

Abstract
Gemcitabine is a widely used chemotherapeutic drug that is administered via intravenous infusion due to a low oral bioavailability of only 10%. This low oral bioavailability is believed to be the result of gemcitabine’s low intestinal permeability and oral absorption, followed by significant presystemic metabolism. In the present study, we sought to define the mechanisms of gemcitabine intestinal permeability, the potential for saturation of intestinal uptake, and the transporter(s) responsible for mediating the oral absorption of drug using in situ single-pass intestinal perfusions in mice. Concentration-dependent studies were performed for gemcitabine over 0.5 to 2000 μM, along with studies of 5 μM gemcitabine in a sodium-containing buffer ± thymidine (which can inhibit concentrative (i.e., CNT1 and CNT3) and equilibrative (i.e., ENT1 and ENT2) nucleoside transporters) or dilazep (which can inhibit ENT1 and ENT2), or in a sodium-free buffer (which can inhibit CNT1 and CNT3). Our findings demonstrated that gemcitabine was, in fact, a high-permeability drug in the intestine at low concentrations, that jejunal uptake of gemcitabine was saturable and mediated almost exclusively by nucleoside transporters, and that jejunal flux was mediated by both high-affinity, low-capacity (Km = 27.4 μM, Vmax= 3.6 pmol/cm2/s) and low-affinity, high-capacity (Km = 700 μM, Vmax= 35.9 pmol/cm2/s) transport systems. Thus, CNTs and ENTs at the apical membrane allow for gemcitabine uptake from the lumen to enterocyte, whereas ENTs at the basolateral membrane allow for gemcitabine efflux from the enterocyte to portal venous blood.
Keywords: Concentrative nucleoside transporters, Equilibrative nucleoside transporters, Gemcitabine, Intestinal permeability, Saturation kinetics
1. Introduction
Gemcitabine (2′,2′-difluoro-2′-deoxycytidine; dFdC) is a pyrimidine nucleoside analogue used in the treatment of various solid tumors [1–4]. Gemcitabine distribution and cellular uptake is mediated by the action of two evolutionarily unrelated transporter families: the concentrative nucleoside transporters (CNTs) and the equilibrative nucleoside transporters (ENTs), belonging to the solute carrier families 28 and 29, respectively [5, 6]. Specifically, gemcitabine is a substrate of the pyrimidine selective transmembrane transporter CNT1 and the broadly selective purine and pyrimidine transmembrane transporters CNT3, ENT1, and ENT2 [7, 8]. CNT1 and CNT3 function as concentrative and unidirectional sodium:substrate cotransporters, whereas ENT1 and ENT2 function as equilibrative and bidirectional sodium-independent transporters [9]. It has also been demonstrated that CNT3 can function as a proton:substrate cotransporter, albeit with altered transport activity and substrate specificity [10].
Following cellular uptake, gemcitabine undergoes phosphorylation events forming the active metabolites gemcitabine diphosphate (dFdCDP) and triphosphate (dFdCTP) [11]. dFdCTP is incorporated into DNA in place of the natural substrate deoxycytidine triphosphate, preventing chain elongation [12] and leading to apoptosis [13]. dFdCDP inhibits ribonucleotide reductase, depleting the pool of deoxynucleotide triphosphates and increasing dFdCTP incorporation into DNA [14]. Additional self-potentiating mechanisms have also been described [15].
Due to a low oral bioavailability of only 10% [16], gemcitabine is administered as an intravenous infusion, typically over 30 min at a dose of 1000 – 1250 mg/m2 once per week [17]. The factors limiting gemcitabine’s oral bioavailability have been explored in humans and mice. One study in humans highlighted the extensive first-pass metabolism of gemcitabine to 2′,2′-difluoro-2′-deoxyuridine (dFdU), via cytidine deaminase, after oral dosing of drug [16]. This study, however, quantified neither the fraction of drug absorbed from the intestinal lumen nor the fraction of drug that escaped presystemic metabolism in the gastrointestinal tract and/or liver. Studies in mice showed that tetrahydrouridine, a potent inhibitor of cytidine deaminase, could increase the oral bioavailability of gemcitabine from 10 to 40% [18]. While showing an improvement in systemic availability of gemcitabine due to enzymatic inactivation, the intestinal permeability and mechanism of oral drug absorption was not studied.
Clearly, an orally administrable form of gemcitabine would benefit patients by providing a noninvasive, patient friendly, and cost-effective alternative to intravenous drug infusions. In particular, the intestinal absorption of gemcitabine has never been investigated systematically, even though gemcitabine is a substrate of several intestinally expressed nucleoside transporters [5]. Moreover, many in vitro systems (e.g., Caco-2) used for perfunctory evaluation of gemcitabine intestinal permeability will likely provide misleading results as low expression levels of nucleoside transporters in these cells prevent an accurate evaluation of potential transporter-mediated uptake [19]. On the other hand, mice and humans show similar intestinal expression of nucleoside transporters [20], suggesting that appropriate mouse models (e.g., in situ intestinal perfusions) would better reflect the oral absorption of gemcitabine in humans as compared to cell culture systems.
With this knowledge gap in mind, the primary goals of this study were to characterize the mechanisms of gemcitabine intestinal permeability, the potential for saturation of intestinal gemcitabine uptake, and the transporter(s) responsible for mediating the oral absorption of gemcitabine using in situ single-pass intestinal perfusions in mice.
2. Materials and methods
2.1. Chemicals
Gemcitabine, dilazep hydrochloride, thymidine, and high-performance liquid chromatography (HPLC) grade acetonitrile and trifluoroacetic acid (TFA) were purchased from Thermo Fisher Scientific (Waltham, MA). The deaminated gemcitabine metabolite dFdU was purchased from Sigma-Aldrich (St. Louis, MO). Radiolabeled gemcitabine (cytosine-2-14C) (55.0 mCi/mmol), subsequently referred to as [14C]-gemcitabine, was purchased from Moravek, Inc. (Brea, CA). CytoScint™ scintillation solution was purchased from MP Biomedicals, LLC (Solon, OH). Hyamine hydroxide was purchased from PerkinElmer (Waltham, MA). All other chemicals were obtained from standard commercial sources.
2.2. Animals
Studies were performed on 8- to 12-week old gender matched C57BL/6 mice. The mice were housed and bred in a temperature-controlled room with 12-hour light/dark cycles and ad libitum access to water and a standard diet (Unit for Laboratory Animal Medicine, University of Michigan, Ann Arbor, MI). Limited validation studies were also performed in 8- to 12-week old female BALB/c mice (Charles River Laboratories, Wilmington, MA). All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
2.3. In situ single-pass intestinal perfusions
Intestinal perfusions were performed as described previously [21–23]. Prior to experimentation, mice were fasted overnight (~ 16 hr) with free access to water. The mice were then anesthetized with sodium pentobarbital (40-60 mg/kg intraperitoneal) and placed on a heating pad to maintain body temperature. The abdominal region was sterilized with 70% ethanol and the intestines exposed via a mid-line abdominal incision. An 8 cm jejunal segment, beginning 2 cm distal from the Ligament of Treitz, was isolated and glass cannulas (2.0 mm outer diameter) were inserted into each end of the segment and secured with silk sutures. The cannulated segment was rinsed with isotonic saline solution (or water in experiments using sodium-free buffer) to remove debris. The mice were then transferred to a 31°C temperature controlled chamber and their abdomen covered with saline-wetted gauze and parafilm to prevent dehydration. Inlet tubing connected the proximal cannula to a 20-mL syringe containing the perfusion solution and positioned in a perfusion pump (PHD Ultra, Harvard Apparatus, South Natick, MA). Outlet tubing connected the distal cannula to a collection vial.
The perfusion solution (pH 6.0) contained 145 mM NaCl, 0.5 mM MgCl2, 1 mM NaH2PO4, 1 mM CaCl2, 3 mM KC1, 5 mM glucose, 5 mM 2-morpholinoethanesulfonic acid (MES), and 100 μM gemcitabine. This solution was perfused through the cannulated jejunal segment at 0.1 ml/min for a total of 90 min in both C57BL/6 and BALB/c mice. After perfusing for 30 min to achieve steady-state conditions, samples of the exiting perfusate were collected at 10 min intervals for the remaining 60 min. These samples were then analyzed using ultra-performance liquid chromatography (UPLC), as described and validated below. Following experimentation, the precise length of the perfused jejunal segment was determined. For experiments in C57BL/6 mice using radiolabeled drug, the perfusion solution contained 100 μM [14C]-gemcitabine (0.5 μCi). These perfusate samples were analyzed by adding 100 μl aliquots to scintillation vials containing 6.0 mL of scintillation solution and quantifying radioactivity using a dual-channel liquid scintillation counter (Beckman LS 6000 SC, Beckman Coulter Inc., Fullerton, CA).
Concentration-dependent uptake studies were conducted in C57BL/6 mice by perfusing gemcitabine through the cannulated intestinal segment at concentrations ranging from 0.5 to 2000 μM. Specificity (i.e., inhibition) studies, also in C57BL/6 mice, were performed by perfusing 5 μM gemcitabine in the absence and presence of 2 mM thymidine or dilazep. An additional specificity study was performed in C57BL/6 mice by perfusing 5 μM gemcitabine in sodium-free buffer, prepared by replacing NaCl and NaH2PO4 in the perfusion solution with equimolar concentrations of N-methyl-D-glucamine and KH2PO4, respectively. Finally, concentration-dependent inhibition studies were performed in C57BL/6 mice where 5 μM gemcitabine was co-perfused over a wide range of inhibitor concentrations for thymidine (0.1 - 2000 μM) and dilazep (0.1 – 2500 μM).
2.4. Blood and intestinal tissue collections
Portal venous blood and intestinal tissue samples were collected from C57BL/6 mice following a standard 90 min perfusion of 5 μM [14C]-gemcitabine (0.5 μCi) alone and in the presence of 2 mM dilazep. Radioactivity was determined in portal venous plasma (nM levels) by adapting a previously described method [22]. Thus, immediately following the perfusion, portal venous blood was collected into tubes containing K3-EDTA and centrifuged for 3 min at 3000 rpm. A 30 μl plasma aliquot was then combined with 6.0 mL of scintillation solution and 20 μL of 0.5 N acetic acid. Likewise, radioactivity in intestinal tissue (pmol/mg) was determined by adapting a previously described method [24]. Following collection of portal venous blood, a jejunal segment was excised, washed with saline for 20 sec, blotted dry on filter paper, weighed, and soaked for 2 days in 0.33 mL of hyamine hydroxide at 37°C. Subsequently, 40 μL of 30% H2O2 was added to the tissue sample which was then incubated for 30 min at 60°C. After cooling to room temperature, 6.0 mL of scintillation solution and 20 μL of 0.5 N acetic acid were added to the sample. Radioactivity in the plasma and tissue samples was then determined using a dual-channel liquid scintillation counter (Beckman LS 6500 SC, Beckman Coulter Inc., Fullerton, CA).
2.5. UPLC analytical method
Inlet and outlet perfusate samples, other than those containing [14C]-gemcitabine, were assayed for gemcitabine, dFdU, and cytosine using a Waters Acquity H-Class UPLC system (Milford, MA) equipped with a photodiode array detector. dFdU was assayed since it is the primary metabolite of gemcitabine [25], whereas cytosine was reported to be formed as a gemcitabine metabolite during mouse intestinal perfusions [26]. The analytes were resolved at 40°C on an Acquity HSS T3 column (2.1 × 100 mm), fitted with an HSS T3 VanGuard precolumn (2.1 × 5 mm). Separation was achieved using a gradient elution method combining water (plus 0.1% TFA) and acetonitrile (plus 0.1% TFA) at a flow rate of 0.4 ml/min. The solvent gradient was initiated at 100% water, which changed linearly to 94% water over 3.0 min, changed linearly to 85% water over the next 2.0 min, and then returned linearly to 100% water over the final 1.0 min. Prior to analysis, perfusate samples were centrifuged for 15 min at 15000 rpm and 5 μl of the supernatant was subsequently injected onto the column via an autosampler. Under these conditions, cytosine (detection wavelength = 284 nm) eluted at ~1.0 min, gemcitabine (detection wavelength = 284 nm) at ~3.0 min, and dFdU (detection wavelength = 265 nm) at ~4.2 min.
2.6. UPLC method validation
The UPLC method was validated with respect to selectivity, linearity, accuracy, and precision. First, selectivity was established by analyzing outlet perfusate samples following perfusion of drug-free perfusion solution and evaluating the potential for endogenous compound interference with the three analytes. Next, linearity was confirmed by preparing calibration standards for gemcitabine (0.25 to 100 μM), cytosine (1 to 100 μM), and dFdU (0.25 to 5 μM). Inter- and intraday accuracy and precision were evaluated by analyzing quality control samples of gemcitabine (0.25, 25 and 100 μM), cytosine (1, 50 and 100 μM), and dFdU (0.25, 1 and 5 μM) in triplicate on three separate days. Average bias was < 6% interday and < 13% intraday, whereas the relative standard deviation was < 9% interday and < 5% intraday.
2.7. Data analysis
The effective permeability (Peff) of gemcitabine was calculated according to the parallel tube complete radial mixing model [27]:
| (1) |
where Qin represents the inlet flow rate of perfusion buffer (0.1 ml/min), the total outlet concentration of gemcitabine and dFdU, after correcting for intestinal water flux, Cin the inlet gemcitabine concentration, R the intestinal radius of the perfused segment (0.1 cm), and L the length of the perfused segment. Using the gravimetrically determined outlet flow rate (Qout), and the measured outlet concentrations of gemcitabine (Cout,gemcitabine) and dFdU (Cout,dFdU), was calculated according to [28]: disp
| (2) |
The steady-state flux (J) of gemcitabine was calculated as:
| (3) |
The concentration-dependent flux of gemcitabine was best fit to an equation consisting of two Michaelis-Menten (i.e., saturable) terms:
| (4) |
where Vmax,1 and Vmax,2 correspond to the maximum uptake rates for transport systems 1 and 2, and Km,1 and Km,2 correspond to the Michaelis constants for transport systems 1 and 2, referenced to inlet drug concentrations.
Concentration-dependent inhibition data were normalized (i.e., gemcitabine flux expressed as percent of control) and fit to Eq. 5 for thymidine inhibition or to Eq. 6 for dilazep inhibition:
| (5) |
| (6) |
where IC50 corresponds to the half maximal inhibitory concentration, I to the inhibitor concentration of thymidine or dilazep, and Bottom to the residual flux of 5 μM gemcitabine at maximum inhibition.
Data were reported as mean ± SE. When comparing two groups, statistical differences were evaluated using an unpaired t-test. Statistical differences between three or more groups were evaluated by one-way ANOVA followed by Dunnett’s test for pairwise comparisons (Prism version 7, GraphPad Software, La Jolla, California). A p value ≤ 0.05 was considered significant. Nonlinear regression was also performed using GraphPad Prism software and the quality of fits was evaluated using the coefficient of determination (r2), visual inspection of the residuals, variation of the parameter estimates, and the corrected Akaike information criterion (AICc) [29].
3. Results
3.1. Verification of experimentally-determined intestinal permeability
Following the intestinal perfusion of 100 μM gemcitabine in C57BL/6 and BALB/c mice, outlet perfusate samples contained gemcitabine and low concentrations of dFdU (less than 3%). Cytosine was absent in outlet samples following perfusion in both strains (Fig. 1). To further support our results (i.e., no other metabolites were being formed), additional intestinal perfusions were performed with 100 μM [14C]-gemcitabine. As shown in Fig. 2, the effective permeability of 100 μM gemcitabine in C57BL/6 mice was not different between the two methods, with average Peff values of 1.14 × 10−4 cm/s when analyzed by UPLC and 1.05 × 10−4 cm/s when analyzed by radioactivity. These results validate the experimental and analytical methods applied in the current study and demonstrate that gemcitabine metabolites are not confounding the findings.
Fig. 1.
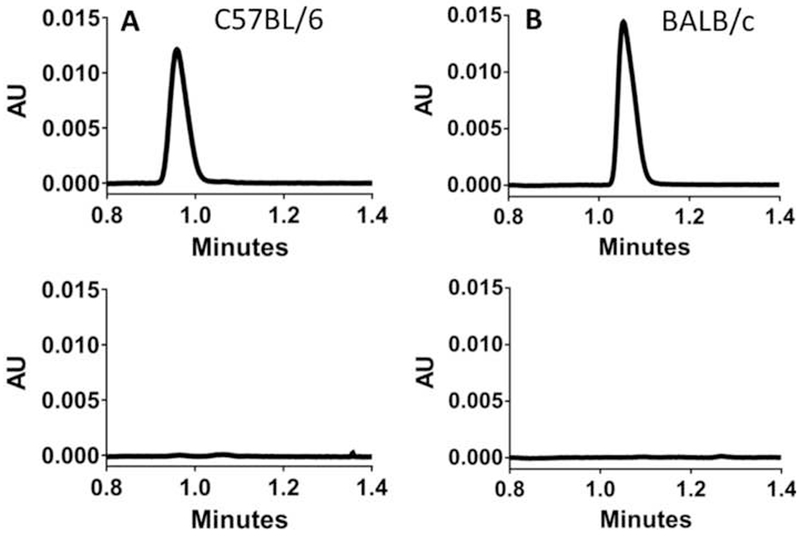
Representative chromatograms demonstrating the absence of cytosine in outlet samples following in situ jejunal perfusion of 100 μM gemcitabine in (A) C57BL/6 mice and (B) BALB/c mice. The top panels show 5 μM cytosine standards and the bottom panels show perfusion outlet samples.
Fig. 2.
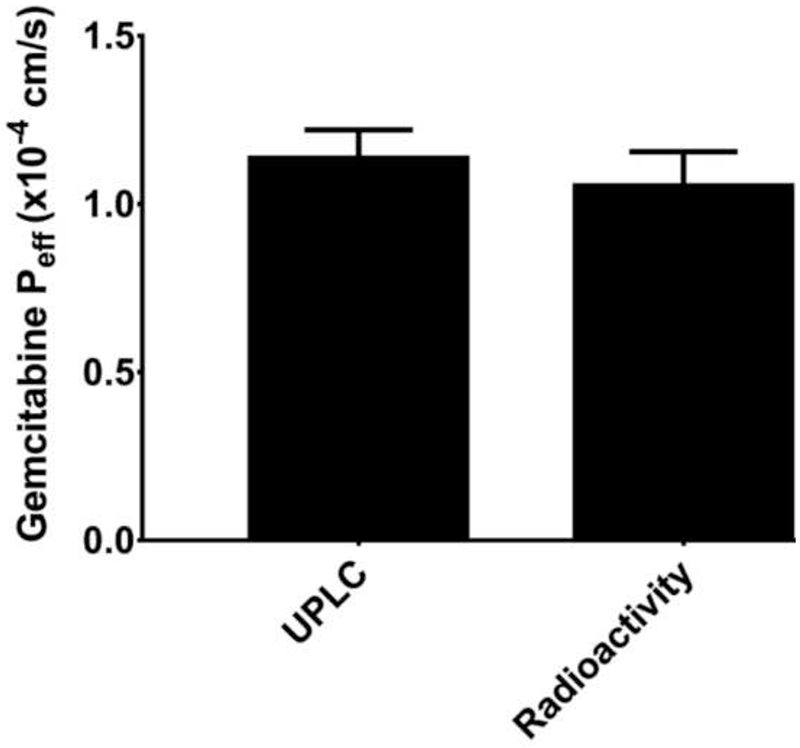
In situ jejunal permeability of gemcitabine in C57BL/6 mice when inlet and outlet concentrations of drug were measured by UPLC (after perfusions of 100 μM unlabeled gemcitabine) or total radioactivity (after perfusions of 100 μM [14C]-gemcitabine). Data are expressed as mean ± SE, n=4. There was no significant difference between the two groups, as determined by unpaired t-test.
3.2. Concentration-dependent uptake studies
To explore the potential for saturation of gemcitabine intestinal uptake, in situ jejunal perfusions were performed in C57BL/6 mice at 14 concentrations ranging from 0.5 μM to 2 mM. As shown in Fig. 3, gemcitabine displayed saturable kinetics, where the flux was best described by two Michaelis-Menten terms (Table 1). Thus, a high-affinity, low-capacity transport system was identified (Vmax,1 and Km,1) along with a low-affinity, high-capacity transport system (Vmax,2 and Km,2). Examination of the data using a Wolf-Augustinsson-Hofstee plot (Fig. 4) shows clear deviation from linearity, providing further evidence that two distinct transport systems are mediating the intestinal uptake of gemcitabine. As a result, subsequent studies were performed using inlet concentrations of 5 μM gemcitabine to ensure linear conditions.
Fig. 3.
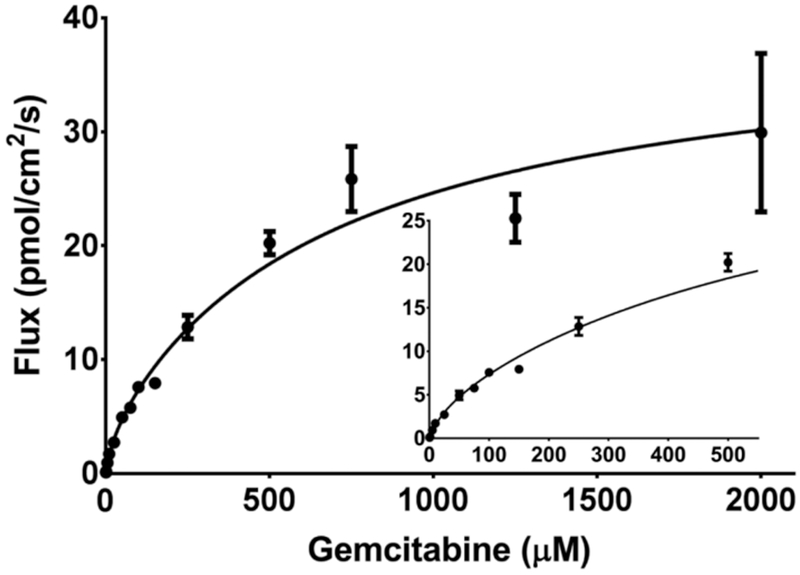
Concentration-dependent flux of gemcitabine during 0.5 μM to 2 mM in situ jejunal perfusions of drug in C57BL/6 mice. The solid line represents the predicted flux when data were fit to two Michaelis-Menten terms. The inset shows the plot at low concentrations of gemcitabine. Data are expressed as mean ± SE, n=4 (error bars may be hidden by the symbol).
Table 1.
Transport kinetics of gemcitabine during in situ jejunal perfusions in C57BL/6 mice
| Parameter | Estimate (mean ± SE) |
|---|---|
| Vmax,1 (pmol/cm2/s) | 3.6 ± 2.1 |
| Km,1 (μM) | 27.4 ± 13.9 |
| Vmax,2 (pmol/cm2/s) | 35.9 ± 5.5 |
| Km,2 (μM) | 700 ± 330 |
| r2 | 0.951 |
Gemcitabine flux was best fit to two saturable Michaelis-Menten terms where Vmax,1 Vmax,2 correspond to the maximum uptake rates for transport systems 1 and 2, and Km,1 and Km,2 correspond to the Michaelis constants for transport systems 1 and 2 (see Equation 4).
Fig. 4.
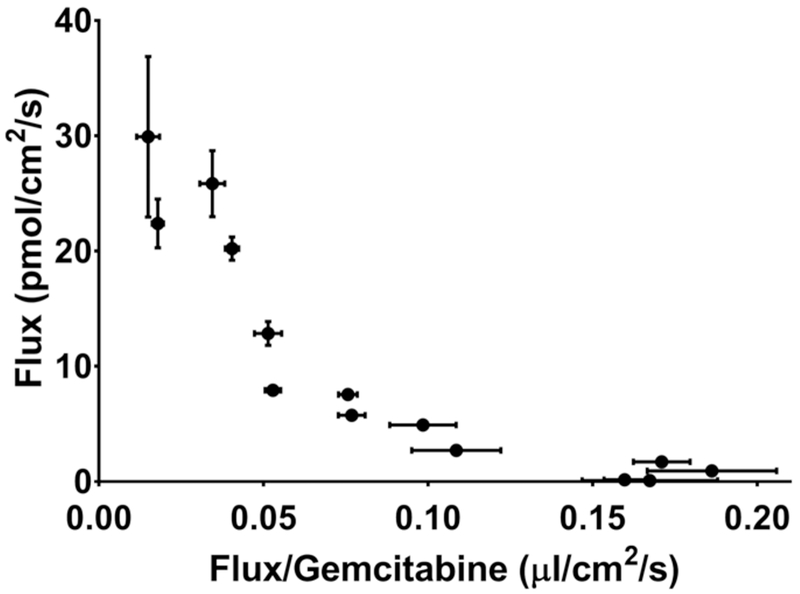
Wolf-Augustinsson-Hofstee analysis of the concentration-dependent flux of gemcitabine during 0.5 μM to 2 mM in situ jejunal perfusions of drug in C57BL/6 mice. Data are expressed as mean ± SE, n=4 (error bars may be hidden by the symbol).
3.3. Inhibition Studies
To elucidate the transporter(s) responsible for mediating the intestinal permeability of gemcitabine in C57BL/6 mice, 5 μM gemcitabine was perfused in a sodium-containing buffer with either 2 mM thymidine (which can broadly inhibit the nucleoside transporters) or dilazep (which can inhibit the nucleoside transporters ENT1 and ENT2), or in a sodium-free buffer (which can inhibit the nucleoside transporters CNT1 and CNT3). As shown in Fig. 5, the jejunal permeability of gemcitabine was reduced by about 95% when in the presence of 2 mM thymidine (i.e., residual permeability = 4.6%) and by more than 65% when drug was perfused in a sodium-free buffer (i.e., residual permeability = 31.7%). Moreover, 2 mM dilazep coperfusion reduced the jejunal permeability of gemcitabine by about 50% (i.e., residual permeability = 50.2%). Additional studies were performed in C57BL/6 mice to examine the concentration-dependent inhibition of 5 μM gemcitabine flux by thymidine and dilazep. As shown in Fig. 6, both thymidine and dilazep inhibited gemcitabine flux with IC50 values of 98.5 μM and 212 μM, respectively. Taken together, these results demonstrate that the intestinal permeability of gemcitabine is mediated almost exclusively by nucleoside transporters, with contributions by both the CNTs and ENTs.
Fig. 5.
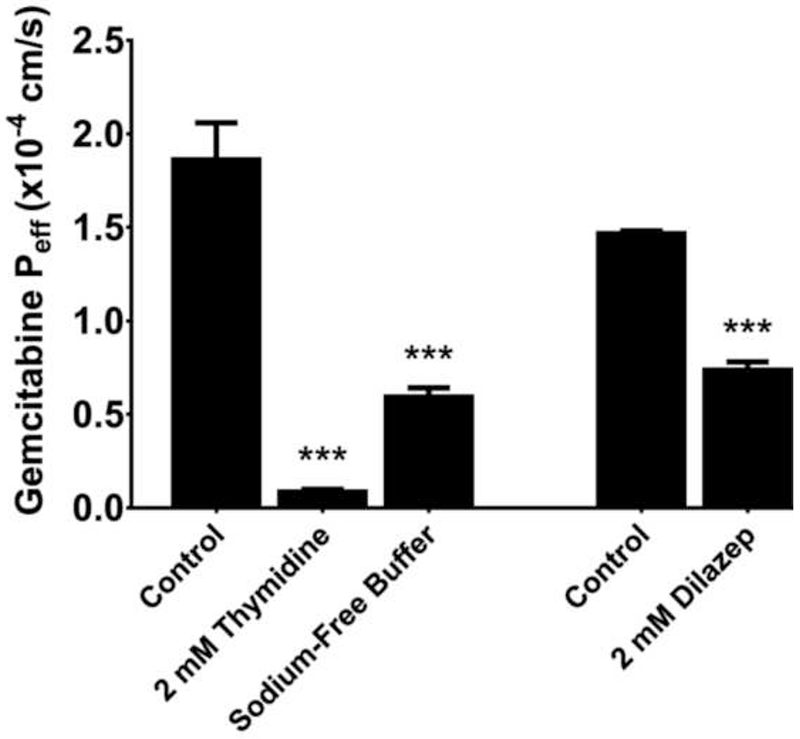
Effect of inhibitors on the intestinal permeability of 5 μM gemcitabine during in situ jejunal perfusions of drug in C57BL/6 mice. Experiments with 2 mM thymidine and sodium-free buffer were performed several months before those with 2 mM dilazep and, as a result, control values were reported for each set of experiments. Data are expressed as mean ± SE, n=4. *** p<0.001 relative to control, as determined by ANOVA followed by Dunnett’s test for thymidine and sodium-free buffer data; and by unpaired t-test for dilazep data. There was no significant difference between the two control groups, as determined by unpaired t-test.
Fig. 6.
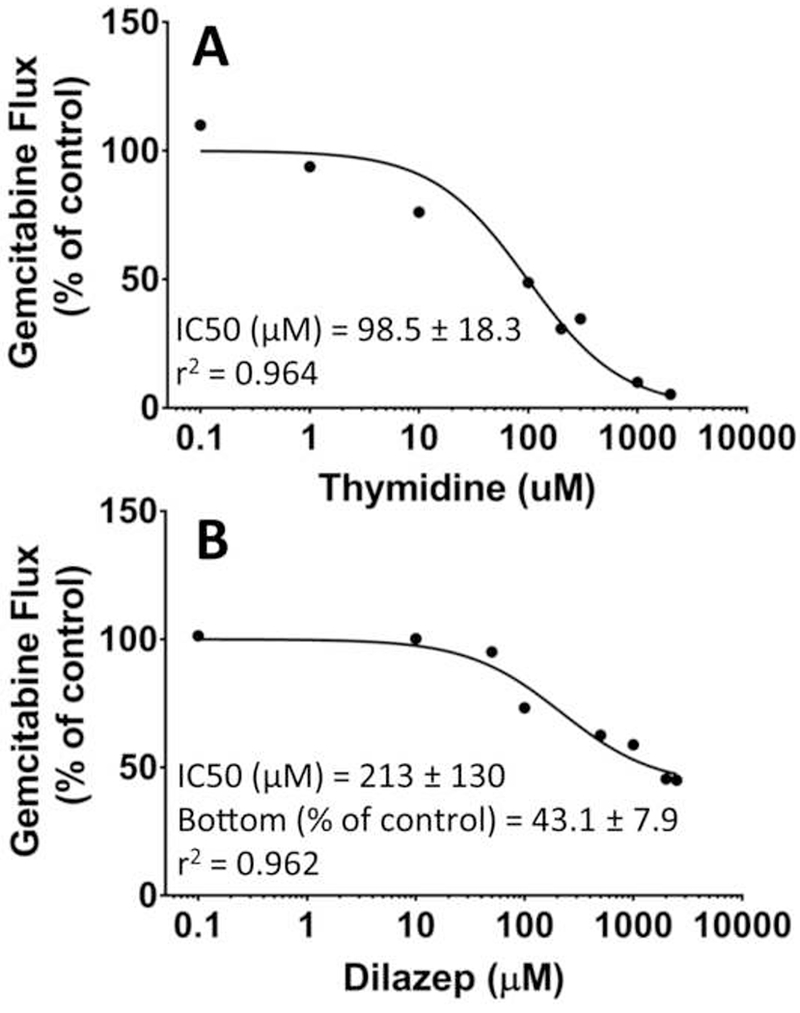
Concentration-dependent inhibition of gemcitabine flux in C57BL/6 mice during in situ jejunal perfusions of 5 μM drug alone and in the presence of (A) 0.1-2000 μM thymidine or (B) 0.1-2500 μM dilazep. The solid line represents the predicted flux when data were fitted to Eq. 5 for thymidine and to Eq. 6 for dilazep. Data are expressed as mean, n=1-4.
3.4. Accumulation of gemcitabine in intestinal tissue and portal venous plasma
In this study, 5 μM [14C]-gemcitabine was perfused in both the absence and presence of 2 mM dilazep (i.e., an ENT1 and ENT2 inhibitor) for 90 min in C57BL/6 mice, at which time jejunal tissue and portal venous blood samples were obtained. As shown in Fig. 7, total gemcitabine radioactivity in intestinal tissue was not altered by coperfusion with dilazep. However, total gemcitabine radioactivity in portal venous plasma decreased by more than 65% (i.e., % control = 32.5%).
Fig. 7.

Total radioactivity of gemcitabine and drug-related species in (A) jejunal tissue and (B) portal venous plasma following in situ jejunal perfusions of 5 μM [14C]-gemcitabine in the absence and presence of 2 mM dilazep in C57BL/6 mice. Data are expressed as mean ± SE, n=4. ***p<0.001, as determined by unpaired t-test. There was so significant difference detected in jejunal tissue radioactivity, as determined by an unpaired t-test.
4. Discussion
Various delivery techniques have been explored to increase the oral bioavailability of gemcitabine including the drug’s formulation as a prodrug [26, 30–32], its incorporation into polymeric microparticulates [33], nanoparticles [34, 35], and a self-microemulsifying drug delivery system [36], and its co-administration with a cytidine deaminase inhibitor [18]. Many of these studies aimed to increase the oral bioavailability of gemcitabine, at least partly, by increasing the drug’s purported low intestinal permeability [26, 30, 32–35]. Our studies indicated that the tenet of gemcitabine having low permeability was unfounded and, as a result, we decided to characterize systemically the drug’s mechanism of absorption, its potential for saturable intestinal uptake, and the mechanism(s) by which gemcitabine may transit through enterocytes. In doing so, our studies revealed several major findings, for the first time, including that: 1) at low concentrations, gemcitabine was a high-permeability drug in the intestine; 2) the jejunal uptake of gemcitabine was saturable and mediated almost exclusively by nucleoside transporters (~ 95%); 3) the jejunal flux of gemcitabine was mediated by two distinct transport systems, one being of high-affinity and low-capacity (i.e., CNTs) and the other being of low-affinity and high-capacity (i.e., ENTs); and 4) apically-expressed CNT(s) and ENT(s) mediate the uptake of gemcitabine into enterocytes, whereas basolaterally-expressed ENT(s) mediate the efflux of gemcitabine from enterocytes into portal venous blood.
The in situ jejunal permeability of gemcitabine was high in C57BL/6 mice (i.e., 1.5-1.9 × 10−4 cm/s at 5 μM, Fig. 5; and 1.1 × 10−4 cm/s at 100 μM, Fig. 2) and on the order of other compounds we studied with high intestinal permeability. For example, the in situ jejunal permeability (Peff) of glycylsarcosine was 1.7 × 10−4 cm/s [37], the Peff of cefadroxil was 0.6-0.8 × 10−4 cm/s [21, 22], the Peff of valacyclovir was 0.9 × 10−4 cm/s [23], and the Peff of 5-aminolevulinic acid was 1.9 × 10−4 cm/s [38]. To place these results in context, Fagerholm et al. reported that Peff values of about >0.7 × 10−4 cm/s in humans and >0.2 × 10−4 cm/s in rats will result in oral drug absorption of 90-100% in vivo [39]. The discrepancy between our current results with gemcitabine using in situ mouse perfusions (i.e., high permeability) and those using in vitro Caco-2 cells (i.e., low permeability) probably reflects the low expression of nucleoside transporters in these cell cultures [19] as well as the unphysiologic nature of in vitro systems lacking a blood supply. Interestingly, Tsume et al. [26] performed similar in situ jejunal perfusion studies with 100 μM gemcitabine in female BALB/c mice, but reported the drug to have low intestinal permeability. Specifically, their reported Peff value of 0.02 × 10−4 cm/s for gemcitabine was >50-fold lower than the value observed in our current study at the same drug concentration in mixed-gender C57BL/6 mice (no gender bias noted). Potential strain differences in gemcitabine Peff were evaluated in the current study, where the Peff 100 μM gemcitabine was not different between C57BL/6 mice (1.14 ± 0.08 × 10−4 cm/s) and BALB/c mice (1.09 ± 0.13 × 10−4 cm/s). Thus, we attribute this “apparent” discrepancy between the current work and the previously published work to the fact that previous investigators [26] corrected gemcitabine Peff for the cytosine metabolite found in their outlet samples. In contrast, no evidence of cytosine was observed in the outlet samples during our jejunal perfusions of gemcitabine in both C57BL/6 and BALB/c mice (Fig. 1).
To further validate the UPLC analytical technique used in the current work (i.e., verify all drug related metabolites were being correctly quantified), perfusions were performed in C57BL/6 mice using radiolabeled gemcitabine. Thus, the total amount of drug and drug-related species in the inlet and outlet samples could be determined by simply measuring radioactivity, without the need to specify the identity of the gemcitabine metabolite(s). The [14C]-label was present on the cytosine moiety of gemcitabine, thereby ensuring detection of any perfused gemcitabine present in the outlet as cytosine. We found no significant difference in the jejunal permeability when samples were analyzed via radioactivity and UPLC (i.e., <10%, Fig. 2), indicating that the UPLC assay was correctly identifying all relevant gemcitabine metabolites and that gemcitabine is, in fact, highly permeable in the intestine. Thus, it appears that previous investigators [26] reported an HPLC peak that was incorrectly identified as a gemcitabine metabolite, causing a substantial underestimation of gemcitabine permeability. No such peak was observed in our assay for gemcitabine and metabolites using UPLC, which has improved resolution over HPLC.
The potential for saturation of gemcitabine’s intestinal uptake along with the drug’s transport kinetics were explored by evaluating the flux of gemcitabine over a large range of perfusate concentrations (i.e., 0.5 μM to 2 mM) in C57BL/6 mice. These experiments showed a clear saturation of intestinal flux (Fig. 3), which was best described by two Michaelis-Menten terms. This mathematical fit (Table 1) suggested that both high-affinity, low-capacity (Km = 27.4 μM, Vmax= 3.6 pmol/cm2/s) and low-affinity, high-capacity (Km = 700 μM, Vmax= 35.9 pmol/cm2/s) transport systems mediated the intestinal uptake of gemcitabine. The existence of two distinct transport systems was further supported by clear nonlinearity in a Wolf-Augustinsson-Hofstee analysis of the concentration-dependent flux (Fig. 4). Based on intrinsic clearance calculations, Vmax/Km, the high- and low-affinity transport systems were predicted to account for 72% and 28% of the uptake, respectively, under linear conditions. However, as shown in Fig. 8, whereas the high-affinity transport system or CNTs dominated at lower μM concentrations, the low-affinity transport system or ENTs dominated at higher millimolar concentrations.
Fig. 8.

Contribution of the high-affinity (CNTs) and low-affinity (ENTs) transport systems toward gemcitabine flux, visualized over the three intestinal concentration ranges of (A) 0 – 100 μM, (B) 0 – 1,000 μM, and (C) 0 – 10,000 μM.
Broad inhibition of nucleoside transporters via co-perfusion of 2 mM thymidine (IC50 = 98.5 μM) reduced gemcitabine permeability by about 95% in C57BL/6 mice, showing that gemcitabine uptake is mediated almost exclusively via nucleoside transporters. Experiments in Xenopus laevis oocytes have reported Km values for gemcitabine transport via hCNTl (Km ~25 μM), hCNT3 (Km ~60 μM), hENTl (Km ~160 μM) and hENT2 (Km ~750 μM) [7, 8]. These published Km values, along with our findings showing the presence of high- and low-affinity transport systems for gemcitabine (Table 1) and that both CNTs and ENTs mediate the intestinal uptake of gemcitabine (Fig. 5), suggest that the high-affinity transport system in the current study corresponds to uptake via CNTs and the low-affinity transport system to uptake via ENTs. Furthermore, inhibition of CNTs in C57BL/6 mice (via perfusion with sodium-free buffer) reduced the intestinal permeability of gemcitabine by 68%, matching closely our predicted contribution of the high-affinity transport system (i.e., 72% of the total transport). Inhibition of ENTs in C57BL/6 mice, via coperfusion with 2 mM dilazep (IC50 = 212 μM), reduced the intestinal permeability of gemcitabine by about 50%, matching closely the predicted maximum dilazep-mediated reduction in gemcitabine flux, based upon fitting the concentration-dependent inhibition data (Fig. 6). Still, dilazep’s reduction in gemcitabine flux was greater than our predicted contribution of the low-affinity transport system (28%), with the source of this disparity remaining unclear at present.
It is widely accepted that CNT1 and CNT3 are expressed apically in the small intestine, but there is some debate regarding the localization of ENT1 and ENT2 [5]. It has been reported that ENT2 is expressed primarily on the apical membrane of Caco-2 cells [40], that ENT1 and ENT2 are expressed on both the apical and basolateral membranes of human small intestine [41], and that ENT1 is expressed only on the basolateral membrane of human intestine [5]. It is important to consider that, although a low-affinity transport system was observed in the concentration-dependent flux data and that dilazep reduced gemcitabine’s intestinal permeability, this does not necessarily imply apical expression of ENT(s). Saturation/inhibition of basolateral ENT(s) could increase intracellular gemcitabine concentrations, leading to increased repartitioning of drug from the enterocyte to lumen, thereby reducing the intestinal permeability of drug. This possibility, however, seems improbable as gemcitabine possesses very low passive permeability (Fig. 5) and thus is unlikely to repartition into the lumen to a significant extent.
Previous studies have explored the duodenal expression of nucleoside transporter mRNA in ICR mice [20] and the longitudinal expression (i.e., stomach, duodenum, jejunum, ileum, large intestine) of nucleoside transporter mRNA in C57BL/6 mice [42]. These studies showed that the gene expression of CNTs and ENTs was similar between mouse and human, that CNTs were expressed primarily in small intestine, and that ENTs were expressed in both the small and large intestines but at much lower levels than CNTs. To further address the localization of ENTs and to better understand their role in mediating the intestinal uptake (i.e., lumen to enterocyte) and efflux (i.e., enterocyte to portal blood) of gemcitabine, intestinal perfusions of 5 μM [14C]-gemcitabine were performed in C57BL/6 mice (± the ENT inhibitor dilazep), after which total radioactivity was determined in jejunal segments and portal venous plasma. Co-perfusions with 2 mM dilazep reduced the intestinal permeability of gemcitabine by about 50% (Fig. 5), showed no change in the cellular accumulation of gemcitabine radioactivity, and reduced the portal venous plasma concentration of gemcitabine radioactivity by 68% (Fig. 7). These observations demonstrate that ENTs mediate, at least in part, the basolateral transport of gemcitabine into portal venous blood as gemcitabine’s efflux from enterocytes was substantially decreased by ENT inhibition. Still, despite the reduction of gemcitabine in portal venous blood, the intracellular jejunal concentration of gemcitabine was not increased when co-perfused with dilazep. This shows that gemcitabine’s reduction in intestinal permeability after ENT inhibition is not due to increased accumulation of gemcitabine in the intestinal tissue and passive repartitioning back into lumen, but a reduction in drug uptake into epithelial cells because of apically-expressed ENTs. A proposed schematic for the transepithelial flux of gemcitabine in small intestine is presented in Fig 9.
Fig. 9.

Proposed mechanism for the absorption of gemcitabine in small intestine. The apical uptake of gemcitabine (i.e., from lumen to enterocytes) is mediated by CNTs and ENTs, whereas the basolateral efflux of gemcitabine and/or gemcitabine metabolites (i.e., from enterocytes to portal venous blood) is mediated by ENTs.
There are currently no oral dosage forms of gemcitabine. Nonetheless, it is anticipated that typical intravenous doses of 1000-1250 mg/m2, given orally, would produce millimolar levels of drug in the intestinal lumen, favoring uptake by the ENTs. However, controlled-release or divided doses would serve to reduce the luminal concentrations of drug, thereby, taking advantage of both the CNTs and ENTs. Still, it would be important to determine how gemcitabine oral formulations and/or dose rates might influence the drug’s presystemic metabolism in the gastrointestinal tract and/or liver, along with that of drug absorption.
In conclusion, novel approaches to develop orally-administered formulations of gemcitabine have been challenged by the drug’s perceived low intestinal permeability and significant presystemic metabolism. Our studies conclusively demonstrate that gemcitabine has, in fact, high permeability in the small intestine via CNT and ENT nucleoside transporters. Although gemcitabine should have low systemic availability after oral dosing, vectorial transport systems identified in the current study suggest that oral dosing (at the appropriate rate) may be advantageous for the selective targeting of gemcitabine to cancers of the intestine and liver. Furthermore, the demonstrated saturability of gemcitabine intestinal uptake highlights the importance of oral dosing rate in determining the fraction absorbed from the intestinal lumen, an important consideration for implementation of strategies aiming to enable oral gemcitabine administration by co-administration of a cytidine deaminase inhibitor.
Acknowledgments
This work was supported, in part, by the National Institutes of Health National Institute of General Medical Sciences grants R01GM115481 (to DES) and T32GM007767 (to BRT).
Abbreviations:
- CNT1
concentrative nucleoside transporter 1
- CNT3
concentrative nucleoside transporter 3
- CNTs
concentrative nucleoside transporters
- dFdC
2′,2′-difluoro-2′-deoxycytidine (or gemcitabine)
- dFdCDP
gemcitabine diphosphate
- dFdCTP
gemcitabine triphosphate
- dFdU
2′,2′-difluoro-2′-deoxyuridine
- ENT1
equilibrative nucleoside transporter 1
- ENT2
equilibrative nucleoside transporter 2
- ENTs
equilibrative nucleoside transporters
- IC50
half maximal inhibitory concentration
- Peff
effective permeability
- UPLC
ultra-performance liquid chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest, financial or otherwise.
References
- [1].Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. [DOI] [PubMed] [Google Scholar]
- [2].Pfisterer J, Plante M, Vergote I, du Bois A, Hirte H, Lacave AJ, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24(29):4699–707. [DOI] [PubMed] [Google Scholar]
- [3].Albain KS, Nag SM, Calderillo-Ruiz G, Jordaan JP, Llombart AC, Pluzanska A, et al. Gemcitabine plus Paclitaxel versus Paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26(24):3950–7. [DOI] [PubMed] [Google Scholar]
- [4].Sandler AB, Nemunaitis J, Denham C, von Pawel J, Cormier Y, Gatzemeier U, et al. Phase III Trial of Gemcitabine Plus Cisplatin Versus Cisplatin Alone in Patients With Locally Advanced or Metastatic Non-Small-Cell Lung Cancer. J Clin Oncol. 2000;18(1):122-. [DOI] [PubMed] [Google Scholar]
- [5].Young JD, Yao SY, Baldwin JM, Cass CE, Baldwin SA. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med. 2013. ;34(2-3): 529–47. [DOI] [PubMed] [Google Scholar]
- [6].Mackey JR, Mani RS, Seiner M, Mowles D, Young JD, Belt JA, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58(19):4349–57. [PubMed] [Google Scholar]
- [7].Mackey JR, Yao SY, Smith KM, Karpinski E, Baldwin SA, Cass CE, et al. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst. 1999;91(21):1876–81. [DOI] [PubMed] [Google Scholar]
- [8].Hu H, Endres CJ, Chang C, Umapathy NS, Lee EW, Fei YJ, et al. Electrophysiological characterization and modeling of the structure activity relationship of the human concentrative nucleoside transporter 3 (hCNT3). Mol Pharmacol. 2006;69(5):1542–53. [DOI] [PubMed] [Google Scholar]
- [9].Parkinson FE, Damaraju VL, Graham K, Yao SY, Baldwin SA, Cass CE, et al. Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr Top Med Chem. 2011;11(8):948–72. [DOI] [PubMed] [Google Scholar]
- [10].Smith KM, Slugoski MD, Loewen SK, Ng AM, Yao SY, Chen XZ, et al. The broadly selective human Na+/nucleoside cotransporter (hCNT3) exhibits novel cation-coupled nucleoside transport characteristics. J Biol Chem. 2005;280(27):25436–49. [DOI] [PubMed] [Google Scholar]
- [11].Heinemann V, Hertel LW, Grindey GB, Plunkett W. Comparison of the cellular pharmacokinetics and toxicity of 2′,2′-difluorodeoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1988;48(14):4024–31. [PubMed] [Google Scholar]
- [12].Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51(22):6110–7. [PubMed] [Google Scholar]
- [13].Huang P, Plunkett W. Fludarabine- and gemcitabine-induced apoptosis: incorporation of analogs into DNA is a critical event. Cancer Chemother Pharmacol. 1995;36(3):181–8. [DOI] [PubMed] [Google Scholar]
- [14].Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, et al. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol. 1990;38(4):567–72. [PubMed] [Google Scholar]
- [15].Plunkett W, Huang P, Xu Y-Z, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- [16].Veltkamp SA, Jansen RS, Callies S, Pluim D, Visseren-Grul CM, Rosing H, et al. Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study. Clin Cancer Res. 2008; 14(11):3477–86. [DOI] [PubMed] [Google Scholar]
- [17].Gemzar [package insert], Eli Lilly and Company, Indianapolis, IN: 2014. [Google Scholar]
- [18].Beumer JH, Eiseman JL, Parise RA, Joseph E, Covey JM, Egorin MJ. Modulation of Gemcitabine (2′,2′-Difluoro-2′-Deoxycytidine) Pharmacokinetics, Metabolism, and Bioavailability in Mice by 3,4,5,6-Tetrahydrouridine. Clin Cancer Res. 2008;14(11):3529–35. [DOI] [PubMed] [Google Scholar]
- [19].Takahashi K, Yoshisue K, Chiba M, Nakanishi T, Tamai I. Involvement of Concentrative Nucleoside Transporter 1 in Intestinal Absorption of Trifluridine Using Human Small Intestinal Epithelial Cells. J Pharm Sci. 2015;104(9):3146–53. [DOI] [PubMed] [Google Scholar]
- [20].Kim H-R, Park S-W, Cho H-J, Chae K-A, Sung J-M, Kim J-S, et al. Comparative gene expression profiles of intestinal transporters in mice, rats and humans. Pharmacol Res. 2007;56(3):224–36. [DOI] [PubMed] [Google Scholar]
- [21].Posada MM, Smith DE. Relevance of PepT1 in the intestinal permeability and oral absorption of cefadroxil. Pharm Res. 2013;30(4): 1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hu Y, Smith DE. Species differences in the pharmacokinetics of cefadroxil as determined in wildtype and humanized PepT1 mice. Biochem Pharmacol. 2016;107:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Epling D, Hu Y, Smith DE. Evaluating the intestinal and oral absorption of the prodrug valacyclovir in wildtype and huPepT1 transgenic mice. Biochem Pharmacol. 2018;155:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ma K, Hu Y, Smith DE. Peptide transporter 1 is responsible for intestinal uptake of the dipeptide glycylsarcosine: studies in everted jejunal rings from wild-type and Pept1 null mice. J Pharm Sci. 2011;100(2):767–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol. 1991;9(3):491–8. [DOI] [PubMed] [Google Scholar]
- [26].Tsume Y, Borras Bermejo B, Amidon GL. The dipeptide monoester prodrugs of floxuridine and gemcitabine-feasibility of orally administrable nucleoside analogs. Pharmaceuticals. 2014;7(2):169–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ho NF, Higuchi WI. Theoretical model studies of intestinal drug absorption. IV. Bile acid transport at premicellar concentrations across diffusion layer-membrane barrier. J Pharm Sci. 1974;63(5):686–90. [DOI] [PubMed] [Google Scholar]
- [28].Sutton SC, Rinaldi MTS, Vukovinsky KE. Comparison of the gravimetric, phenol red, and 14C-PEG-3350 methods to determine water absorption in the rat single-pass intestinal perfusion model. AAPS PharmSci. 2001;3(3):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hurvich CM, Tsai C-L. Regression and time series model selection in small samples. Biometrika. 1989;76(2):297–307. [Google Scholar]
- [30].Tsume Y, Incecayir T, Song X, Hilfinger JM, Amidon GL. The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acids: enhanced membrane permeability and enzymatic stability. Eur J Pharm Biopharm. 2014;86(3):514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wickremsinhe E, Bao J, Smith R, Burton R, Dow S, Perkins E. Preclinical absorption, distribution, metabolism, and excretion of an oral amide prodrug of gemcitabine designed to deliver prolonged systemic exposure. Pharmaceutics. 2013;5(2):261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang G, Chen H, Zhao D, Ding D, Sun M, Kou L, et al. Combination of l-Carnitine with Lipophilic Linkage-Donating Gemcitabine Derivatives as Intestinal Novel Organic Cation Transporter 2-Targeting Oral Prodrugs. J Med Chem. 2017;60(6):2552–61. [DOI] [PubMed] [Google Scholar]
- [33].Lim JH, You SK, Baek JS, Hwang CJ, Na YG, Shin SC, et al. Preparation and evaluation of polymeric microparticulates for improving cellular uptake of gemcitabine. Int J Nanomed. 2012;7:2307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Derakhshandeh K, Fathi S. Role of chitosan nanoparticles in the oral absorption of Gemcitabine. Int J Pharm. 2012;437(1):172–7. [DOI] [PubMed] [Google Scholar]
- [35].Chen G, Svirskis D, Lu W, Ying M, Huang Y, Wen J. N-trimethyl chitosan nanoparticles and CSKSSDYQC peptide: N-trimethyl chitosan conjugates enhance the oral bioavailability of gemcitabine to treat breast cancer. J Controlled Release. 2018;277:142–53. [DOI] [PubMed] [Google Scholar]
- [36].Hao WH, Wang JJ, Hsueh SP, Hsu PJ, Chang LC, Hsu CS, et al. In vitro and in vivo studies of pharmacokinetics and antitumor efficacy of D07001-F4, an oral gemcitabine formulation. Cancer Chemother Pharmacol. 2013;71(2):379–88. [DOI] [PubMed] [Google Scholar]
- [37].Jappar D, Wu SP, Hu Y, Smith DE. Significance and regional dependency of peptide transporter (PEPT) 1 in the intestinal permeability of glycylsarcosine: in situ single-pass perfusion studies in wild-type and Pept1 knockout mice. Drug Metab Dispos. 2010;38(10):1740–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xie Y, Hu Y, Smith DE. The proton-coupled oligopeptide transporter 1 plays a major role in the intestinal permeability and absorption of 5-aminolevulinic acid. Br J Pharmacol. 2016;173(1):167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fagerholm U, Johansson M, Lennernas H. Comparison between permeability coefficients in rat and human jejunum. Pharm Res. 1996;13(9):1336–42. [DOI] [PubMed] [Google Scholar]
- [40].Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136(2):607–18. [DOI] [PubMed] [Google Scholar]
- [41].Govindarajan R, Bakken AH, Hudkins KL, Lai Y, Casado FJ, Pastor-Anglada M, et al. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol. 2007;293(5):R1809–22. [DOI] [PubMed] [Google Scholar]
- [42].Lu H, Chen C, Klaassen C. Tissue distribution of concentrative and equilibrative nucleoside transporters in male and female rats and mice. Drug Metab Dispos. 2004;32(12): 1455–61. [DOI] [PubMed] [Google Scholar]