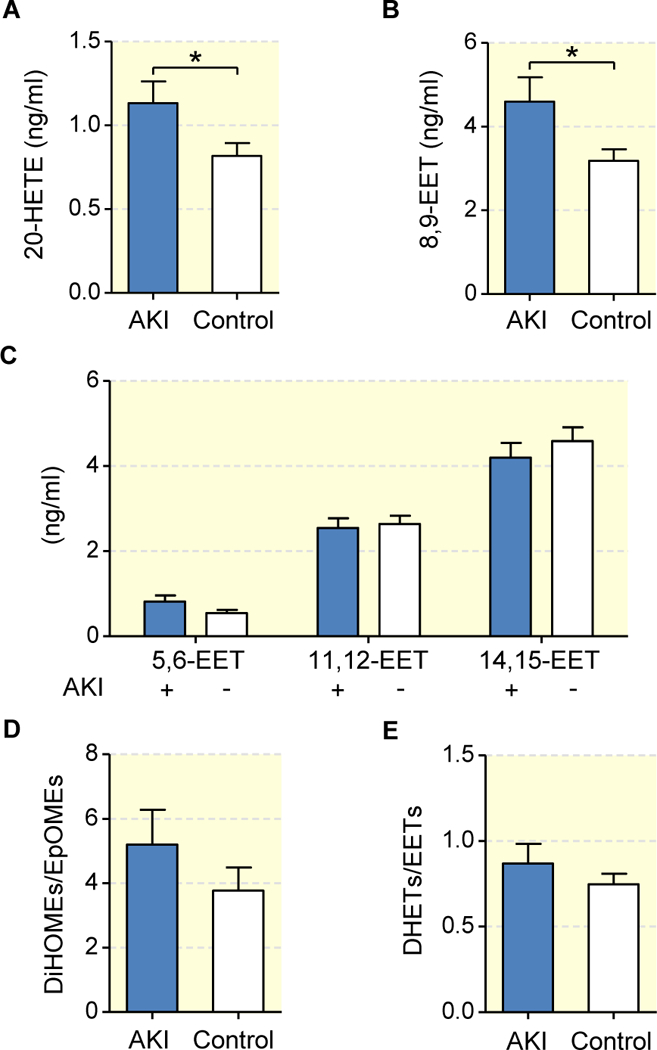
Shown are the pre-operational total plasma levels of 20-HETE (a), 8,9-EET (b) and other regioisomeric EETs (c) as well as the total DiHOME/ EpOME-(d) and DHET/EET-ratios (e) in patients who underwent open heart surgery with (n=21) and without (n=39) developing postoperative AKI. Data are given as mean ± SEM. Statistically significant differences (t-test) were observed as indicated: *(p<0.05).
DISCUSSION
Our experimental study shows that pretreating the kidney with a synthetic analog of the endogenous CYP-eicosanoids 11,12- and 14,15-EET significantly alleviates I/R-induced renal injury in a rat model of AKI. Our translational pilot study indicates that individual differences in CYP-eicosanoid formation may contribute to the risk of developing AKI in patients undergoing open cardiac surgery.
Measuring the levels of free and esterified CYP-eicosanoids in the rat kidney, we found that EETs, unlike 20-HETE, did not accumulate during ischemia. Accordingly, ischemia induced an imbalance of these metabolites compared with physiological conditions. EETs and 20-HETE play opposing roles in the regulation of vascular tone, inflammation and apoptosis14. Therefore, we propose that this imbalance may contribute to the initiation of AKI. Using the same model (uninephrectomized Lewis rats), we showed previously that renal I/R-injury can be significantly ameliorated by inhibiting the generation and action of 20-HETE9. In comparison, our present data show that an even higher extent of protection can be achieved using a synthetic EET analog with the aim of compensating for the lack of ischemia-induced endogenous EET accumulation.
EETs and 20-HETE are synthesized from free AA by CYP epoxygenases and hydroxylases, respectively13,15,40. Once produced, 20-HETE and EETs are partially re-esterified into phospholipids13,15,41. This membrane pool is accessible to phospholipases activated during ischemia and, thus, provides a potential source of free 20-HETE and EETs even when de novo synthesis is limited due to hypoxia. In the kidney, ischemia induces membrane translocation and activation of Ca2+-dependent phospholipase A2 (PLA2) activities5. Studies in mice deficient in cPLA2α revealed that this enzyme significantly contributed to I/R-injury of the heart and brain6–8. Recently, the PLA2 inhibitor quinacrine was shown to protect against glycerol-induced AKI in rats42. Our data demonstrate that ischemia preferentially increases the renal levels of free 20-HETE and DHETs, but not EETs. A similar imbalance presumably also occurs in the ischemic heart as indicated by the different levels of these metabolites in the coronary venous plasma before and after coronary artery occlusion43. The differential accumulation of free EETs and 20-HETE may be primarily due to differences in their further metabolic fate. Free EETs are rapidly metabolized to DHETs by the sEH, an enzyme that does not require molecular oxygen and may be thus active also in the ischemic kidney44. In contrast, major routes of 20-HETE metabolism are oxygen-dependent because they are initiated by cyclooxygenases12,45 and CYP enzymes29. Thus, hypoxia may limit the further metabolism of 20-HETE, but not of EETs. In the EET analog used in this study, the labile epoxide typical of natural EETs is replaced by a metabolically and chemically more stable urea bioisostere. Furthermore, this EET analog may have dual activity, i.e., it does not only mimic the biological activities of the naturally occurring isoform (14,15-EET) but can also act as a weak sEH inhibitor as shown in vitro for the hydrolysis of an artificial substrate by the human recombinant sEH enzyme32. However, we found that the EET analog neither affected the ischemia-induced accumulation of free DHETs and 20-HETE nor increased the levels of free EETs. Thus, it is highly unlikely that the EET analog exerted its beneficial effects in our rat model of AKI by inhibiting the sEH.
Sustained medullary vasoconstriction that may, at least in part, be associated with endothelial dysfunction occurs in the reperfusion phase. It delays the recovery of medullary perfusion and oxygenation and, thus, contributes to I/R-induced renal injury9,46–48. Using MRI for monitoring the time course of renal blood oxygenation, we found that the EET analog improved cortical and outer medullary reoxygenation during the early reperfusion phase. EETs mediate vasodilation of renal arterioles by stimulating calcium-activated potassium (BK) channels in vascular smooth muscle cells19,20. Moreover, EETs inhibit salt reabsorption in proximal and distal tubules by inhibiting the Na+/H+-exchanger and the epithelial Na+-channel respectively12,13,49. Thus, the EET analog might have improved renal reoxygenation by both increasing microvascular oxygen supply and reducing tubular oxygen consumption. As changes in T2* are related to the local concentration of deoxyhemoglobin, they largely reflect variations in blood oxygenation, but may also be influenced by vasomotion and hematocrit50. EET-mediated vasodilation increases the blood volume fraction and, hence, increases renal deoxyhemoglobin concentration (i.e. decreases T2*) even at unchanged blood oxygenation. The observed EET-induced increase in T2* might therefore underestimate the actual improvement of blood reoxygenation.
The EET analog efficiently stimulated Akt phosphorylation at Thr-308 as well as Ser-473 indicating activation of both the PI3K and mTORC2 pathways. Activation of the PI3K/Akt survival pathway is an essential component of EET-mediated protection against apoptosis in endothelial cells and cardiomyocytes, and also plays a central role in EET-mediated protection of the heart against I/R-injury23,24,51,52. Phosphorylation of Akt at Thr-308 is required to mediate phosphorylation and inactivation of GSK-3β and other pro-apoptotic mediators53. In line with this general mechanism, we found that the EET analog markedly stimulated GSK-3β inactivation and reduced tubular epithelial cell apoptosis. Recently, increased GSK-3β phosphorylation was also shown in a mouse model of ischemic AKI after pretreating the animals i.p. with the natural metabolite 14,15-EET54. The key role of GSK-3β in promoting apoptosis after renal ischemia has been identified recently and substantiated by showing that direct pharmacologic inhibition of GSK-3β ameliorated I/R-induced tubular damage and decline of renal function55. The direct involvement of mTORC2-mediated phosphorylation of Akt at Ser-473 in renal I/R-injury has not been studied yet. Phosphorylation of Akt at Ser-473 is essential for stabilization of actin cytoskeleton56 and endothelial viability in response to hypoxic stress57. Moreover, the mTORC2/Akt pathway might be related to induction of autophagy56, a mechanism increasingly recognized to protect tubular cells from degeneration and acute ischemic injury58–60.
Our data also show that the EET analog significantly reduced I/R-induced inflammatory cell infiltration. This finding is in line with the capacity of EETs to inhibit pro-inflammatory activation of endothelial cells by repressing NF-κB activation21. EET mediated anti-inflammatory effects were also shown in chronic models such as streptozotocin-induced diabetic mice31 or DOCA-salt hypertensive mice61. In these studies, renal protection was achieved by pharmacological inhibition or genetic deletion of the sEH enzyme. These measures elevate endogenous EET levels through decreasing EET degradation. Compared to the extensive studies on designing and using sEH inhibitors35,62, the development of metabolically robust EET analogs feasible for in vivo application is only at the beginning32–34,63. A potential advantage of EET analogs is that they directly compensate for an EET deficiency36. In contrast, the beneficial effects of sEH inhibitors largely depend on endogenous EET production and, thus, on the expression and activity of CYP epoxygenases under the given pathological conditions. Providing first evidence for their therapeutic potential, synthetic EET analogs rescued the metabolic syndrome phenotype of HO-2-null mice64, prevented adiposity and vascular dysfunction in rats fed a high fat diet65, attenuated cisplatin nephrotoxicity66 in rats, and decreased renal inflammation and injury in ANG II hypertension67. To our knowledge, the present study is the first demonstrating that EET analogs efficiently protect against renal I/R-injury.
Our study has several limitations related to the fact that we used a uninephrectomized model and administered the EET analog before inducing ischemia. Unilateral nephrectomy was shown to diminish ischemic AKI through enhanced perfusion and reduced pro-inflammatory and pro-fibrotic responses.68 The preventive setting revealed a crucial role of CYP-eicosanoids in AKI initiation; however, further studies are needed to prove the therapeutic potential of EET analogs in decreasing renal injury after the ischemic event had occurred.
We believe that these findings on the role of EETs and 20-HETE in experimental renal I/R-injury might have a clinical perspective considering that AKI is a frequent complication of cardiovascular surgery and renal transplantation, i.e. in clinical settings, where preventive treatment appears possible and highly desirable. Noteworthy, a renal transplantation study showed that the extent of 20-HETE release immediately following allograft reperfusion is negatively associated with post-transplant allograft function69. Furthermore, a recent candidate gene approach revealed that the risk of developing AKI following cardiac surgery is increased in patients who carry the 55Arg allele of the sEH gene that confers increased EET hydrolase activities70. Our pilot translational study suggests that increased 20-HETE and 8,9-EET levels might predispose to developing AKI following open cardiac surgery; however, proving the actual utility of these CYP eicosanoids as predictive biomarkers for AKI will now require appropriately designed prospective studies. Whereas the increased 20-HETE levels are in line with the detrimental role of 20-HETE in our unilateral model of experimental renal I/R injury9,10, the finding of increased 8,9-EET levels was rather unexpected. Among the various human CYP epoxygenases, CYP2C9 has the highest catalytic capacity of producing 8,9-EET71. Increased vascular expression of CYP2C9 has been suggested to occur in patients with coronary artery disease72. Providing a possible link between increased 8,9-EET levels and AKI, 8,9-EET can be further metabolized by COX enzymes to yield metabolites that reduce the glomerular filtration rate and may contribute to inflammatory glomerular diseases73,74.
Taken together, our results show that ischemia induces an imbalance between 20-HETE and EETs in the kidney and suggest that this imbalance plays a pivotal role in setting the stage for the cascade of events leading to renal I/R-injury. EET analogs compensating for endogenous EET deficiency in the initiation phase of renal I/R-injury may provide novel therapeutic options for the prevention of ischemic AKI.
MATERIALS AND METHODS
Animals
Inbred male Lewis rats were purchased from Harlan-Winkelmann (Borchen, Germany) and housed under standard conditions. All experimental procedures were approved by the local authority (Landesamt für Gesundheit und Soziales Berlin) and were consistent with the guidelines of the Charité-Universitätsmedizin Berlin for the use of laboratory animals.
Surgical procedure
Rats were 7–9 weeks of age and weighed 210–260 g. All rats (n = 5–8 per group) underwent midline laparotomy and right nephrectomy under isoflurane-induced anesthesia. Right kidneys served as control (native group) for determination of CYP-eicosanoids and for the analysis of pro-survival signaling. EET analog (60 μg) or its vehicle (1% DMSO in saline) was infused into the remaining left kidney by single intraaortic injection (100 μl) between two short-time aortic clamps placed above and below the level of the left renal artery. The synthetic EET analog was synthesized as described previously32. The left kidney was exposed to 45 min of warm ischemia by clamping the renal pedicle 5 min after drug administration. During ischemia, animals were kept at a constant core body temperature (37°C ± 0.5°C). Kidneys were harvested immediately after ischemia (0 h reperfusion group) or 2 hours post-reperfusion (2 h reperfusion group). Additional rats were allowed to recover (I/R + 48 hours reperfusion group) and animals with uninephrectomy, but without I/R served as control (sham group). These rats received a single injection of buprenorphine followed by tramadol via tap water for analgesia. Rats were housed in metabolic cages for urine collection over a period of 24 h beginning one day after surgery. Rats were anesthetized with isoflurane (2%) for final tissue sampling and termination. Blood samples were collected by puncture of the V. cava and the remaining left kidneys were harvested48 hours post-reperfusion. The kidneys were separated, frozen in liquid nitrogen or formalin-fixed.
Renal function and histology
Creatinine in serum and urine, and serum urea were measured with an automated system. Formalin-fixed and paraffin-embedded renal sections were stained with hematoxylin and eosin to assess the severity of necrotic damage. Acute tubular necrosis (ATN) score was graded from 0 to 3 corresponding to none, mild, moderate, or severe necrosis by two investigators (UH and GB) in a blinded fashion, as described previously38. Cell swelling, vacuolization, nuclear pyknosis, necrosis and cellular infiltrate were evaluated. Renal apoptosis was examined using a commercially available in situ cell death detection kit, TMR red (Roche, Grenzach-Wyhlen, Germany) following manufacturer’s instructions. A minimum of 10 randomly chosen image fields of cortex and outer medulla at 400x magnification were evaluated using AxioVision digital microscopy system (Zeiss, Jena, Germany). Positive cell staining was expressed as percentage of TUNEL-positive area per field of view (FOV). For assessment of monocyte/macrophage infiltration, tissue sections were stained with anti-ED1 antibody (Serotec, Oxford, UK) using APAAP method (Dako, Glostrup, Denmark). The number of ED1-positive cells was scored in 10 randomly chosen FOVs in cortex and outer medulla. Results were expressed as mean ± SEM of cells per FOV at 400x magnification. Quantitative morphometric analysis was conducted by two investigators (UH and GB) in a blinded fashion.
Analysis of CYP-eicosanoids in kidney samples
Rats were prepared as described above. The left kidneys were subjected to 45 min of warm ischemia and removed either immediately (0 h reperfusion) or after a subsequent 2 h reperfusion phase. The harvested kidneys were immediately snap-frozen in liquid nitrogen. One quarter of the kidney was pulverized in liquid nitrogen (Biopulverizer, BiospecProducts,Bartlesville, OK, USA) and 30 mg aliquots were used for subsequent analysis of CYP-eicosanoids. The pulverized samples were thawed after adding 1 ml of methanol/water (1:1) containing 2 mM ZnSO4 and the mix of deuterated internal standards. ZnSO4 was used to block sEH activity in the renal samples75 and, thus, to protect free EETs and the corresponding internal standard from hydrolysis during the extraction procedure. Solid phase extraction was performed using Varian Bond Elut Certify II columns, either directly after adding 2 ml of 1 M sodium acetate buffer pH 6.0 to determine free metabolite levels, or after alkaline hydrolysis to determine total (free + esterified) metabolite levels as described previously71. Quantification of 20-HETE, EETs and DHETs by liquid chromatography tandem mass spectrometry (LC-MS/MS; Lipidomix GmbH, Berlin, Germany) was performed as described previously71. The EET analog was quantified simultaneously extending the multiple reaction monitoring protocol to include the transition of its parent ion to the diagnostic fragment ion (339.3→238) at a collision energy of 8 V. 10 ng of each 20-HETE-d6,14,15-EET-d8, and 14,15-DHET-d11 (Cayman Chemicals, Ann Arbor, MI, USA) served as internal standards and the amounts of 20-HETE, 5,6-, 8,9-, 11,12- and 14–15-EET and of the corresponding regioisomeric DHETs present in the renal samples were calculated from respective calibration curves. Results are shown as ng 20-HETE, EETs (sum of the four regioisomeric EETs) or DHETs (sum of the four regioisomeric DHETs) per g kidney weight.
Western blot analysis
The following primary antibodies were used for Western blot analyses: anti-phospho-Akt Ser473 (#4060), anti-phospho -Akt Thr308 (#2965), anti-phospho-GSK-3β Ser9 (#5558) and anti-total-AKT (#9272), anti-total-GSK-3β (#9832) from Cell Signaling Technology; GAPDH (#5G4) from HyTest (Finland). Corresponding secondary antibodies included donkey anti-rabbit HRP-IgG (#711–035-152) and donkey anti-mouse HRP-IgG (#711–035-151) from Jackson ImmunoResearch. Kidney samples (one eighth of a kidney) were homogenized in RIPA buffer supplemented with a protease inhibitor cocktail (Roche Applied Science) and phosphatase inhibitors (10 mM β-Glycerophosphate, 5mM NaF, 1mM Na3VO4, 1 mM Na-pyrophosphate). Protein samples (50 μg of total protein) were separated by 10% SDS-PAGE and transferred onto PVDF membranes (GE Healthcare, Amersham, UK). The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-Tween) and incubated first with the primary antibodies against the phosphorylated form to be detected (1:1000 dilutions) as well as against GAPDH (1:20000) overnight at 4°C. After washing with TBS-Tween, the membranes were incubated with the appropriate secondary antibody - donkey anti-rabbit (1:15000) or donkey anti-mouse (1:25000) - for 1h at RT. Chemiluminescent detection and image analysis were performed with ECL substrate (SuperSignal West Pico/Dura, Thermo Scientific, Rockford, IL, USA) and the G:BOX Chemi XL 1.4 imaging system (Syngene, Cambridge, UK). After visualization, the membranes were stripped using glycine-based Stripping buffer for 30 min at 52°C, and re-probed with antibodies (1:1000 dilutions) against total Akt and total GSK-3ß, respectively.
MRI monitoring of renal oxygenation
Male Lewis rats (n = 7–10 per group), aged 2–3 months, weighing 250–300 g underwent experimental I/R injury inside a 9.4 Tesla small animal MR system (Biospec 94/20, Bruker Biospin, Ettlingen, Germany) using our previously described protocol.39 Briefly, the rats were anesthetized using urethane (20% in saline, 6 ml/kg body mass) and kept at a constant core body temperature of 37°C during surgery and MRI monitoring. Animals underwent midline laparotomy and right nephrectomy. For intrarenal administration of the EET analog or vehicle, an aortic catheter was placed with its tip directly at the left renal branch. The rats were transferred into the MR scanner and renal blood oxygenation was continuously monitored by T2* mapping with a temporal resolution of ~ 3 min. Ischemia was induced by closing a remotely controlled hydraulic occluder around the renal artery and vein for 45 minutes.39 Interruption of renal blood flow was confirmed by time-of-flight (TOF) MR angiography of the kidney. Ischemia was followed by a reperfusion phase of ~100 minutes.
Circulating CYP-eicosanoid levels in patients with and without development of AKI after cardiac surgery
A subset of preoperative plasma samples collected from patients of the “Sodium Bicarbonate in Cardiac Surgery Study (Bic-MC)” (Trial registration: ClinicalTrials.gov NCT00672334) were analyzed for circulating CYP-eicosanoid levels37. The study was designed as a multicenter, double-blinded (patients, clinical and research personnel), randomized controlled trial and enrolled 350 adult patients undergoing open heart surgery with the use of cardiopulmonary bypass. At induction of anesthesia, patients received either 24 hours of intravenous infusion of sodium bicarbonate (5.1 mmol/kg) or sodium chloride (5.1 mmol/kg). The primary endpoint was the proportion of patients developing acute kidney injury which was defined a priori as an increase in serum creatinine concentration greater than 25% or 0.5 mg/dl (44 mmol/l) from baseline to peak value at any time within the first 5 d after cardiopulmonary bypass. Included were cardiac surgical patients in whom the use of cardiopulmonary bypass was planned and having one or more of the following risk factors for postoperative acute kidney injury: age above 70 years, pre-existing renal impairment (preoperative plasma creatinine concentration 120 μmol/l), New York Heart Association class III/IV or impaired left ventricular function (left ventricular ejection fraction < 35%), valvular surgery or concomitant valvular and coronary artery bypass graft surgery, redo cardiac surgery, insulin-dependent Type 2 diabetes mellitus. The analyzed plasma sample subset was derived from the non-bicarbonate treated control arm of the study collected at the German Heart Center (Berlin, Germany) (n=81) and included all available patients developing postoperative AKI,defined according to AKI Network criteria76(AKI-group, n=21) as well as age- and gender-matched patients without postoperative AKI (control group; n=39).The plasma levels of CYP-eicosanoids were determined after alkaline hydrolysis and solid phase extraction by LC-MS/MS as described previously71.
Statistical Analysis
The results were expressed as mean ± standard error of mean (SEM). With the exception of the MRI data, statistical analysis was performed by One-way-ANOVA (with Bonferroni-post-hoc-test) or t-test (if two groups were compared only) using GraphPad Prism 5 software (GraphPad Inc., La Jolla, USA). P<0.05 was considered significant. For MRI data, T2* derived from nine regions-of-interest 39 per kidney were statistically analyzed using SPSS (version 20, IBM Deutschland GmbH, Germany). Following one-sample Kolmogorov-Smirnov test to confirm normal distribution and Levene’s test to confirm equality of variances two-tailed t-tests were performed for differences between the EET analog group and the vehicle group, comparing the average over the last 5 time points during reperfusion. A probability value of ≤0.05 was considered significant.
ACKNOWLEDGEMENTS
We thank Christel Andrée and Ramona Zummach for excellent technical assistance. This study was supported in part by grants of the DFG-German Research Foundation (FOR 1368), the MDC Clinical Research Cooperation Program, and by USPHS NIH (GM31278) and the Robert A. Welch Foundation to JRF. Ye Zhu was supported by NSFC (NO.81600529) and (17ykpy64).
Footnotes
DISCLOSURES
The authors are not aware of conflicts of interest and have nothing to disclose.
REFERENCES
- 1.Lameire N, Van Biesen W, Vanholder R. Acute kidney injury. Lancet. 2008;372(9653):1863–1865. [DOI] [PubMed] [Google Scholar]
- 2.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012;82(5):516–524. [DOI] [PubMed] [Google Scholar]
- 3.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY. Temporal changes in incidence of dialysis-requiring AKI. Journal of the American Society of Nephrology : JASN. 2013;24(1):37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nature reviews Nephrology. 2014;10(4):193–207. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura H, Nemenoff RA, Gronich JH, Bonventre JV. Subcellular characteristics of phospholipase A2 activity in the rat kidney. Enhanced cytosolic, mitochondrial, and microsomal phospholipase A2 enzymatic activity after renal ischemia and reperfusion. J Clin Invest. 1991;87(5):1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonventre JV, Huang Z, Taheri MR, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390(6660):622–625. [DOI] [PubMed] [Google Scholar]
- 7.Tabuchi S, Uozumi N, Ishii S, Shimizu Y, Watanabe T, Shimizu T. Mice deficient in cytosolic phospholipase A2 are less susceptible to cerebral ischemia/reperfusion injury. Acta neurochirurgica Supplement. 2003;86:169–172. [DOI] [PubMed] [Google Scholar]
- 8.Saito Y, Watanabe K, Fujioka D, et al. Disruption of group IVA cytosolic phospholipase A(2) attenuates myocardial ischemia-reperfusion injury partly through inhibition of TNF-alpha-mediated pathway. Am J Physiol Heart Circ Physiol. 2012;302(10):H2018–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoff U, Lukitsch I, Chaykovska L, et al. Inhibition of 20-HETE synthesis and action protects the kidney from ischemia/reperfusion injury. Kidney Int. 2011;79(1):57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roman RJ, Akbulut T, Park F, Regner KR. 20-HETE in acute kidney injury. Kidney Int. 2011;79(1):10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regner KR, Zuk A, Van Why SK, et al. Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int. 2009;75(5):511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGiff JC, Quilley J. 20-HETE and the kidney: resolution of old problems and new beginnings. Am J Physiol. 1999;277(3 Pt 2):R607–623. [DOI] [PubMed] [Google Scholar]
- 13.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82(1):131–185. [DOI] [PubMed] [Google Scholar]
- 14.Imig JD. Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function. Prostaglandins Other Lipid Mediat. 2013;104–105:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41(2):163–181. [PubMed] [Google Scholar]
- 16.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276(39):36059–36062. [DOI] [PubMed] [Google Scholar]
- 17.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292(3):C996–1012. [DOI] [PubMed] [Google Scholar]
- 18.Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and Blood Pressure Regulation: Clinical Implications. Cardiol Rev. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. Journal of the American Society of Nephrology : JASN. 1996;7(11):2364–2370. [DOI] [PubMed] [Google Scholar]
- 20.Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007;49(3):590–596. [DOI] [PubMed] [Google Scholar]
- 21.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang B, Graham L, Dikalov S, et al. Overexpression of cytochrome P450 CYP2J2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol Pharmacol. 2001;60(2):310–320. [DOI] [PubMed] [Google Scholar]
- 23.Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, et al. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am J Physiol Heart Circ Physiol. 2008;294(2):H724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jamieson KL, Endo T, Darwesh AM, Samokhvalov V, Seubert JM. Cytochrome P450-derived eicosanoids and heart function. Pharmacol Ther. 2017;179:47–83. [DOI] [PubMed] [Google Scholar]
- 25.Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH. Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J Clin Invest. 1994;94(6):2414–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaergel E, Muller DN, Honeck H, et al. P450-dependent arachidonic acid metabolism and angiotensin II-induced renal damage. Hypertension. 2002;40(3):273–279. [DOI] [PubMed] [Google Scholar]
- 27.Zhao X, Pollock DM, Zeldin DC, Imig JD. Salt-sensitive hypertension after exposure to angiotensin is associated with inability to upregulate renal epoxygenases. Hypertension. 2003;42(4):775–780. [DOI] [PubMed] [Google Scholar]
- 28.Carroll MA. Role of the adenosine(2A) receptor-epoxyeicosatrienoic acid pathway in the development of salt-sensitive hypertension. Prostaglandins Other Lipid Mediat. 2012;98(3–4):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller DN, Theuer J, Shagdarsuren E, et al. A peroxisome proliferator-activated receptor-alpha activator induces renal CYP2C23 activity and protects from angiotensin II-induced renal injury. Am J Pathol. 2004;164(2):521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CR, Imig JD, Edin ML, et al. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24(10):3770–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol. 2011;301(5):R1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falck JR, Kodela R, Manne R, et al. 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J Med Chem. 2009;52(16):5069–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imig JD, Elmarakby A, Nithipatikom K, et al. Development of epoxyeicosatrienoic acid analogs with in vivo anti-hypertensive actions. Frontiers in physiology. 2010;1:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell WB, Imig JD, Schmitz JM, Falck JR. Orally Active Epoxyeicosatrienoic Acid Analogs. J Cardiovasc Pharmacol. 2017;70(4):211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imig JD. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol Ther. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schunck WH, Konkel A, Fischer R, Weylandt KH. Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharmacol Ther. 2018;183:177–204. [DOI] [PubMed] [Google Scholar]
- 37.Haase M, Haase-Fielitz A, Plass M, et al. Prophylactic perioperative sodium bicarbonate to prevent acute kidney injury following open heart surgery: a multicenter double-blinded randomized controlled trial. PLoS medicine. 2013;10(4):e1001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dragun D, Hoff U, Park JK, et al. Prolonged cold preservation augments vascular injury independent of renal transplant immunogenicity and function. Kidney Int. 2001;60(3):1173–1181. [DOI] [PubMed] [Google Scholar]
- 39.Pohlmann A, Hentschel J, Fechner M, et al. High temporal resolution parametric MRI monitoring of the initial ischemia/reperfusion phase in experimental acute kidney injury. PLoS One. 2013;8(2):e57411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta. 2011;1814(1):210–222. [DOI] [PubMed] [Google Scholar]
- 41.Carroll MA, Balazy M, Huang DD, Rybalova S, Falck JR, McGiff JC. Cytochrome P450-derived renal HETEs: storage and release. Kidney Int. 1997;51(6):1696–1702. [DOI] [PubMed] [Google Scholar]
- 42.Al Asmari AK, Al Sadoon KT, Obaid AA, Yesunayagam D, Tariq M. Protective effect of quinacrine against glycerol-induced acute kidney injury in rats. BMC nephrology. 2017;18(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nithipatikom K, DiCamelli RF, Kohler S, et al. Determination of cytochrome P450 metabolites of arachidonic acid in coronary venous plasma during ischemia and reperfusion in dogs. Anal Biochem. 2001;292(1):115–124. [DOI] [PubMed] [Google Scholar]
- 44.Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene. 2013;526(2):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng MK, McGiff JC, Carroll MA. Renal arterial 20-hydroxyeicosatetraenoic acid levels: regulation by cyclooxygenase. Am J Physiol Renal Physiol. 2003;284(3):F474–479. [DOI] [PubMed] [Google Scholar]
- 46.Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int. 2004;66(2):496–499. [DOI] [PubMed] [Google Scholar]
- 47.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72(2):151–156. [DOI] [PubMed] [Google Scholar]
- 48.Cantow K, Flemming B, Ladwig-Wiegard M, Persson PB, Seeliger E. Low dose nitrite improves reoxygenation following renal ischemia in rats. Scientific reports. 2017;7(1):14597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei Y, Sun P, Wang Z, Yang B, Carroll MA, Wang WH. Adenosine inhibits ENaC via cytochrome P-450 epoxygenase-dependent metabolites of arachidonic acid. Am J Physiol Renal Physiol. 2006;290(5):F1163–1168. [DOI] [PubMed] [Google Scholar]
- 50.Niendorf T, Pohlmann A, Arakelyan K, et al. How bold is blood oxygenation level-dependent (BOLD) magnetic resonance imaging of the kidney? Opportunities, challenges and future directions. Acta physiologica. 2015;213(1):19–38. [DOI] [PubMed] [Google Scholar]
- 51.Yang S, Lin L, Chen JX, et al. Cytochrome P-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-alpha via MAPK and PI3K/Akt signaling pathways. Am J Physiol Heart Circ Physiol. 2007;293(1):H142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Batchu SN, Chaudhary KR, El-Sikhry H, et al. Role of PI3Kalpha and sarcolemmal ATP-sensitive potassium channels in epoxyeicosatrienoic acid mediated cardioprotection. J Mol Cell Cardiol. 2012;53(1):43–52. [DOI] [PubMed] [Google Scholar]
- 53.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80(1):29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng BQ, Luo Y, Kang X, et al. Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3beta signaling. Proc Natl Acad Sci U S A. 2017;114(47):12608–12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z, Havasi A, Gall J, et al. GSK3beta promotes apoptosis after renal ischemic injury. Journal of the American Society of Nephrology : JASN. 2010;21(2):284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature cell biology. 2004;6(11):1122–1128. [DOI] [PubMed] [Google Scholar]
- 57.Barilli A, Visigalli R, Sala R, et al. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc Res. 2008;78(3):563–571. [DOI] [PubMed] [Google Scholar]
- 58.Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. Journal of the American Society of Nephrology : JASN. 2011;22(5):902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82(12):1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature reviews Nephrology. 2017;13(11):681–696. [DOI] [PubMed] [Google Scholar]
- 61.Manhiani M, Quigley JE, Knight SF, et al. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297(3):F740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8(10):794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sudhahar V, Shaw S, Imig JD. Epoxyeicosatrienoic acid analogs and vascular function. Current medicinal chemistry. 2010;17(12):1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sodhi K, Inoue K, Gotlinger KH, et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther. 2009;331(3):906–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG. EET agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in Bach 1 and an increase in HO-1 levels. Prostaglandins Other Lipid Mediat. 2012;98(3–4):133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khan AH, Falck JR, Manthati VL, Campbell WB, Imig JD. Epoxyeicosatrienoic acid analog attenuates angiotensin II hypertension and kidney injury. Frontiers in pharmacology. 2014;5:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kierulf-Lassen C, Nielsen PM, Qi H, et al. Unilateral nephrectomy diminishes ischemic acute kidney injury through enhanced perfusion and reduced pro-inflammatory and pro-fibrotic responses. PLoS One. 2017;12(12):e0190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dolegowska B, Blogowski W, Domanski L. Is it possible to predict the early post-transplant allograft function using 20-HETE measurements? A preliminary report. Transplant international : official journal of the European Society for Organ Transplantation. 2009;22(5):546–553. [DOI] [PubMed] [Google Scholar]
- 70.Shuey MM, Billings FTt, Wei S, et al. Association of gain-of-function EPHX2 polymorphism Lys55Arg with acute kidney injury following cardiac surgery. PLoS One. 2017;12(5):e0175292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnold C, Markovic M, Blossey K, et al. Arachidonic Acid-Metabolizing Cytochrome P450 Enzymes Are Targets of Omega-3 Fatty Acids. J Biol Chem. 2010;285(43):32720–31733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fichtlscherer S, Dimmeler S, Breuer S, Busse R, Zeiher AM, Fleming I. Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation. 2004;109(2):178–183. [DOI] [PubMed] [Google Scholar]
- 73.Katoh T, Takahashi K, Capdevila J, et al. Glomerular stereospecific synthesis and hemodynamic actions of 8,9-epoxyeicosatrienoic acid in rat kidney. Am J Physiol. 1991;261(4 Pt 2):F578–586. [DOI] [PubMed] [Google Scholar]
- 74.Homma T, Zhang JY, Shimizu T, Prakash C, Blair IA, Harris RC. Cyclooxygenase-derived metabolites of 8,9-epoxyeicosatrienoic acid are potent mitogens for cultured rat glomerular mesangial cells. Biochem Biophys Res Commun. 1993;191(1):282–288. [DOI] [PubMed] [Google Scholar]
- 75.Draper AJ, Hammock BD. Inhibition of soluble and microsomal epoxide hydrolase by zinc and other metals. Toxicological sciences : an official journal of the Society of Toxicology. 1999;52(1):26–32. [DOI] [PubMed] [Google Scholar]
- 76.Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Critical care. 2007;11(2):R31. [DOI] [PMC free article] [PubMed] [Google Scholar]