

Abstract
Several studies have unraveled the negative role of Akt1 in advanced cancers, including metastatic prostate cancer (mPCa). Hence, understanding the consequences of targeting Akt1 in the mPCa and identifying its downstream novel targets is essential. We studied how Akt1 deletion in PC3 and DU145 cells activates the Nodal pathway and promotes PCa epithelial-to-mesenchymal transition (EMT) and metastasis. Here we show that Akt1 loss increases Nodal expression in PCa cells accompanied by activation of FoxO1/3a, and EMT markers Snail and N-cadherin as well as loss of epithelial marker E-cadherin. Treatment of PCa cells with FoxO inhibitor AS1842856 abrogated the Nodal expression in Akt1 deleted PCa cells. Akt1 deficient PCa cells exhibited enhanced cell migration and invasion in vitro and lung metastasis in vivo, which were attenuated by treatment with Nodal pathway inhibitor SB505124. Interestingly, Nodal mRNA analysis from two genomic studies in cBioportal showed a positive correlation between Nodal expression and Gleason score indicating the positive role of Nodal in human mPCa. Collectively, our data demonstrate Akt1-FoxO-Nodal pathway as an important mediator of PCa metastasis and present Nodal as a potential target to treat mPCa patients.
Keywords: Akt1, FoxO, Metastasis, Nodal, Prostate cancer
Graphical Abstract
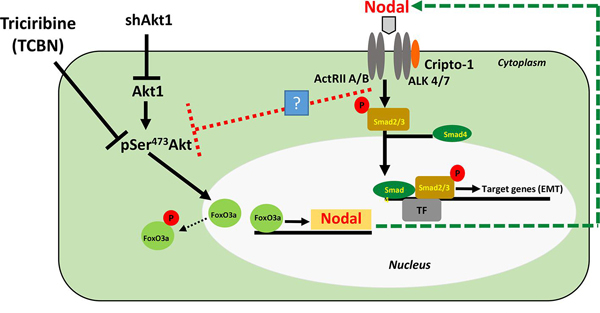
Prostate cancer cell Akt1 loss promotes epithelial-to-mesenchymal transition (EMT) via FoxO-mediated increased Nodal expression and ALK4/7 activation in vitro and metastasis to the mouse lungs.
1. Introduction
Despite the early diagnosis and significant advances in treatments, prostate cancer (PCa) still ranks the second and third on cancer-related mortalities in men in the United States and Europe, respectively [1, 2]. Recent statistics reveal that the higher mortality of PCa is mainly due to its metastasis to the bone, lungs, and brain [3, 4]. Whereas the 5-year survival rate of non-metastatic PCa patients has always been >99 %, the 5-year survival rate of metastatic PCa (mPCa) patients has been further declined to ≤ 30% [1]. Uncertainty in the molecular mechanisms mediating cancer cell dissemination to distant organs is a major roadblock in the effective management of mPCa [4]. In-depth molecular characterization and identification of novel, druggable targets will pave the way for future therapies in mPCa.
Several investigators over the past 2 decades have demonstrated the integral role of Akt (Protein kinase B) in multiple cellular processes such as, survival, proliferation, growth, invasion, and migration, that are implicated in tumorigenesis and cancer malignancy [5–7]. As a result, many Akt targeting drugs have been developed, tested and a few of these have entered the clinical trials [8]. In prostate cancer, we have demonstrated that Akt is necessary for cancer cell motility [9], survival [10–12], proliferation [11, 13], invasion [9, 14], transendothelial migration [14]. Several preclinical studies have also demonstrated an isoform-specific effect of Akt on cell migration and invasion, which are essential steps in the metastasis. Whereas Akt2 promoted the invasive phenotype of breast and ovarian cancer cells [15, 16], Akt1 was intriguingly found to abrogate cell migration and invasion by inhibiting epithelial-to-mesenchymal transition (EMT) in breast cancer [15, 17–20]. Recently, several reports in various cancer types have emerged explaining an unexpected, counteractive role of Akt1 in the advanced cancers [19, 21–24], including our findings in PCa [25–27]. Despite the controversial role of Akt1 in the advanced PCa [28], knowing how Akt1 orchestrates this process is crucial to optimize the current therapies and pave the way for potential new therapies for patients with mPCa. One of the genes that was significantly elevated in our previous gene arrays from the mice experiments on PCa is ‘Nodal’ [25]. Hence we hypothesized that Nodal pathway activation downstream of Akt1 suppression is involved in the promotion of PCa cell EMT and metastasis.
Nodal, a secreted protein belonging of TGFβ superfamily, is known to be expressed during embryogenesis, where it plays a vital role in inhibiting embryonic cell differentiation and maintains human stem cell pluripotency [29, 30]. In concert with another related signaling partner Lefty, Nodal regulates physiological cell migration to determine anterior-posterior and left-right axises asymmetry during vertebral development [31]. Although Nodal was thought to be absent in adulthood, it is normally expressed in the mammary glands, endometrium and placenta, and a specific population of pancreas and liver cells [32]. Nodal transmits signals by binding to a heterodimeric receptor complex of Activin-like kinase type II receptors (ActRIIA/ActRIIB) and Activin-like kinase type I receptors (ALK4/ALK7) [33]. Activation of ALK4 or ALK7 by type II receptors and a co-receptor Cripto-1 results in the phosphorylation of Smad2/3, which further interacts with Smad4 to enter the nucleus and regulate the expression of the target genes [34]. The activity of Nodal pathway is kept in check by the endogenously-secreted, extracellularly-acting inhibitors Lefty A, Lefty B and Cerberus, which are transcribed in response to Nodal signaling, thus providing a negative feedback mechanism [32, 33]. Lefy A and B block this pathway by binding to Nodal and/or Cripto-1, whereas Cerberus exerts its effect by binding to free Nodal [33].
In this study, we investigated the molecular switch that induces EMT and metastasis in PCa cells following Akt1 suppression. Based on our results, although Akt1 inhibition activates caspases to induce apoptosis in a selected population of the androgen-independent PCa cells, activation of the FoxO3a-Nodal pathway induced downstream of Akt1 suppression in apoptosis resistant population of PCa cells, plays a significant role in promoting PCa cell EMT in vitro and lung metastasis in vivo. Our results demonstrate that the Nodal-induced cell migration and invasion are mediated through the canonical Smad2/3-dependent pathway and SB505124, a specific antagonist of Nodal receptors ALK4/ALK7, abrogated Nodal-induced Smad2 phosphorylation, PCa cell migration, invasion and metastasis to the lungs. The data from ‘the cancer genome atlas (TCGA)’ study also indicated that the expression levels of Nodal in PCa patients is directly proportional to their Gleason score. Taken together, these results clearly demonstrate that the activation of Nodal pathway downstream of Akt1 suppression promotes PCa cell EMT in vitro and metastasis in vivo and therefore targeting Nodal pathway using SB505124 could serve as a potential therapeutic strategy for the treatment of mPCa.
2. Materials and Methods
2.1. Cell culture, gene silencing by shRNA, antibodies and other reagents
Human PC3 and DU145 cell lines were purchased from ATCC (Manassas, VA), cultured in DMEM high glucose medium (Hyclone, Logan, UT) with 10% Fetal bovine serum (FBS, Atlanta Biologicals, Atlanta, GA), 100 U/ml penicillin, and 100 mg/ml streptomycin in a humidified incubator at 37°C and 5% CO2. Cells were routinely passaged and when they were 80–90% confluent, transfection was carried out using SMARTvector 2.0 Lentivirus ShControl (non-targeting) and ShAkt1 (ACGCTTAACCTTTCCGCTG) from Dharmacon (Lafayette, CO), followed by selection with puromycin (0.6 ng/ml, Sigma Millipore, St. Louis, MO). Primary antibodies against Akt1 (Cat #2938), pSer473Akt (Cat #4060), pThr308Akt (Cat #2965), pFoxO1/3a (Cat #9464), pFoxO3a (Cat #9465), FoxO3a (Cat #2497), FoxO1 (Cat #2880), pSmad2/3 (Cat #8828), Smad2/3 (Cat #8685), Snail (Cat #3879), E-cadherin (Cat #3195), and N-cadherin (Cat #4061) were purchased from Cell Signaling (Danvers, MA). Nodal antibodies (Cat # SC-28913 and SC-373910) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). β-actin antibody (Cat #A5441) was purchased from Sigma (St. Louis, MO). Triciribine (TCBN) and SB505124 were purchased from Selleckchem (Houston, TX), and FoxO1/3a inhibitor (AS1842856) was purchased from Calbiochem (San Diego, CA). All other reagents were purchased from Fisher Scientific (Hanover Park, IL).
2.2. Western blotting
Western blotting was performed as published previously [25]. Briefly, the cell lysates were prepared using 1X RIPA lysis buffer (Millipore, Temecula, CA) supplemented with protease and phosphatase inhibitor tablets (Roche Applied Science, Indianapolis, IN). Protein concentration was measured by the DC protein assay (Bio-Rad Laboratories, Hercules, CA) and approximately 30–60 µg of cell lysates in Laemmli buffer were used. Densitometry was performed using NIH ImageJ software.
2.3. Migration assay
Cell migration assay was performed as explained previously [9]. Cells were grown on 6-well plates until reaching 60–70% confluence. Scratch was made in the cell monolayer using 1ml pipette tip followed by a one-time wash with 1X PBS. Cells were incubated with SB505124 (20 μM) or 0.5 % DMSO in DMEM containing 5% FBS for 24 hours. This optimal dose used in the study did not affect the viability. Images of scratches were taken immediately after scratching (0 hours) and 24 hours after treatment. The rate of migration was measured as a percentage of scratch filling using the equation ([1-T24/T0] X 100), where T24 is the area at the end point (24 hours) and T0 is the area measured immediately after making the scratches.
2.4. Matrigel® invasion assay
Twenty-four Transwell® permeable plate supports with 8.0 μm polycarbonate membrane and Matrigel® were purchased from Corning Life Sciences (Tewksbury, MA). A concentration of 5mg/ml of Matrigel was used for coating supports. Matrigel® invasion assay was performed as published previously [35]. Briefly, cells were seeded in 6 well plates, washed one time with 1X PBS and treated with either SB505124 (20 µM) or 0.5 % DMSO (diluted with 0.9% saline) for 24 hours. The medium was aspirated, and cells were washed once with 1X PBS, detached using sterile 20 mM EDTA in PBS and washed once with 0.9% saline. Cells were re-suspended in serum-free medium with/without treatment based on the final treatment. Using Countess automated cell counter (Invitrogen), 100,000 cells were seeded on to the Matrigel® in the upper chamber of the transwell plates filled with 100 µl of serum-free medium. Cells that invaded the matrigel and reached the bottom layers of the supports after 24 hours incubation were fixed using 3.7 % paraformaldehyde then stained with 0.5% crystal violet solution. Three bright field images of each insert were taken using an inverted microscope and three blinded reviewers counted the invaded cells. The average number of invaded cells from every three images was calculated and considered for the analysis.
2.5. In vivo nude mouse xenograft model
All animal procedures were performed according to the protocol approved by the Institutional Animal Care and Use Committee at the Charlie Norwood Veterans Affairs Medical Center, Augusta, Georgia (protocol 15–08-083). Tumor xenografts were implanted as explained previously [27]. ShAkt1 and ShCtrl DU145 cells were grown to 60–70% confluence in T75 flasks. Cells were washed once with 1X PBS, detached using trypsin and re-suspended in 0.9% saline. A volume of 150–200 µl cell suspension containing 1×106 cells was injected through the tail vein into 8-week-old nude mice (athymic nude mice; Harlan, Indianapolis, IN). Animals in each group were injected (i.p.) with either 10mg/kg SB505124 dissolved in 75% DMSO (diluted with 0.9% saline) daily for 15 days or with a vehicle only (n=8 in each group). Mice weight was monitored every 3 days up to day 16. On day 16, mice were euthanized and one lobe of the right lungs (post-caval lobe) was collected and snap-frozen for H&E staining while the rest were stained with 15% India ink through intratracheal injection (5 ml). Stained lungs were carefully resected and rinsed in Fekete’s solution (300 ml 70% ethanol, 30 ml 37% formaldehyde, 5 ml glacial acetic acid), then placed in fresh Fekete’s solution overnight. Lung tissue would remain stained with India ink whereas lung tumor nodules would become unstained and can be visualized by the naked eye. The number of lung nodules was counted by three blinded reviewers and the lung colonization in all groups and the average of their scores were considered for the analysis.
2.6. The cancer genome atlas (TCGA) study analysis
Genomic data analysis from databases such as TCGA has the advantage of determining the changes in the expression of signaling molecules using bigger sample size as compared to the smaller sample size analysis in a conventional laboratory setup. This is more important in prostate cancer tissues where the cell [population is heterogeneous and variations high. Hence, we downloaded the data on the expression of Nodal and corresponding clinical information for 498 prostate cancer patients from the TCGA database [36] from cBioPortal (https://www.cbioportal.org/). According to the Gleason Score, patients were stratified into 3 cohorts, Gleason Score of 6–7 (N=292), 8 (N=64) and 9–10 (N=142). Changes in the Nodal mRNA expression was compared to the Gleason score. The same approach was performed on another study on the Prostate Adenocarcinoma [37] with 150 prostate cancer patients. Based on the Gleason Score known for 139 patients from this study, patients were stratified into 3 cohorts, Gleason Score of 6–7 (N=117), Gleason Score of 8 (N=11) and Gleason Score of 9 (N=11), and similar analysis on their mRNA expression of Nodal was conducted. Furthermore, the association between Nodal mRNA expression and the clinical characteristics of patients in both studies was investigated.
2.7. Statistical analysis
All the data are presented as the mean ± standard error of the mean (SEM) to determine significant differences between treatments and control values. We have used One-way ANOVA for groups of 3 or more and Student’s two-tailed t-test for studies including 2 independent groups. Statistical analysis was performed using GraphPad Prism version 6.01 software and results are considered significant when p-value < 0.05.
3. Results
3.1. The nodal expression is increased in Akt1 knockdown androgen-insensitive human PCa cells associated with increased EMT markers and enhanced Smad2/3 pathway
In a recent study, we showed that the pharmacological inhibition of Akt1 using TCBN in the tumor-bearing TRansgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model promoted PCa EMT and metastasis [25, 26]. A gene array analysis of the prostate tumors from these mice revealed an increased expression of several EMT markers, including Nodal when compared to the control prostates [25]. To investigate this using human PCa cell lines, we silenced the expression of Akt1 gene using ShRNAs in PC3 and DU145 cells. Akt1 gene silencing resulted in a significant increase in the Nodal protein expression in both DU145 and PC3 cells (Figure 1A–C). This was accompanied by a complete loss of epithelial marker E-cadherin and increased expression of EMT markers Snail and N-cadherin (Figure 1A–C). Since Akt1 is essential for cell survival, its loss was associated with apoptosis indicated by enhanced cleaved caspase-3 expression (Figure 1A–C). We next determined if increased Nodal expression in ShAkt1 PC3 and DU145 cells results in the activation of the canonical pathway. Our analysis showed a significant increase in the phosphorylated Smad2 correlating to the Nodal expression levels in ShAkt1 DU145 and PC3 cells with no net changes in the total Smad2/3 expression (Figure 1D–E), thus suggesting that Nodal expression as a result of Akt1 suppression promotes EMT in PCa cells through the canonical Smad2/3 pathway.
Figure 1. Akt1 silencing promotes Nodal expression and EMT in PC3 and DU145 PCa cells.

(A) Representative Western blot images of cell lysates obtained from control (ShControl) and Akt1-silenced (ShAkt1) DU145 and PC3 cells showing changes in the expression of Nodal, EMT markers (N-cadherin and Snail), apoptotic marker cleaved caspase-3, epithelial marker (E-cadherin), phosphorylated Akt and Akt1. (B-C) Bar graphs showing band densitometry analysis of Western blot images from cell lysates obtained from control (ShControl) and Akt1-silenced (ShAkt1) DU145 and PC3 cells showing significant increase in the expression of Nodal, EMT markers (N-cadherin and Snail) and apoptotic marker cleaved caspase-3 associated with decreased expression of epithelial marker (E-cadherin), phosphorylated Akt and Akt1 normalized to loading control β-actin, respectively (n=3). (D) Representative Western blot images of cell lysates obtained from control (ShControl) and Akt1-silenced (ShAkt1) DU145 and PC3 cells showing changes in the expression of phosphorylated and total expression of Smad2/3. (E) Bar graphs showing band densitometry analysis of Western blot images from cell lysates obtained from control (ShControl) and Akt1-silenced (ShAkt1) DU145 and PC3 cells showing a significant increase in the expression of phosphorylated Smad2 normalized to loading control β-actin, respectively, with no changes in the expression of total Smad2 (n=3). Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
3.2. Increased Nodal expression in ShAkt1 PCa cells is reliant on FoxO3a signaling
It is well established that Akt1 regulates FoxO1/3a transcriptional activity via direct phosphorylation followed by a nuclear exit and subsequent proteasomal degradation [38]. Therefore, it was in our interest to determine whether FoxOs have a role in enhancing Nodal expression in PCa cells. Treatment with Akt inhibitor TCBN significantly increased Nodal expression in DU145 cells in a dose-dependent manner (Figure 2A–B). The increase in Nodal expression in TCBN-treated DU145 and PC3 cells was associated with decreased phosphorylated FoxO3a and increased total FoxO3a, thereby correlating the activity and nuclear presence of FoxO3a (Figure 2C–F) with the increased Nodal expression. Similarly, Akt1 gene silencing in PC3 and DU145 cells also resulted in a significant decrease in the phosphorylation and increase in the total expression of FoxO3a in DU145 cells (Figure 3A–B). Interestingly, treatment with FoxO inhibitor AS1842856 resulted in a significant decrease in the Nodal expression in ShAkt1 as well as ShCtrl PC3 and in ShAkt1 DU145 cells (Figure 3C–D) indicating an important role of FoxO transcription factors in the regulation of Nodal expression in PCa cells.
Figure 2. Pharmacological inhibition of Akt1 activated FoxO1/3a and promotes Nodal expression in PCa cells.

(A-B) Representative Western blot images and respective bar graph of band densitometry analysis on the effect of Akt inhibitor TCBN treatment (72 hours) on Nodal expression in DU145 cells (n=3). (C-D) Representative Western blot images and respective bar graph of band densitometry analysis on the effect of Akt inhibitor TCBN treatment (72 hours) on phosphorylation and total expression of FoxO3a in DU145 cells (n=3). (E-F) Representative Western blot images and respective bar graph of band densitometry analysis on the effect of Akt inhibitor TCBN treatment (72 hours) on phosphorylation and total expression of FoxO3a as well as Nodal in PC3 cells (n=3). Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
Figure 3. Increased Nodal expression in Akt1-deficient PCa cells is attenuated by treatment with FoxO inhibitor AS1842765.

(A-B) Representative Western blot images and bar graph of the band densitometry analysis of ShControl and ShAkt1 DU145 and PC3 cells probed for phosphorylated and total expression of FoxO1 and FoxO3a showing a significant decrease in FoxO phosphorylation in ShAkt1 PCa cells compared to ShControl, respectively (n=6). (C-D) Representative Western blot images and bar graph of the band densitometry analysis of ShControl and ShAkt1 DU145 and PC3 cells probed for Nodal expression showing significant increase in Nodal expression in ShAkt1 PCa cells and a significant decrease in Nodal expression in ShAkt1 PCa cells by treatment with FoxO1/3a inhibitor AS1842765 (10 µM; 72 hours; n=3). Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
3.3. The Nodal pathway inhibition in vitro blunted the migration and invasion of ShAkt1 PCa cells
Increased expression of Nodal correlating to the increased expression of EMT markers in ShAkt1 PCa cells suggested a role for the Nodal pathway in the regulation of cell motility and invasion, the features of aggressive, metastatic cancer cells. To test this, we determined the effect of Nodal pathway inhibitor SB505124 on migration and invasion of ShCtrl and ShAkt1 DU145 and PC3 cells. Our analysis indicated a significant increase in the migration rate of ShAkt1 DU145 (Figure 4A–B) and PC3 cells (Figure 4E–F) compared to their respective controls in a cell monolayer scratch assay. This was accompanied by an increase in the phosphorylation levels of Smad2 in ShAkt1 DU145 (Figure 4C–D) and PC3 cells (Figure 4G–H). Increased cell migration of ShAkt1 DU145 and PC3 cells was significantly inhibited by treatment with Nodal pathway inhibitor SB505124 (Figure 4A–B and 4E–F, respectively), which was accompanied by a significant reduction in the expression levels of phosphorylated Smad2 (Figure 4C–D and 4G–H, respectively).
Figure 4. Nodal pathway inhibition blunts Smad2/3 phosphorylation and abrogates enhanced migration of Akt1-deficient DU145 and PC3 cells.

(A) Representative images from the scratch assay (0 and 24 hours) in ShControl and ShAkt1 DU145 cell monolayers showing the migration of PCa cells in the presence and absence of Nodal pathway inhibitor SB505124 after 24 hours. (B) Bar graph showing the significant inhibitory effect of Nodal pathway inhibitor SB505124 on ShAkt1 DU145 cell migration compared to ShControl cells (n=3). (C) Representative Western blot images of cell lysates obtained from ShControl and ShAkt1 DU145 cells showing changes in the expression of phosphorylated and total expression of Smad2 in the presence and absence of Nodal pathway inhibitor SB505124. (D) Bar graphs showing the band densitometry analysis of Western blot images from cell lysates obtained from ShControl and ShAkt1 DU145 cells showing a significant decrease in the expression of phosphorylated Smad2 in ShAkt1 cell lysates by treatment with Nodal pathway inhibitor SB505124 (n=3). (E) Representative images from the scratch assay (0 and 24 hours) in ShControl and ShAkt1 PC3 cell monolayers showing the migration of PCa cells in the presence and absence of Nodal pathway inhibitor SB505124 after 24 hours. (F) Bar graph showing the significant inhibitory effect of Nodal pathway inhibitor SB505124 on ShAkt1 PC3 cell migration compared to ShControl cells (n=3). (G) Representative Western blot images of cell lysates obtained from ShControl and ShAkt1 PC3 cells showing changes in the expression of phosphorylated and total expression of Smad2 in the presence and absence of Nodal pathway inhibitor SB505124. (H) Bar graphs showing the band densitometry analysis of Western blot images from cell lysates obtained from ShControl and ShAkt1 PC3 cells showing a significant decrease in the expression of phosphorylated Smad2 in ShAkt1 cell lysates by treatment with Nodal pathway inhibitor SB505124 (n=3). Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
In the next step, we determined whether inhibition of Nodal pathway would affect the rate of invasion of PCa cells. Our analysis indicated a significant increase in the invasion of ShAkt1 DU145 (Figure 5A–B) and PC3 cells (Figure 5C–D) compared to their respective controls in a Matrigel® based Transwell invasion assay. Enhanced invasion of ShAkt1 DU145 and PC3 cells were significantly inhibited by treatment with Nodal pathway inhibitor SB505124 (Figure 5A–B and 5C–D, respectively). Together, these results clearly demonstrated the importance of Nodal signaling in the regulation of PCa cell migration and invasion downstream of Akt1 suppression.
Figure 5. Nodal pathway inhibition abrogates enhanced invasion of Akt1-deficient DU145 and PC3 cells.

(A) Representative images from the Matrigel Boyden chamber invasion (0 and 24 hours) by ShControl and ShAkt1 DU145 cells showing the invasion of PCa cells in the presence and absence of Nodal pathway inhibitor SB505124 after 24 hours. (B) Bar graph showing the significant inhibitory effect of Nodal pathway inhibitor SB505124 on ShAkt1 DU145 cell invasion compared to ShControl cells (n=3). (C) Representative images from the Matrigel Boyden chamber invasion (0 and 24 hours) by ShControl and ShAkt1 PC3 cells showing the invasion of PCa cells in the presence and absence of Nodal pathway inhibitor SB505124 after 24 hours. (D) Bar graph showing the significant inhibitory effect of Nodal pathway inhibitor SB505124 on ShAkt1 PC3 cell invasion compared to ShControl cells (n=3). Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
3.4. The Nodal pathway inhibition in vivo attenuated lung metastasis of ShAkt1 PCa cells
The efficacy of Nodal pathway inhibitor SB505124 to suppress enhanced migration and invasion of ShAkt1 PCa cells suggested the utility of SB505124 to inhibit PCa metastasis in vivo. To test this, we performed lung metastasis assay by administering ShCtrl and ShAkt1 DU145 cells via tail-vein (1×106 cells suspended in 150–200 µl saline) to athymic nude mice with and without treatment by the Nodal pathway inhibitor SB505124 (10 mg/kg daily and for 15 days) or vehicle (DMSO) via i.p. route. Whereas nodules were not clearly visible in the lungs of ShCtrl DU145 cell administered mice, numerous DU145 cell tumor nodules were visible in the lungs of ShAkt1 DU145 cells administered mice, visualized by India ink staining (Figure 6A). Treatment with SB505124 resulted in a significant reduction in the number of ShAkt1 DU145 tumor nodules in the mouse lungs as compared to the DMSO treated controls (Figure 6A–B). In order to confirm this further, we analyzed the lung sections from a different set of mice for the presence of DU145 cell tumor colonies. Our analysis indicated that the mice administered with ShAkt1 DU145 cells had >5-fold higher number of tumor colonies compared to the mice administered with ShCtrl DU145 cells, and it was significantly inhibited by treatment with SB505124 (Figure 6C–D). This treatment, however, had no effect on the animal weight. Finally, we determined if SB505124 has any direct effect on PCa tumor growth. To do this, we implanted ShAkt1 DU145 cell tumor xenografts in athymic nude mice and treated them with DMSO (vehicle) or SB505124. Our results indicated that the Nodal pathway inhibitor has no significant effect on the growth of ShAkt1 DU145 cell tumor xenografts in vivo (Figure 6E–G). The dose administered in vivo did not affect cell viability in the tumor xenografts as evidenced by no changes in the tumor size.
Figure 6. Nodal pathway inhibition blunts enhanced mouse lung metastasis of Akt1-deficient DU145 cells.

(A-B) Representative images of athymic nude mouse lungs infused with India ink through trachea and bar graph showing a significant increase in the number of PCa tumor nodules upon administration of ShAkt1 DU145 cells compared to ShControl, which was significantly inhibited by treatment with Nodal pathway inhibitor SB505124 (10 mg/kg/day; Day 15; n=6). (C-D). Representative H&E stained lung section images showing a significant increase in lung colonization of ShAkt1 DU145 compared to ShControl DU145 cells, which was significantly attenuated by a 15-day treatment with SB505124 (n=4). (E-G) Representative images of ShAkt1 DU145 tumor xenografts implanted (s.c.) in athymic nude mice and bar graphs showing no significant inhibition of tumor weight and volume by Nodal pathway inhibition (Day 15; n=6), respectively. Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
3.5. Changes in Nodal expression positively correlates with the Gleason score of human PCa
Since Nodal expression was significantly increased in PCa cells and its pharmacological suppression attenuated PCa cell migration and invasion in vitro, and metastasis in vivo, we determined if there is a correlation between increased Nodal expression and the stage of PCa. To do this, we collected the information available from cBioportal on changes in the Nodal expression in PCa patients enrolled in the TCGA studies, based on their Gleason score. The prostate adenocarcinoma patient characteristics of TCGA provisional study (n=498) and the MSKCC genomic analysis of prostatic adenocarcinoma study (n=150) are shown in Tables 1 and 2, respectively. When the patients were stratified based on the age, Nodal mean mRNA expression was significantly higher in the group with age >60 years compared to age ≤ 60 (3.99 vs. 2.48, respectively; p = 0.0376). Interestingly, when patient data were stratified based on the Gleason Score, Nodal expression was higher in patients with a higher Gleason score (2.6 in Gleason score 6–7 vs. 4.659 in Gleason score 9–10; p = 0.042, respectively) (Figure 7A; Table 1). Similarly, Nodal expression was significantly upregulated in the higher Gleason Score (9) compared to lower Gleason Score (6–7) group (6.521 vs. 6.15; p = 0.0025, respectively) in TCGA MSKCC study (Figure 7B and Table 2). These results indicate that the Nodal expression in PCa is directly proportional to the stage of the disease progression.
Table 1:
(A) Table showing the clinical characteristics of 498 patients of the TCGA study [Prostate adenocarcinoma (TCGA, Provisional)] extracted from cBioportal. (B) Table showing the associations between the mRNA expression levels of Nodal and clinical characteristics indicating that Nodal expression is significantly upregulated with higher age (>60 years) and Gleason score (9–10) compared to lower age (≤ 60 years) and Gleason score (6–7), respectively.
| Characteristics | No. of patients (N=498) | % | Characteristics (Number of patients) | Mean Nodal mRNA expression | p value |
|---|---|---|---|---|---|
| Age [Range] | 223 | 44.78 | Age [Range] | 2.48 | 0.0376 |
| ≤ 60 [41–60 yrs] | 275 | 55.22 | ≤ 60 [41–60 yrs] | 3.99 | |
| > 60 [61–78 yrs] | > 60 [61–78 yrs] | ||||
| Gleason score | 45 | 9.04 | Gleason score | 2.60 | 0.042 (GS 6–7 vs. GS 9–10) |
| 6 | 247 | 49.59 | 6–7 (292) | 3.62 | |
| 7 | 64 | 12.85 | 8 (64) | 4.66 | |
| 8 | 138 | 27.71 | 9–10 (142) | ||
| 9 | 4 | 0.80 | |||
| 10 | |||||
| T stage [pT] | 177 | 35.54 | T stage [pT] | 3.19 | 0.5167 |
| T1 | 174 | 34.94 | T1–2 (351) | 2.56 | |
| T2 | 53 | 10.64 | T3–4 (55) | ||
| T3 | 2 | 0.40 | |||
| T4 | 1 | 0.20 | |||
| Unknown | 91 | 18.27 | |||
| NA | |||||
| N stage [pN] | 345 | 69.28 | N stage [pN] | 3.06 | 0.3711 |
| N0 | 80 | 16.06 | N0 (345) | 3.89 | |
| N1 | 73 | 14.66 | N1 (80) | ||
| NA | |||||
| M stage [pM] | 456 | 91.57 | M stage [pM] | 3.326 | 0.2296 |
| M0 | 3 | 0.60 | M0 (456) | 9.072 | |
| M1 | 39 | 7.83 | M1 (3) | ||
| NA | |||||
| Survival status | 488 | 97.99 | Survival status | 3.325 | 0.8937 |
| Alive | 10 | 2.01 | Alive (488) | 2.980 | |
| Dead | Dead (10) | ||||
| |
Overall survival | 3.398 | 0.8253 | ||
| Median | 3.238 | ||||
| |
≤ 30.49 mths | ||||
| |
(249)> 30.49 mths (249) | ||||
Table 2:
(A) Table showing the available clinical characteristics of 150 patients of the TCGA study [Prostate adenocarcinoma (MSKCC, cancer cell 2010)] extracted from cBioportal. (B) Table showing the associations between the mRNA expression levels of Nodal and clinical characteristics indicating that Nodal expression is significantly upregulated with higher (9) compared to lower Gleason score (6–7).
| Characteristics | No. of patients (N=150) | % |
|---|---|---|
| Age [years] | Unknown | |
| Gleason score | 41 | 27.33 |
| 6 | 76 | 50.67 |
| 7 | 11 | 7.33 |
| 8 | 11 | 7.33 |
| 9 | 0 | 0 |
| 10 | 11 | 7.33 |
| NA | ||
| T stage [pT] | 80 | 53.33 |
| T1 | 58 | 38.67 |
| T2 | 6 | 4 |
| T3 | 1 | 0.67 |
| T4 | 5 | 3.33 |
| NA | ||
| N stage [pN] | Unknown | |
| M stage [pM] | Unknown | |
| Survival status | Unknown | |
| Characteristics (Number of patients) | Mean Nodal mRNA expression | P value |
| Gleason score | 6.15 | 0.0062 (GS 6–7 vs.GS 9) |
| 6–7 (117) | 6.19 | |
| 8 (11) | 6.52 | |
| 9–10 (11) | ||
| T stage [pT] | 6.184 | 0.5592 |
| T1–2 (138) | 6.104 | |
| T3–4 (7) | ||
| Disease free Survival | 6.22 | 0.2557 |
| Median | 6.15 | |
| ≤ 45.37 mths (70) | ||
| > 45.37 mths (70) | ||
| Overall survival | Unknown | |
Figure 7. Genomic data on Nodal expression in human PCa tissues positively correlates with the Gleason score and cancer metastasis.

(A). Comparison between prostate cancer tissues with different Gleason scores (known for 498 out of the 501 patients) shows that Nodal expression is significantly upregulated with higher (9–10) compared to lower Gleason score (6–7). (B) Comparison between prostate cancer tissues with different Gleason scores (known for only 135 out of the 150 patients) shows that Nodal expression is significantly upregulated with higher (9) compared to lower Gleason score (6–7). Data are presented as mean ± SEM. *p<0.05; **p<0.01; #p<0.001.
4. Discussion
The oncogene Akt, although promotes tumor survival, proliferation, and growth, its suppression has been implicated in promoting tumor cell EMT, motility, and invasion in vitro [17, 19, 24, 39, 40] and metastasis in vivo in several cancer types such as the breast, prostate, head and neck, and non-small cell lung cancers [19, 23–25]. Recent studies in our laboratory have identified that Akt suppression in advanced PCa promotes EMT and metastasis [25, 26]. Interestingly, endothelial-specific loss of Akt1 alone promoted PCa metastasis to the mouse lungs [27], indicating that the endothelial cells, by opening its endothelial-barrier [41], also might contribute to the increased PCa metastasis upon pharmacological Akt inhibition. Although promotion of EMT with Akt1 gene deletion has been implicated as a common feature in all these studies, reports on the mechanisms by which Akt1 suppression promotes EMT in different cancer types have been disparate suggesting that our understanding on how Akt1 suppression promotes cancer metastasis is far from complete. From our gene array studies in the TRAMP mouse prostates, we found a significantly higher expression of Nodal with Akt suppression as compared to Akt intact prostates [25]. Based on this, we hypothesized that activation of Nodal pathway as a result of Akt1 inhibition promotes PCa cell EMT and the pharmacological inhibition of this pathway attenuates Akt1 deficient PCa cell metastasis in vivo.
In the current study, we report for the first time that Akt1 is directly involved in the regulation of Nodal expression. Genetic deletion of Akt1 or pharmacological suppression of total Akt that promoted PCa cell EMT and metastasis also resulted in increased Nodal expression. This was accompanied by reduced FoxO1/3a phosphorylation, enhanced FoxO3a expression, and their transcriptional activation. The ability of FoxO inhibitors to blunt the effect of Akt1 suppression on Nodal expression indicated that Nodal is under the transcriptional control of FoxO1/3a. The effect of Nodal pathway inhibitor to mitigate the enhanced cell migration and invasion observed in Akt1 silenced PC3 and DU145 cells through the suppression of canonical Smad2/3 pathway indicated the involvement of Nodal in promoting EMT in these cells. The efficacy of Nodal pathway inhibitor to suppress Akt1-deficient DU145 cell lung metastasis in mice with no significant effect on the growth of DU145 cell tumor xenografts demonstrated the specific role of Nodal in promoting PCa metastasis in the advanced stages. The direct relationship between the increased Nodal mRNA expression in PCa patient samples correlating to their Gleason score and age as analyzed from the TCGA database further strengthened our conclusions that Nodal is a specific mediator of PCa metastasis.
The ability of PCa cells to migrate and invade as a result of EMT also determines its metastatic potential [25, 35]. Therefore, it is necessary to confirm a direct link between the observed changes in the Nodal expression in the Akt1-deficient PCa cells and their increased migration and invasion. Whereas the Nodal promoted cell invasion via increased MMP2 expression in glioma [42], it promoted PCa EMT via increased N-cadherin, Vimentin and Snail, and it enhanced MMP2 and CXCR4 expression in pancreatic cancer cells [43]. Nodal also activated cell migration and invasion via increased MMP2 and MMP9 expression in bladder cancer cells [44]. Nodal has also been demonstrated to effect through the Smad2/3-independent mechanisms such as the promotion of angiogenesis via ERK1/2 and HIF1α dependent VEGF signaling in glioma [45] and stimulation of aggressive melanoma by inducing EMT through increased MMP2 and MMP9 expression and Snail stabilization-mediated by GSK-3β pathway [46]. Nodal promotes EMT-like phenotype through p38 MAPK activation in choriocarcinoma and breast cancer cells [47] and confers stemness and malignancy via activating Wnt/β-catenin-Oct4 axis in PCa and lung cancer [48]. The results from our study in PCa demonstrate that Akt1 inhibition in Pca cells enhances Nodal expression through activation of downstream FoxO1/3a transcriptional factors and promotes Pca cell migration, invasion in vitro and metastasis in vivo. Therefore, suppression of Nodal pathway by SB505124 inhibits the canonical Smad2/3 signaling and thus attenuates the enhanced motility and invasion of Akt1 silenced PCa cells.
Despite the wealth of literature with mechanisms demonstrating Nodal functions, we still do not have a clear understanding of Nodal expression regulation in cancer cells. A recent study linked an elevated expression of Nodal to the promotion of cell motility in breast cancer and invasive ductal carcinoma via MAP kinase-interacting serine/threonine kinase-1 activation [49]. In a distinct scenario, our data are in agreement with the above reports and indicates a reciprocal relationship between Akt1 activity and Nodal expression, where gene silencing or pharmacological inhibition of the former in the highly metastatic PCa cell lines resulted in increased expressionof the latter. In this study, we also provide the novel evidence on the role of Akt1-FoxO signaling in enhancing PCa cell-derived Nodal expression, in turn, promoting EMT and metastasis. In support of this, Fu and Peng reported an interesting observation in ovarian cancer cells, where Nodal pathway arbitrated a negative feedback loop on Akt1 activity thus reducing FoxO3a phosphorylation and enhancing its mRNA as well as protein expression through the canonical Smad pathway [50]. Although there is a potential role for FoxO1 in regulating Nodal expression, the predominance of FoxO3a accompanied with very low levels of FoxO1 in the PCa cells indicated that the majority of the observed effects are regulated by FoxO3a.
Although Nodal has been shown to induce apoptosis in ovarian cancer cells [51], it activated the canonical Smad2/3 pathway to induce vascular mimicry and promoted the expression of EMT markers such as the Snail and Slug in breast cancer cells [52]. Even in PCa, we observed increased cleaved caspase-3 levels in Akt1 silenced PC3 and DU145 cells correlating to the Nodal expression indicating that some level of apoptosis is associated with Nodal expression. A significant increase in the apoptosis of tumor cells was also observed in Akt1 deficient TRAMP mice [25]. Despite this, Nodal expression was directly linked to the promotion of PCa cell EMT, migration, and invasion.
Nodal has been reported for its potential role in several cancers, including PCa [48, 53–55]. Interestingly, Nodal mRNA expression was directly proportional to the age, Gleason score and the metastasis stage of PCa in our analysis of the TCGA study data obtained from cBioportal. Our findings from rodent studies along with the patient data on Nodal expression strongly suggest the potential therapeutic use of Nodal inhibitors to inhibit mPCa. Our results corroborate a previous report linking increased Nodal staining intensity to high-grade prostate tumor biopsies compared to low-grade samples [54]. In summary, whereas the clinical data signify a positive correlation between Nodal expression and the stage of PCa, our overall findings clearly demonstrate the important role of Nodal pathway activation as a consequence of Akt1 suppression leading to the activation of FoxO1/3a in promoting EMT and metastasis in the advanced PCa. Furthermore, our study present FoxO along with Nodal as two therapeutic targets in the treatment of mPCa.
ACKNOWLEDGMENTS
Funds were provided by the National Institutes of Health grants R01HL103952 and UL1TR002378 to PRS. This work has been accomplished using the resources and facilities at Charlie Norwood Medical Center in Augusta, GA. The funders had no role in the study design, data collection, analysis and decision to publish the data. The contents of the manuscript do not represent the views of the Department of Veteran Affairs or the United States Government.
Footnotes
CONFLICT OF INTEREST
The authors declare that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019, CA Cancer J Clin 69(1) (2019) 7–34. [DOI] [PubMed] [Google Scholar]
- [2].Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C, Negri E, European cancer mortality predictions for the year 2018 with focus on colorectal cancer, Ann Oncol 29(4) (2018) 1016–1022. [DOI] [PubMed] [Google Scholar]
- [3].Turajlic S, Swanton C, Metastasis as an evolutionary process, Science 352(6282) (2016) 169–75. [DOI] [PubMed] [Google Scholar]
- [4].Guan X, Cancer metastases: challenges and opportunities, Acta Pharm Sin B 5(5) (2015) 402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Testa JR, Tsichlis PN, AKT signaling in normal and malignant cells, Oncogene 24(50) (2005) 7391–3. [DOI] [PubMed] [Google Scholar]
- [6].Altomare DA, Testa JR, Perturbations of the AKT signaling pathway in human cancer, Oncogene 24(50) (2005) 7455–64. [DOI] [PubMed] [Google Scholar]
- [7].Song M, Bode AM, Dong Z, Lee MH, AKT as a Therapeutic Target for Cancer, Cancer Res (2019). [DOI] [PubMed]
- [8].Liu P, Cheng H, Roberts TM, Zhao JJ, Targeting the phosphoinositide 3-kinase pathway in cancer, Nat Rev Drug Discov 8(8) (2009) 627–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Goc A, Liu J, Byzova TV, Somanath PR, Akt1 mediates prostate cancer cell microinvasion and chemotaxis to metastatic stimuli via integrin beta(3) affinity modulation, Br J Cancer 107(4) (2012) 713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR, Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression, J Pharmacol Exp Ther 336(2) (2011) 496–505. [DOI] [PubMed] [Google Scholar]
- [11].Goc A, Al-Husein B, Kochuparambil ST, Liu J, Heston WW, Somanath PR, PI3 kinase integrates Akt and MAP kinase signaling pathways in the regulation of prostate cancer, International Journal of Oncology 38(1) (2010). [PubMed] [Google Scholar]
- [12].Goc A, Kochuparambil ST, Al-Husein B, Al-Azayzih A, Mohammad S, Somanath PR, Simultaneous modulation of the intrinsic and extrinsic pathways by simvastatin in mediating prostate cancer cell apoptosis, BMC Cancer 12 (2012) 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alhusban A, Al-Azayzih A, Goc A, Gao F, Fagan SC, Somanath PR, Clinically relevant doses of candesartan inhibit growth of prostate tumor xenografts in vivo through modulation of tumor angiogenesis, J Pharmacol Exp Ther 350(3) (2014) 635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Al-Husein B, Goc A, Somanath PR, Suppression of interactions between prostate tumor cell-surface integrin and endothelial ICAM-1 by simvastatin inhibits micrometastasis, J Cell Physiol 228(11) (2013) 2139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ, Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression, Cancer Res 69(12) (2009) 5057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ, Overexpression of AKT2:protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells, Cancer Res 63(1) (2003) 196–206. [PubMed] [Google Scholar]
- [17].Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ, Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion, Cancer Res 64(9) (2004) 3171–8. [DOI] [PubMed] [Google Scholar]
- [18].Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS, Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition, The Journal of cell biology 171(6) (2005) 1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y, Li LY, Lai CC, Chang SS, Hsu YH, Sun HL, Kim J, Yamaguchi H, Lee DF, Wang H, Wang Y, Chou CK, Hsu JM, Lai YJ, LaBaff AM, Ding Q, Ko HW, Tsai FJ, Tsai CH, Hortobagyi GN, Hung MC, AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation, Cancer Res 76(6) (2016) 1451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Riggio M, Perrone MC, Polo ML, Rodriguez MJ, May M, Abba M, Lanari C, Novaro V, AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins, Sci Rep 7 (2017) 44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN, MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells, Sci Signal 2(92) (2009) ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, Guzman G, Hay N, Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms, Cancer Cell 29(4) (2016) 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rao G, Pierobon M, Kim IK, Hsu WH, Deng J, Moon YW, Petricoin EF, Zhang YW, Wang Y, Giaccone G, Inhibition of AKT1 signaling promotes invasion and metastasis of non-small cell lung cancer cells with K-RAS or EGFR mutations, Sci Rep 7(1) (2017) 7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brolih S, Parks SK, Vial V, Durivault J, Mostosi L, Pouyssegur J, Pages G, Picco V, AKT1 restricts the invasive capacity of head and neck carcinoma cells harboring a constitutively active PI3 kinase activity, BMC Cancer 18(1) (2018) 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gao F, Alwhaibi A, Sabbineni H, Verma A, Eldahshan W, Somanath PR, Suppression of Akt1-beta-catenin pathway in advanced prostate cancer promotes TGFbeta1-mediated epithelial to mesenchymal transition and metastasis, Cancer Lett 402 (2017) 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alwhaibi A, Gao F, Artham S, Hsia BM, Mondal A, Kolhe R, Somanath PR, Modulation in the microRNA repertoire is responsible for the stage-specific effects of Akt suppression on murine neuroendocrine prostate cancer, Heliyon 4(9) (2018) e00796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gao F, Alwhaibi A, Artham S, Verma A, Somanath PR, Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover, Br J Cancer 118(11) (2018) 1464–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Alwhaibi A, Verma A, Adil MS, Somanath PR, The unconventional role of Akt1 in the advanced cancers and in diabetes-promoted carcinogenesis, Pharmacological research 145 (2019) 104270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].James D, Levine AJ, Besser D, Hemmati-Brivanlou A, TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells, Development 132(6) (2005) 1273–82. [DOI] [PubMed] [Google Scholar]
- [30].Vallier L, Alexander M, Pedersen RA, Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells, J Cell Sci 118(Pt 19) (2005) 4495–509. [DOI] [PubMed] [Google Scholar]
- [31].Schier AF, Nodal signaling in vertebrate development, Annu Rev Cell Dev Biol 19 (2003) 589–621. [DOI] [PubMed] [Google Scholar]
- [32].Quail DF, Siegers GM, Jewer M, Postovit LM, Nodal signalling in embryogenesis and tumourigenesis, Int J Biochem Cell Biol 45(4) (2013) 885–98. [DOI] [PubMed] [Google Scholar]
- [33].Kalyan A, Carneiro BA, Chandra S, Kaplan J, Chae YK, Matsangou M, Hendrix MJC, Giles F, Nodal Signaling as a Developmental Therapeutics Target in Oncology, Mol Cancer Ther 16(5) (2017) 787–792. [DOI] [PubMed] [Google Scholar]
- [34].Bianco C, Strizzi L, Ebert A, Chang C, Rehman A, Normanno N, Guedez L, Salloum R, Ginsburg E, Sun Y, Khan N, Hirota M, Wallace-Jones B, Wechselberger C, Vonderhaar BK, Tosato G, Stetler-Stevenson WG, Sanicola M, Salomon DS, Role of human cripto-1 in tumor angiogenesis, J Natl Cancer Inst 97(2) (2005) 132–41. [DOI] [PubMed] [Google Scholar]
- [35].Al-Azayzih A, Gao F, Somanath PR, P21 activated kinase-1 mediates transforming growth factor beta1-induced prostate cancer cell epithelial to mesenchymal transition, Biochim Biophys Acta 1853(5) (2015) 1229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cancer N Genome Atlas Research, The Molecular Taxonomy of Primary Prostate Cancer, Cell 163(4) (2015) 1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL, Integrative genomic profiling of human prostate cancer, Cancer Cell 18(1) (2010) 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nho RS, Hergert P, FoxO3a and disease progression, World J Biol Chem 5(3) (2014) 346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A, Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT, Molecular cell 20(4) (2005) 539–50. [DOI] [PubMed] [Google Scholar]
- [40].Chen L, Kang QH, Chen Y, Zhang YH, Li Q, Xie SQ, Wang CJ, Distinct roles of Akt1 in regulating proliferation, migration and invasion in HepG2 and HCT 116 cells, Oncology reports 31(2) (2014) 737–44. [DOI] [PubMed] [Google Scholar]
- [41].Gao F, Artham S, Sabbineni H, Al-Azayzih A, Peng XD, Hay N, Adams RH, Byzova TV, Somanath PR, Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover, Cell Mol Life Sci 73(20) (2016) 3917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee CC, Jan HJ, Lai JH, Ma HI, Hueng DY, Lee YC, Cheng YY, Liu LW, Wei HW, Lee HM, Nodal promotes growth and invasion in human gliomas, Oncogene 29(21) (2010) 3110–23. [DOI] [PubMed] [Google Scholar]
- [43].Duan W, Li R, Ma J, Lei J, Xu Q, Jiang Z, Nan L, Li X, Wang Z, Huo X, Han L, Wu Z, Wu E, Ma Q, Overexpression of Nodal induces a metastatic phenotype in pancreatic cancer cells via the Smad2:3 pathway, Oncotarget 6(3) (2015) 1490–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li Y, Zhong W, Zhu M, Hu S, Su X, Nodal regulates bladder cancer cell migration and invasion via the ALK/Smad signaling pathway, Onco Targets Ther 11 (2018) 6589–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hueng DY, Lin GJ, Huang SH, Liu LW, Ju DT, Chen YW, Sytwu HK, Chang C, Huang SM, Yeh YS, Lee HM, Ma HI, Inhibition of Nodal suppresses angiogenesis and growth of human gliomas, J Neurooncol 104(1) (2011) 21–31. [DOI] [PubMed] [Google Scholar]
- [46].Fang R, Zhang G, Guo Q, Ning F, Wang H, Cai S, Du J, Nodal promotes aggressive phenotype via Snail-mediated epithelial-mesenchymal transition in murine melanoma, Cancer Lett 333(1) (2013) 66–75. [DOI] [PubMed] [Google Scholar]
- [47].Quail DF, Zhang G, Findlay SD, Hess DA, Postovit LM, Nodal promotes invasive phenotypes via a mitogen-activated protein kinase-dependent pathway, Oncogene 33(4) (2014) 461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Qi YF, Wu L, Li ZQ, Wu ML, Wang HF, Chan KY, Lu LL, Cai SH, Wang HS, Du J, Nodal signaling modulates the expression of Oct-4 via nuclear translocation of beta-catenin in lung and prostate cancer cells, Arch Biochem Biophys 608 (2016) 34–41. [DOI] [PubMed] [Google Scholar]
- [49].Guo Q, Li VZ, Nichol JN, Huang F, Yang W, Preston SEJ, Talat Z, Lefrere H, Yu H, Zhang G, Basik M, Goncalves C, Zhan Y, Plourde D, Su J, Torres J, Marques M, Al Habyan S, Bijian K, Amant F, Witcher M, Behbod F, McCaffrey L, Alaoui-Jamali MA, Giannakopoulos NV, Brackstone M, Postovit LM, Del Rincon SV, Miller WH, MNK1/NODAL signaling promotes invasive progression of breast ductal carcinoma in situ, Cancer Res (2019). [DOI] [PMC free article] [PubMed]
- [50].Fu G, Peng C, Nodal enhances the activity of FoxO3a and its synergistic interaction with Smads to regulate cyclin G2 transcription in ovarian cancer cells, Oncogene 30(37) (2011) 3953–66. [DOI] [PubMed] [Google Scholar]
- [51].Xu G, Zhong Y, Munir S, Yang BB, Tsang BK, Peng C, Nodal induces apoptosis and inhibits proliferation in human epithelial ovarian cancer cells via activin receptor-like kinase 7, J Clin Endocrinol Metab 89(11) (2004) 5523–34. [DOI] [PubMed] [Google Scholar]
- [52].Gong W, Sun B, Zhao X, Zhang D, Sun J, Liu T, Gu Q, Dong X, Liu F, Wang Y, Lin X, Li Y, Nodal signaling promotes vasculogenic mimicry formation in breast cancer via the Smad2:3 pathway, Oncotarget 7(43) (2016) 70152–70167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vo BT, Khan SA, Expression of nodal and nodal receptors in prostate stem cells and prostate cancer cells: autocrine effects on cell proliferation and migration, Prostate 71(10) (2011) 1084–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lawrence MG, Margaryan NV, Loessner D, Collins A, Kerr KM, Turner M, Seftor EA, Stephens CR, Lai J, BioResource APC, Postovit LM, Clements JA, Hendrix MJ, Reactivation of embryonic nodal signaling is associated with tumor progression and promotes the growth of prostate cancer cells, Prostate 71(11) (2011) 1198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vo BT, Cody B, Cao Y, Khan SA, Differential role of Sloan-Kettering Institute (Ski) protein in Nodal and transforming growth factor-beta (TGF-beta)-induced Smad signaling in prostate cancer cells, Carcinogenesis 33(11) (2012) 2054–64. [DOI] [PMC free article] [PubMed] [Google Scholar]