

Abstract
ELL2 is a potential tumor suppressor in prostate cancer. ELL2 knockout in mice induced mPIN, the putative precursor of prostate cancer and ELL2 knockdown enhanced proliferation in cultured prostate cancer cells. To explore the mechanism of ELL2 action in prostate cancer, we investigated the role of Birc3, an apoptosis inhibitor, in prostate cancer cells and the regulation of its expression by ELL2. ELL2 knockdown enhanced Birc3 expression in LNCaP and C4-2 cell line models. BrdU assay showed that Birc3 knockdown inhibited proliferation, ELL2 knockdown enhanced proliferation, and Birc3 knockdown counteracted ELL2 knockdown-induced proliferation in LNCaP cells. Trypan blue assay suggested that Birc3 knockout did not induce cell death in LNCaP cells. These findings suggested that Birc3 is a downstream gene of ELL2 and may play a role in driving prostate cancer proliferation.
Keywords: Birc3, ELL2, prostate cancer, LNCaP, western blot, qPCR, BrdU
Introduction
Prostate cancer is one of the most common malignant tumors in men, particularly in aging men. As life expectancy increases, the incidence and mortality rates of prostate cancer are expected to rise. It was estimated that there were 164,690 new cases of prostate cancer and 29,430 death from this disease in 2018 in the United States, which makes prostate cancer the most frequently diagnosed cancer and the second leading cause for cancer-related death in US males [1]. However, the pathogenesis and molecular mechanisms of prostate cancer remain incompletely understood. Defining the mechanisms involved in prostate cancer initiation and progression may lead to better prevention and/or treatment of this disease.
The eleven-nineteen-Lys-rich leukemia (ELL) family consists of ELL, ELL2 and ELL3. ELL proteins are RNA Pol II elongation factors that can trigger basal transcription, promote the catalytic rate of transcription in vitro, stabilize RNA Pol II, and assemble the other super elongation complex (SEC) members [2]. ELL2 is involved in multiple biological functions [3-7]; and was identified as an essential component of the SEC and the little elongation complex (LEC) [8,9]. ELL2 can bind to MED26 [10], which plays a role in the regulation of transcription activity. ELL2 is also closely associated with ELL-associated factor 2 (EAF2) [11], and both ELL2 and EAF2 have been shown to act as potential prostate cancer tumor suppressors [12-17]. Prostate-specific deletion of ELL2 in mice induced prostatic lesions, such as murine prostatic intraepithelial neoplasia (mPIN), increased vascular density, and cell proliferation [12]. ELL2 expression was significantly decreased in high Gleason grade prostate tumor specimens and metastasis lesions [14]. ELL2 and EAF2 have been shown to physically interact with two important tumor suppressors in prostate cancer, p53 and Rb [14,18,19]. These findings suggested a tumor suppressive role of ELL2 in the prostate. However, the mechanisms of ELL2 action in the prostate are unclear.
Baculoviral IAP repeat-containing protein3 (Birc3), also called cellular inhibitor of apoptosis (cIAP2), belongs to the IAP family of proteins that contains a BIR domain. There are eight proteins in human IAP family: NAIP (BIRC1), c-IAP1 (BIRC2), c-IAP2 (BIRC3), XIAP (BIRC4), survivin (BIRC5), Apollon/Bruce (BIRC6), ML-IAP (BIRC7 or livin) and ILP-2 (BIRC8) [20]. Initially, IAPs were found as components of the tumor necrosis factor receptor 2 (TNFR2) complex via binding to TRAF1 and TRAF2 [21]. cIAPs were thought to protect cells from apoptosis through inactivating caspase such as caspase-3 and -7 directly or indirectly. Down-regulation of Birc3 enhances apoptosis by activating caspase-7. cIAP proteins could function as ubiquitin E3 ligases, via their RING domains to mediate proteasomal degradation of their target proteins, such as RIP1, TRAF2, and NF-kB-inducing kinase (NIK) [22]. Overexpressed Birc3 or disruption of Birc3 in malignant tumor could contribute to disease progression and chemotherapy resistance [23-28]. In chronic lymphocytic leukemia, patients with mutant Birc3 suffered worse overall response to fludarabine-based chemotherapy compared to those with wild-type Birc3 [29]. Jiang, et al., [24] reported Birc3 was up-regulated in gallbladder cancer, while a significant association was found between lymph node metastasis, prognosis and overexpression of Birc3. Birc3 was also reported to elevate in mouse and human osteosarcomas, which played an important role in anti-apoptosis and enhance of tumor growth of tumor cells [26]. Also, mRNA expression analyses demonstrated that the p53+/- osteosarcomas and rhabdomyosarcomas mouse model exhibited high expression of Birc3 mRNA, as well as p53ME-/- and p53ME-/- RbME-/- mammary carcinoma xenograft mice [26,30]. It was reported that Birc3 could regulate the chemotherapy sensitivity of prostate cancer [28,31]. Our recent studies showed a trend of Birc3 up-regulation by knockdown of EAF2, a binding partner of ELL2 [32]. Overall, there are few reported studies on Birc3 in prostate cancer, and its role in prostate carcinogenesis is not clear.
In this study, we explored ELL2 regulation of Birc3 expression and potential role of Birc3 in prostate cancer cell proliferation and death. Our findings suggest that Birc3 plays a potentially important role in ELL2 signaling and in prostate cancer progression.
Materials and methods
Cell culture, knockdown and western blotting
The LNCaP, VCaP, and 22Rv1 cell lines were purchased from ATCC. C4-2 was from Dr. Leland W.K. Chung (Cedars-Sinai, Los Angeles, CA) and LN95 was from Dr. Jun Luo (Johns Hopkins University, Baltimore, MD). LNCaP, 22Rv1 and C4-2 were cultured in RPMI-1640 (10-040-CV, Corning Cellgro, Corning, NY), while VCaP was maintained in DMEM (12-604F, Lonza, Basel, Switzerland). The LN95 cells were cultured in phenol red-free RPMI-1640 medium supplemented with 10% charcoal-stripped FBS. All medium was complemented with 10% fetal bovine serum (FBS), 1% glutamine plus 1% penicillin/streptomycin (10378016, Gibco, Waltham, MA). Cells were incubated at 37°C in the atmosphere of 85% humidity with 5% CO2. The medium was changed every two days.
For knockdown, cells were seeded in 6-well plate at 24 h before knockdown, of which the density was 3×105/well. The siRNA for Birc3, ELL2 and control (D-001810-10-50, Dharmacon, Lafayette, CO) were obtained from Dharmacon. The sequences of the siRNA oligonucleotides were Birc3-1: sense, 5’-UAAGGGAAGAGGAGAGAGAA-3’; antisense, 5’-UUCUCUCUCCUCUUCCCUUA-3’; Birc3-2: sense, 5’-CAAGGUGUGAGUACUUGAUAAGAAT-3’, antisense, 5’-AUUCUUAUCAAGUACUCACACCUUGGA-3’, siELL2-1: sense, 5’-CUCACAUCCUCCUCAGAUUGUAAAU-3’, antisense, 5’-AUUUACAAUCUGAGGAGGAUGUGAGAU-3’, siELL2-2: sense, 5’-CUCACAUCCUCCUCAGAUUGUAAAT-3’, antisense, 5’-AUUUACAAUCUGAGGAGGAUGUGAGAU-3’. DharmaFECT Transfection Reagent (T-2001-02, Dharmacon) was used for siRNA delivery. LNCaP cells in each well were incubated in antibiotics-free medium for 0.5-1 hour prior to the knockdown initiation. Cells were transfected with 25 nM siRNA, using 5 μl DharmaFECT Transfection Reagent in antibiotics-free, bovine serum-free medium. Nontarget siControl was added to each single knockdown well to guarantee the identical siRNA (25 nM) amount in every group. After transfection (24 hour), the medium was replaced and cells were incubated for an additional 24 hours for RNA extraction or 48 hours for western blotting.
For Western blotting, cells were lysed in 70 μl lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton x-100, 1% Sodium deoxycholate, 0.1% SDS, 1 mM EDTA plus protease inhibitors cocktail (P8340, SIGMA, St. Louis, MO) in each well after washing twice in PBS. The concentration of all protein samples was determined using BCA protein quantification assay and a total of 30 μg protein for each sample was loaded into 8% SDS polyacrylamide gel for electrophoresis. After protein electro-transfer, membranes were probed using antibodies against ELL2 (A302-505A, Bethyl Laboratories, Montgomery, TX), Birc3 (3130, Cell Signaling Technology, Danvers, MA), or GAPDH (sc-47724, Santa Cruz Biotechnology, Dallas, Texas). Western blot signals were detected using ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Hercules, CA) after incubation with primary antibodies at 4°C overnight and subsequently with peroxidase-conjugated secondary antibody.
Real-time reverse transcription quantitative PCR (qPCR)
Total RNA was purified using the RNeasy Mini kit (74104, Qiagen, Germantown, MD) according to the manufacturer’s instruction. The concentration of purified RNA was measured by NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). For reverse transcription, 500 ng of RNA was applied with 2 μl of PrimeScript™ RT Master Mix (RR036A, Takara Bio, Mountain View, CA) in a total volume of 10 μl. Expressions levels of the genes Birc3, ELL2, and GAPDH were determined using StepOne Real-Time PCR System (Life Technologies, Waltham, MA) with Real-time qPCR Master Mix (639676, Takara Bio, Mountain View, CA). The primer sequences for qPCR were as followed: ELL2: Forward Primer, TGACTGCATCCAGCAAACAT; Reverse Primer, TCGTTTGTTGCACACACTGTAA. Birc3: Forward Primer, GACTCAGGTGTTGGGAATCTG; Reverse Primer, GACTCAGGTGTTGGGAATCTG. GAPDH: sense, CATGTTCGTCATGGGTGTGA; antisense, GGTGCTAAGCAGTTGGTGGT. The PCR reactions were carried out in a total volume of 20 μl containing 10 μl TB Green Advantage qPCR Premix (2×), 0.4 μl PCR Forward Primer (10 µM), 0.4 μl PCR Reverse Primer (10 µM), 0.4 μl ROX Reference Dye LSR (50×), 2 μl Template, and 6.8 μl dH2O. The expression of GAPDH was set as an internal control.
BrdU assay
Cells plated in 24-well plates were treated with 50 nM siRNA for 72 hr and labeled with bromodeoxyuridine (BrdU) for 16 h subsequently. Afterwards, cells were fixed by Carnoy’s fixative (3:1 methanol:glacial acetic acid) for 20 minutes at -20°C, incubated with 2 M HCl for 1 h at 37°C, and then washed three times using 0.1 M boric acid. Cells were then treated with 3% hydrogen peroxide for 20 minutes at 37°C and blocked in 10% goat serum for 1 hour at 37°C. Anti-BrdU antibody (MoBU-1, Santa Cruz Biotechnology, Dallas, Texas) was used to probe BrdU-labeled cells incubated overnight at 4°C, and cells were incubated with CY3-labeled goat anti-mouse secondary antibody (A 10521, Life Technologies, Waltham, MA) for 1 hour at 37°C. Finally, 1 μM SYTOX Green (S7020, Life Technologies, Waltham, MA) or DAPI (D9542, Sigma-Aldrich, St. Louis, MO) was used to stain the nuclei for 15 minutes at room temperature. Images were obtained by a fluorescence microscope (TE2000-U, Nikon, Tokyo, Japan) equipped with a Hamamatsu Orca-Flash4.0LT Digital Camera (C11440, Hamamatsu Corp, Sewickley, PA) and NIS-Elements Documentation v 4.6 software (Nikon Instruments, Inc., Melville, NY). The ratio of BrdU-labeled cells to SYTOX GREEN-stained cells, reflecting the percentage of BrdU-positive cells was determined.
Trypan blue exclusion test
Cells were seeded at a density of 3×105 cells/well in 6-well plates and transfected with indicated siRNAs. Cells were harvested 72 hours after the transfection using Trypsin digestion. The harvested cells were stained with Trypan blue solution (15250061, Gibco, Waltham, MA) at a ratio of 9:1 and counted using a hemocytometer to determine the percentage of dead cells by counting the number of unstained live cells and stained dead cells.
Statistics
Statistical analysis was conducted using SPSS software 19.0 (IBM, Armonk, NY). Data were presented as mean ± standard deviation (SD). GraphPad Prism version 6 was used for graphics (GraphPad Software, San Diego, CA, USA). The differences between groups were analyzed using Student’s t-test. A difference was considered statistically significant when P value was less than 0.05.
Results
ELL2 knockdown up-regulated Birc3 expression in LNCaP and C4-2 prostate cancer cell lines
To explore if ELL2 could modulate Birc3 expression, we tested the effect of ELL2 knockdown on the expression of Birc3 in C4-2 and LNCaP, two widely used prostate cancer cell lines. Knockdown of ELL2 using two different siRNA targeting different regions of ELL2 mRNA caused up-regulation of Birc3 mRNA in both C4-2 and LNCaP cells (Figure 1). Birc3 mRNA level relative to GAPDH mRNA in C4-2 was at 0.0002-0.0004, more than 10 times lower than that in LNCaP cells. Western blot analysis showed that Birc3 protein level was also much lower in C4-2 relative to LNCaP (Figure 2). In fact, LNCaP cells expressed the highest Birc3 protein level among all the tested prostate cancer cell lines (Figure 2). Therefore, we used the LNCaP model in this study to test the role of endogenous Birc3, particularly in response to ELL2 knockdown.
Figure 1.
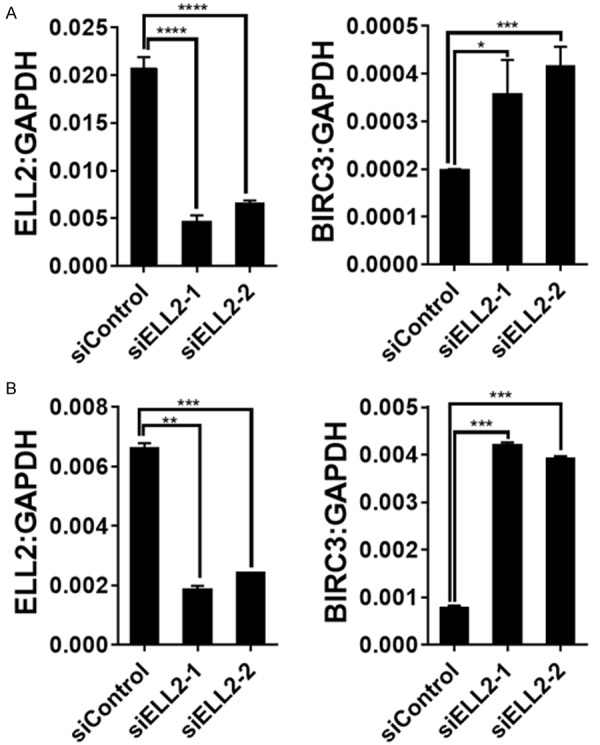
The relative mRNA expression levels of Birc3 and ELL2 in ELL2-knockdown C4-2 (A) and LNCaP (B) were measured by quantitative RT-PCR. The data are normalized to GAPDH and shown as the 2^ΔCt values.
Figure 2.
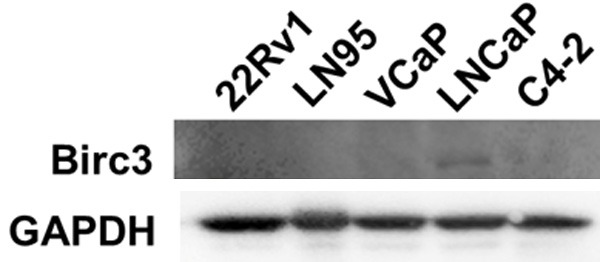
The protein expression level of Birc3 in different prostate cancer cell lines was measured by Western-blotting. GAPDH served as a loading control.
Birc3 knockdown inhibited LNCaP cell proliferation
To explore the potential function of Birc3 in prostate cancer cells, we knocked down Birc3 expression using two different siRNAs targeting Birc3 and a control siRNA and then performed BrdU assay to evaluate the effect of Birc3 loss on LNCaP cell proliferation. Figure 3 showed that both siBirc3-1 and siBirc3-2 treatment caused a reduction in BrdU-positive cells as compared to siControl treatment. This observation suggested a stimulating role of Birc3 in LNCaP cell proliferation.
Figure 3.
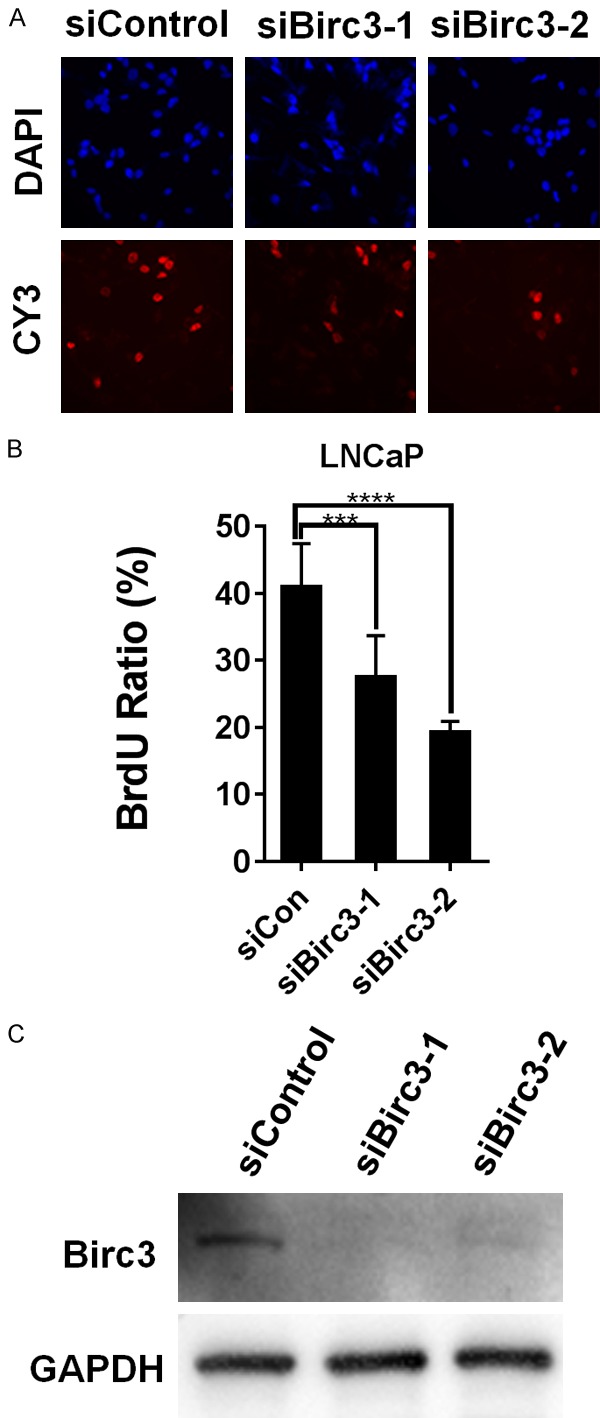
Knockdown of Birc3 can strongly decrease LNCaP cell proliferation. (A) LNCaP cells were transfected with either two different siRNA targeting Birc3 or nontarget siRNA (siCon), cell proliferation rates were determined using BrdU Incorporation assay. Upper panels show nuclear staining with DAPI (Blue), and lower panels show BrdU-positive nuclei stained with CY3 (red). (B) Quantification column graph of BrdU incorporation shown in (A) as mean precentage of BrdU-positive cells relative to the total number of cells. (C) Knockdown efficiency confirmed by Western-blotting. GAPDH served as a loading control.
Birc3 knockdown counteracted cell proliferation induced by ELL2 loss
Previously, we showed that ELL2 knockdown enhanced proliferation in LNCaP cells [12]. Since ELL2 knockdown up-regulated Birc3 expression and Birc3 appeared to play a growth-promoting role in LNCaP cells (Figures 1 and 3), up-regulation of Birc3 by ELL2 knockdown is likely to contribute to the increased LNCaP proliferation upon ELL2 knockdown. To examine the role of Birc3 in LNCaP growth stimulation by ELL2 loss, LNCaP cells were transfected with siControl, siELL2-1/2, siBirc3-1/2, or both siELL2 and siBirc3. As expected, with knockdown by two different sets of siRNA targeting ELL2 and Birc3, siELL2 enhanced cell proliferation and siBirc3 reduced proliferation in the absence or presence of siELL2 (Figure 4). As a control, the western-blot analysis showed that ELL2 knockdown up-regulated Birc3 protein level and siBirc3 decreased Birc3 induction by ELL2 knockdown (Figure 4E, 4F). The above finding suggested that ELL2 knockdown induction of LNCaP cell proliferation is in part mediated through Birc3 up-regulation.
Figure 4.
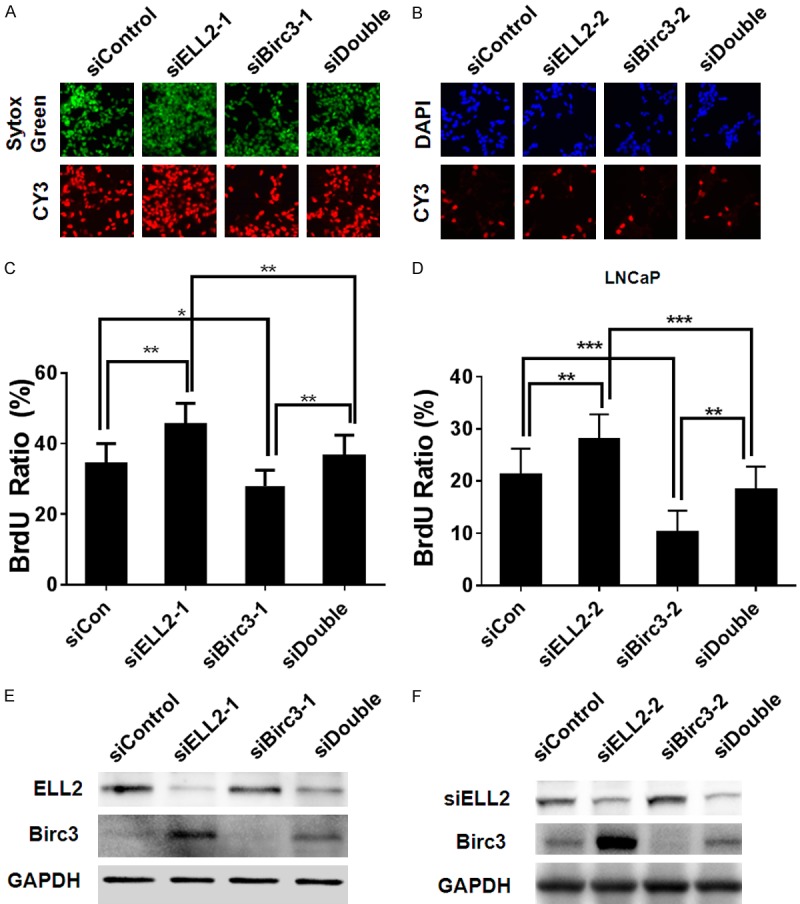
Effect of Birc3 and ELL2 knockdown on LNCaP proliferation. (A) BrdU applied in LNCaP cells transfected with different nontarget siRNA (siCon), siELL2, siBirc3, or concurrent ELL2 and Birc3 knockdown (siDouble). Upper panel shows nuclear staining with Sytox Green (green), and lower panel shows BrdU-positive nuclei stained with CY3 (red). (B) Upper panel shows nuclear staining with DAPI (blue), and lower panel shows BrdU-positive nuclei stained with CY3 (red). (C) Quantification column graphs of BrdU experiment shown in (A) as mean precentage of BrdU-positive cells relative to the total number of cells. (D) Quantification of BrdU experiment shown in (B). (E, F) Knockdown efficiency confirmed by Western-blotting. The expression of Birc3 protein in LNCaP cell was suppressed by siRNA targeted to Birc3. GAPDH served as a loading control.
Birc3 knockdown does not induce cell death in LNCaP cells
Since Birc3 is an anti-apoptosis protein, we tested if Birc3 knockdown could affect LNCaP cell death. Using trypan blue staining, Figure 5A showed that knockdown of ELL2 and Birc3 individually or in combination had no detectable effect on LNCaP cell death. Also, the knockdown of ELL2 and Birc3 individually or in combination had minimal effect on caspase 7 protein level and did not induce the cleaved caspase 7 (Figure 5B). This finding suggested that Birc3 does not have significant influence on prostate cancer cell death.
Figure 5.
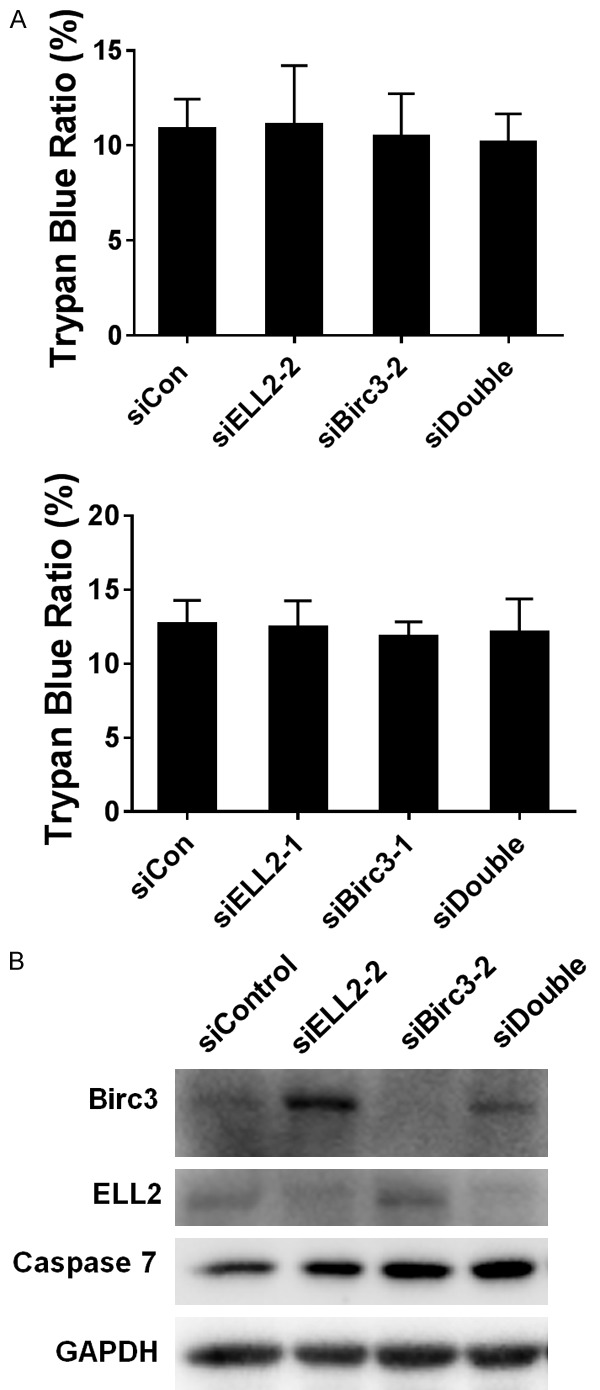
Birc3 and ELL2 knockdown does not induce cell death in LNCaP cells. A. Cell death of the LNCaP cell line after ELL2, Birc3 and combination knockdown. Cell death was determined by trypan-blue exclusion of the treated cells. Control and siRNA-transfected cells are indicated below the bars. B. Caspase-7 expression change during siRNA treatment. LNCaP cell was transfected with siRNA for 72 h, and then caspase-7 was analyzed by Western blotting. GAPDH was used as a loading control.
Discussion
The present study presented evidence that ELL2 knockdown induced Birc3 mRNA and protein expression and that Birc3 knockdown inhibited cell proliferation in the LNCaP cell line model. These observations suggested a growth-promoting role of Birc3 in prostate cancer progression and in ELL2 suppression of prostate cancer cell proliferation.
ELL2 is a potential tumor suppressor in the prostate because prostate-specific deletion of ELL2 in mice induced prostatic epithelial hyperplasia and mPIN and Ell2 knockdown in cultured LNCaP cells enhanced cell proliferation [12]. However, the downstream signaling pathways mediating ELL2 suppression of prostate cancer are not clear. Birc3 is involved in multiple processes, including facilitation of cell viability, radio/chemotherapy resistance and patient survive [24,33-36]. The observation that cells with ELL2 and Birc3 double knockdown proliferated slower than the cells with ELL2 knockdown alone suggested Birc3 up-regulation is required for ELL2 knockdown-stimulated cell proliferation. This suggested that Birc3 is one down-stream gene of ELL2 capable of mediating ELL2 suppression of LNCaP prostate cancer cell proliferation. However, additional pathways are also involved in ELL2 suppression of LNCaP cell proliferation, because cells with ELL2 and Birc3 double knockdown exhibited higher proliferation rate than Birc3 knockdown alone (Figure 4). Since Birc3 expression level was high in LNCaP but low in other prostate cancer cell lines (Figure 2), Birc3 is likely to be important in a subset of prostate cancers.
Birc3 plays an important role as an anti-apoptosis factor through direct binding to Caspase-3, -7 and -9 and promoting ubiquitination of TRAF2, TRAF3 to repress apoptosis pathways and molecules [37]. However, our studies did not find Birc3 regulation of cell death and activation of Caspase-7 in LNCaP cells. Instead, Birc3 knockdown inhibited LNCaP cell proliferation (Figure 3). This suggested that Birc3 plays an important role in regulating cell proliferation rather than apoptosis in LNCaP cells. Previous studies showed that Birc3 enhances cell growth using MTT or CKK8 assays [24,38,39], which suggested that Birc3 could regulate cell growth but did not rule out the possibility that Birc3 enhancement of cell growth is mediated through inhibition of apoptosis. Our finding provided strong evidence for the role of Birc3 in regulation of cell proliferation, in addition to its role as an apoptosis inhibitor. It is possible that Birc3 may regulate proliferation in other cells and future studies will be needed to determine the importance and mechanisms of Birc3 regulation of cell proliferation.
In summary, this study showed that Birc3, an apoptosis inhibitor, can regulate prostate cancer cell proliferation, and is involved in ELL2 regulation of prostate cancer cell proliferation. Birc3 is overexpressed in a subset of prostate cancers and is likely to play an important role in these prostate cancers. Birc3 may be a therapeutic target for these prostate cancers.
Acknowledgements
We are grateful to Aiyuan Zhang and Jianhua Zhou for technical support. This project was supported in part by NIH grant R01 CA186780 (ZW) and R50 CA211242 (LEP). Zhi Wang received a scholarship from the China Scholarship Council.
Disclosure of conflict of interest
The first author, Zhi Wang, who performed the entirety of this study in Pittsburgh, is a Visiting Scholar to the University of Pittsburgh from Xiangya Hospital of Central South University in China.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Byun JS, Fufa TD, Wakano C, Fernandez A, Haggerty CM, Sung MH, Gardner K. ELL facilitates RNA polymerase II pause site entry and release. Nat Commun. 2012;3:633. doi: 10.1038/ncomms1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou J, Feng X, Ban B, Liu J, Wang Z, Xiao W. Elongation factor ELL (Eleven-Nineteen Lysine-rich Leukemia) acts as a transcription factor for direct thrombospondin-1 regulation. J Biol Chem. 2009;284:19142–19152. doi: 10.1074/jbc.M109.010439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qi S, Li Z, Schulze-Gahmen U, Stjepanovic G, Zhou Q, Hurley JH. Structural basis for ELL2 and AFF4 activation of HIV-1 proviral transcription. Nat Commun. 2017;8:14076. doi: 10.1038/ncomms14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martincic K, Alkan SA, Cheatle A, Borghesi L, Milcarek C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat Immunol. 2009;10:1102–1109. doi: 10.1038/ni.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park KS, Bayles I, Szlachta-McGinn A, Paul J, Boiko J, Santos P, Liu J, Wang Z, Borghesi L, Milcarek C. Transcription elongation factor ELL2 drives Ig secretory-specific mRNA production and the unfolded protein response. J Immunol. 2014;193:4663–4674. doi: 10.4049/jimmunol.1401608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu M, Hsu J, Chan C, Li Z, Zhou Q. The ubiquitin ligase Siah1 controls ELL2 stability and formation of super elongation complexes to modulate gene transcription. Mol Cell. 2012;46:325–334. doi: 10.1016/j.molcel.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012;32:2608–2617. doi: 10.1128/MCB.00182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, Florens L, Seidel CW, Lin C, Smith ER, Shilatifard A, Conaway RC, Conaway JW. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell. 2011;146:92–104. doi: 10.1016/j.cell.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong SE, Banks CA, Shilatifard A, Conaway JW, Conaway RC. ELL-associated factors 1 and 2 are positive regulators of RNA polymerase II elongation factor ELL. Proc Natl Acad Sci U S A. 2005;102:10094–10098. doi: 10.1073/pnas.0503017102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pascal LE, Masoodi KZ, Liu J, Qiu X, Song Q, Wang Y, Zang Y, Yang T, Wang Y, Rigatti LH, Chandran U, Colli LM, Vencio RZN, Lu Y, Zhang J, Wang Z. Conditional deletion of ELL2 induces murine prostate intraepithelial neoplasia. J Endocrinol. 2017;235:123–136. doi: 10.1530/JOE-17-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong M, Pascal LE, Cheng E, Masoodi KZ, Chen W, Green A, Cross BW, Parrinello E, Rigatti LH, Wang Z. Concurrent EAF2 and ELL2 loss phenocopies individual EAF2 or ELL2 loss in prostate cancer cells and murine prostate. Am J Clin Exp Urol. 2018;6:234–244. [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu X, Pascal LE, Song Q, Zang Y, Ai J, O’Malley KJ, Nelson JB, Wang Z. Physical and Functional Interactions between ELL2 and RB in the Suppression of Prostate Cancer Cell Proliferation, Migration, and Invasion. Neoplasia (United States) 2017;19:207–215. doi: 10.1016/j.neo.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang T, Jing Y, Dong J, Yu X, Zhong M, Pascal LE, Wang D, Zhang Z, Qiao B, Wang Z. Regulation of ELL2 stability and polyubiquitination by EAF2 in prostate cancer cells. Prostate. 2018;78:1201–1212. doi: 10.1002/pros.23695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao W, Zhang Q, Jiang F, Pins M, Kozlowski JM, Wang Z. Suppression of prostate tumor growth by U19, a novel testosterone-regulated apoptosis inducer. Cancer Res. 2003;63:4698–4704. [PubMed] [Google Scholar]
- 17.Xiao W, Zhang Q, Habermacher G, Yang X, Zhang AY, Cai X, Hahn J, Liu J, Pins M, Doglio L, Dhir R, Gingrich J, Wang Z. U19/Eaf2 knockout causes lung adenocarcinoma, B-cell lymphoma, hepatocellular carcinoma and prostatic intraepithelial neoplasia. Oncogene. 2008;27:1536–1544. doi: 10.1038/sj.onc.1210786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Pascal LE, Zhong M, Ai J, Wang D, Jing Y, Pilch J, Song Q, Rigatti LH, Graham LE, Nelson JB, Parwani AV, Wang Z. Combined loss of EAF2 and p53 induces prostate carcinogenesis in male mice. Endocrinology. 2017;158:4189–4205. doi: 10.1210/en.2017-00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su F, Pascal LE, Xiao W, Wang Z. Tumor suppressor U19/EAF2 regulates thrombospondin-1 expression via p53. Oncogene. 2010;29:421–431. doi: 10.1038/onc.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Almagro MC, Vucic D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp Oncol. 2012;34:200–211. [PubMed] [Google Scholar]
- 21.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 22.Kim JY, Morgan M, Kim DG, Lee JY, Bai L, Lin Y, Liu ZG, Kim YS. TNFalpha induced noncanonical NF-kappaB activation is attenuated by RIP1 through stabilization of TRAF2. J Cell Sci. 2011;124:647–656. doi: 10.1242/jcs.075770. [DOI] [PubMed] [Google Scholar]
- 23.Miura K, Karasawa H, Sasaki I. cIAP2 as a therapeutic target in colorectal cancer and other malignancies. Expert Opin Ther Targets. 2009;13:1333–1345. doi: 10.1517/14728220903277256. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X, Li C, Lin B, Hong H, Jiang L, Zhu S, Wang X, Tang N, Li X, She F, Chen Y. cIAP2 promotes gallbladder cancer invasion and lymphangiogenesis by activating the NF-kappaB pathway. Cancer Sci. 2017;108:1144–1156. doi: 10.1111/cas.13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu HH, Wu JY, Cheng YW, Chen CY, Lee MC, Goan YG, Lee H. cIAP2 upregulated by E6 oncoprotein via epidermal growth factor receptor/phosphatidylinositol 3-kinase/AKT pathway confers resistance to cisplatin in human papillomavirus 16/18-infected lung cancer. Clin Cancer Res. 2010;16:5200–5210. doi: 10.1158/1078-0432.CCR-10-0020. [DOI] [PubMed] [Google Scholar]
- 26.Ma O, Cai WW, Zender L, Dayaram T, Shen J, Herron AJ, Lowe SW, Man TK, Lau CC, Donehower LA. MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res. 2009;69:2559–2567. doi: 10.1158/0008-5472.CAN-08-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagata M, Nakayama H, Tanaka T, Yoshida R, Yoshitake Y, Fukuma D, Kawahara K, Nakagawa Y, Ota K, Hiraki A, Shinohara M. Overexpression of cIAP2 contributes to 5-FU resistance and a poor prognosis in oral squamous cell carcinoma. Br J Cancer. 2011;105:1322–1330. doi: 10.1038/bjc.2011.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung YH, Lim EJ, Heo J, Kwon TK, Kim YH. Tunicamycin sensitizes human prostate cells to TRAIL-induced apoptosis by upregulation of TRAIL receptors and downregulation of cIAP2. Int J Oncol. 2012;40:1941–1948. doi: 10.3892/ijo.2012.1402. [DOI] [PubMed] [Google Scholar]
- 29.Chiaretti S, Marinelli M, Del Giudice I, Bonina S, Piciocchi A, Messina M, Vignetti M, Rossi D, Di Maio V, Mauro FR, Guarini A, Gaidano G, Foa R. NOTCH1, SF3B1, BIRC3 and TP53 mutations in patients with chronic lymphocytic leukemia undergoing first-line treatment: correlation with biological parameters and response to treatment. Leuk Lymphoma. 2014;55:2785–2792. doi: 10.3109/10428194.2014.898760. [DOI] [PubMed] [Google Scholar]
- 30.Cheng L, Zhou Z, Flesken-Nikitin A, Toshkov IA, Wang W, Camps J, Ried T, Nikitin AY. Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene. 2010;29:5700–5711. doi: 10.1038/onc.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gill C, Dowling C, O’Neill AJ, Watson RW. Effects of cIAP-1, cIAP-2 and XIAP triple knockdown on prostate cancer cell susceptibility to apoptosis, cell survival and proliferation. Mol Cancer. 2009;8:39. doi: 10.1186/1476-4598-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascal LE, Wang Y, Zhong M, Wang D, Chakka AB, Yang Z, Li F, Song Q, Rigatti LH, Chaparala S, Chandran U, Parwani AV, Wang Z. EAF2 and p53 Co-Regulate STAT3 Activation in Prostate Cancer. Neoplasia. 2018;20:351–363. doi: 10.1016/j.neo.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gressot LV, Doucette T, Yang Y, Fuller GN, Manyam G, Rao A, Latha K, Rao G. Analysis of the inhibitors of apoptosis identifies BIRC3 as a facilitator of malignant progression in glioma. Oncotarget. 2017;8:12695–12704. doi: 10.18632/oncotarget.8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholson J, Jevons SJ, Groselj B, Ellermann S, Konietzny R, Kerr M, Kessler BM, Kiltie AE. E3 Ligase cIAP2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res. 2017;77:3027–3039. doi: 10.1158/0008-5472.CAN-16-3232. [DOI] [PubMed] [Google Scholar]
- 35.Carr D, Lau R, Hnatykiw AD, Ward GCD, Daneshmand M, Cabrita MA, Pratt MAC. cIAP2 is an independent signaling and survival factor during mammary lactational involution and tumorigenesis. J Mammary Gland Biol Neoplasia. 2018;23:109–123. doi: 10.1007/s10911-018-9398-y. [DOI] [PubMed] [Google Scholar]
- 36.Su W, Jiang X, Chen M, Huang M, Tang N, Wang X, Li X, She F, Chen Y. cIAP1 promotes proliferation and migration and prevents apoptosis in gallbladder cancer in vitro. Biosci Rep. 2019;39 doi: 10.1042/BSR20182266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- 38.Hu X, Meng Y, Xu L, Qiu L, Wei M, Su D, Qi X, Wang Z, Yang S, Liu C, Han J. Cul4 E3 ubiquitin ligase regulates ovarian cancer drug resistance by targeting the antiapoptotic protein BIRC3. Cell Death Dis. 2019;10:104. doi: 10.1038/s41419-018-1200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lau R, Niu MY, Pratt MA. cIAP2 represses IKKalpha/beta-mediated activation of MDM2 to prevent p53 degradation. Cell Cycle. 2012;11:4009–4019. doi: 10.4161/cc.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]