

Abstract
Hyperuricemia has been identified as an independent risk factor for chronic kidney disease (CKD) and is associated with the progression of kidney diseases. It remains unknown whether enhancer of zeste homolog 2 (EZH2), a histone H3 lysine 27 methyltransferase, can regulate metabolism of serum uric acid and progression of renal injury induced by hyperuricemia. In this study, we demonstrated that blockade of EZH2 with 3-DZNeP, a selective EZH2 inhibitor, or silencing of EZH2 with siRNA inhibited uric acid-induced renal fibroblast activation and phosphorylation of Smad3, epidermal growth factor receptor (EGFR), and extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) in cultured renal fibroblasts. Inhibition of EZH2 also suppressed proliferation of renal fibroblasts and epithelial-mesenchymal transition of tubular cells. In a mouse model of renal injury induced by hyperuricemia, EZH2 and trimethylation of histone H3 at lysine27 expression levels were enhanced, which was coincident with renal damage and increased expression of lipocalin-2 and cleaved caspase-3. Inhibition of EZH2 with 3-DZNeP blocked all these responses. Furthermore, 3-DZNeP treatment decreased the level of serum uric acid and xanthine oxidase activity, alleviated renal interstitial fibrosis, inhibited activation of transforming growth factor-β/Smad3, EGFR/ERK1/2, and nuclear factor-κB signaling pathways, as well as reduced expression of multiple chemokines/cytokines. Collectively, EZH2 inhibition can reduce the level of serum uric acid and alleviate renal injury and fibrosis through a mechanism associated with inhibition of multiple signaling pathways. Targeting EZH2 may be a novel strategy for the treatment of hyperuricemia-induced CKD.
Keywords: 3-deazaneplanocin A, enhancer of zeste homolog 2, hyperuricemia, renal fibrosis, renal injury
INTRODUCTION
Recent studies have suggested that elevated level of serum uric acid is an independent risk factor for multiple metabolic disorders, such as hypertension, gout, and renal disease (25, 40, 67, 73). Several pilot studies also indicated that lowering serum uric acid can alleviate renal pathological injury and delay the progression of renal damage at the early time (22, 26, 33, 43, 73). These studies suggested that hyperuricemia contributes to the development and progression of renal damage and chronic kidney disease (CKD).
Uric acid is the end product of purine metabolism in humans, which is catalyzed by xanthine oxidase (XOD) (2). The mechanisms by which hyperuricemia leads to renal damage are still incompletely understood. It has been documented that uric acid-induced renal injury occurs through urate crystal-dependent and -independent pathways (21). Urate crystal-mediated renal injury refers to urate crystal deposition in renal interstitium that triggers inflammation, renal tubular cell apoptosis, and fibrosis, eventually leading to renal failure (21, 33, 71). Uric acid calculus also causes obstructive renal damage and a series of complications such as infection, bleeding, hydropsy, and malignant transformation (47).
Urate crystal-independent pathway is associated with multiple pathological processes, including oxidative stress, tubular epithelial cell transition, activation of renin-angiotensin aldosterone system, and so on (17, 36, 49, 59) In addition, uric acid can activate transcription factor, such as nuclear factor-κB (NF-κB) and induce production of some chemokines/chemokines-like tumor necrotic factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1) (24, 39). Uric acid can also induce activation of transforming growth factor-β (TGF-β) receptors and phosphorylation and nuclear translocation of Smad3 (35). Activated Smad3, together with Smad4, triggers the transcription of TGF-β1-targeted genes (8). On the other hand, TGF-β1 can induce activation of EGF receptor (EGFR) and some downstream signaling pathways, including the extracellular signal-regulated kinases 1/2 (ERK1/2) pathway, and drive the expression of targeted genes, leading to development of glomerular sclerosis and tubulointerstitial fibrosis (23, 29, 33).
Increasing evidence indicates that expression of profibrotic genes is also regulated by epigenetics. Epigenetics is a discipline that studies how the gene function is interpreted by the cell without changing the sequence of DNA (3, 56). There are several types of epigenetic modifications, such as DNA methylation, microRNA, and histone modifications. Histone modifications can occur in both histone proteins and nonhistone proteins, which include acetylation, methylation, phosphorylation, sumoylation, and ubiquitinylation. Our previous studies and others demonstrated that histone acetylation and methylation are closely related to the progression and development of renal fibrosis (7, 41, 44). However, it remains poorly understood whether histone methylation is involved in renal injury induced by hyperuricemia.
Like histone acetylation, histone methylation is positively regulated by histone methyltransferases and negatively regulated by demethylases. The histone methyltransferase enhancer of zeste homolog 2 (EZH2), as the catalytic part of polycomb repressive complex 2, triggering trimethylation of histone H3 at lysine27 (H3K27me3), has been involved in tumorigenesis and fibrogenesis (28). Overexpression of EZH2 is associated with a wide variety of cancerous tissue types, including prostate cancer, breast cancer, bladder cancer, and lung cancer (54, 70). Downregulating EZH2 can effectively inhibit cell proliferation and tumor angiogenesis, reverse epithelial-to-mesenchymal transition (EMT), and prevent tumor progression (15, 19, 38). Inhibition of EZH2 also inhibits the TGF-β1-induced Smad2/3 nuclear translocation and reduces differentiation of human lung fibroblasts into myofibroblasts (65).
Recently, our research group demonstrated that pharmacologic blockage of EZH2 with 3-DZNeP, a carbocyclic analog of adenosine, inhibits TGF-β1-induced activation of renal interstitial fibroblasts in vitro and abrogates deposition of extracellular matrix (ECM) proteins and expression of α-smooth muscle actin (α-SMA) in the obstructed kidney (72). However, it remains unknown whether EZH2 mediates the development of hyperuricemia-induced renal injury. In this study, we investigated the effect of EZH2 inhibition on the activation and proliferation of cultured renal interstitial fibroblasts and renal injury induced by hyperuricemia.
MATERIALS AND METHODS
Antibodies and reagents.
3-Deazaneplanocin A (3-DZNeP) was purchased from Selleckchem (Houston, TX). Antibodies to EZH2, H3K27me3, cleaved caspase-3, p-EGFR, p-ERK1/2, ERK1/2, phosphorylated Smad3 (p-Smad3), and Smad3 were purchased from Cell Signaling Technology (Dancers, MA). Antibodies to MCP-1, TNF-α, p-NF-κB (p65), NF-κB (p65), collagen I (A2), TGF-βRI, EGFR, β-actin, and EZH2 small interfering RNA (siRNA) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). TNF-α ELISA kit and antibody to lipocalin-2 (Lcn2) were from R&D Systems (Minneapolis, MN). Uric acid, α-SMA, DMSO, secondary antibodies for Western blotting, and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). XOD biochemical reagent kit was purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The Cell Counting Kit-8 (CCK-8) proliferation assay kit was purchased from Beyotime Biotechnology (Haimen, China).
Cell culture and treatment.
Both rat renal interstitial fibroblast (NRK-49F) and human tubular epithelial (HK2) cells were obtained from ATCC (Manassas, VA). NRK-49F and HK2 cells were cultured and propagated in DMEM with F-12 containing 10% FBS, and 1% penicillin and streptomycin in an atmosphere of 5% CO2-95% air at 37°C. The newly recovered cells were propagated for at least three generations before they were used in experiments. Our previous study showed that stimulation with uric acid dose dependently (0, 200, 400, and 800 μM) increased expression of α-SMA and collagen, with the maximum induction at a concentration of 800 μM (33). On this basis, we selected 800 μM of uric acid to examine the effect of 3-DZNeP on uric acid-induced renal interstitial fibroblast activation (33). NRK-49F and HK2 cells were starved for 24 h with DMEM containing 0.5% FBS before they were exposed to uric acid in the presence or absence of 3-DZNeP (1, 5, and 10 μM). At 36 h after the treatment, cells were harvested for immunoblot analysis. All of the in vitro experiments were repeated for at least three times.
siRNA transfection.
The siRNA oligonucleotides targeted especially for EZH2 were used in this study. NRK-49F were seeded to 30–40% confluence in antibiotic-free medium and grown for 24 h and then were transfected with EZH2 siRNA (100 pmol) with Lipofectamine 2000. In parallel, scrambled siRNA (100 pmol) was used as control for off-target changes in NRK-49F. At 24 h after transfection, cells were treated with uric acid (800 μM) for an additional 36 h before being harvested for the experiments.
Animals and treatment.
Male C57/black mice (Shanghai Super-B&K Laboratory Animal) weighing 20–25 g were housed under a 12:12-h light-dark cycle with food and water supplied ad libitum. To establish a mouse model of hyperuricemia, the animals were injected intraperitoneally with uric acid (250 mg/kg) in distilled water every day for 21 days. To investigate the effect of 3-DZNeP on renal injury associated with hyperuricemia, 3-DZNeP (1 mg/kg) in vehicle (DMSO) was given intraperitoneally immediately after uric acid injection. The sham group was injected with an equal volume of DMSO as a control. Six mice were used in each group. At the end of 21 days, kidney samples were harvested for protein analysis and histological examination. Blood was taken for the measurement of serum uric acid and XOD. The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Tongji University.
Renal function assay.
Levels of serum uric acid and serum creatinine were examined by automatic biochemistry assay (P800; Modular).
Analysis of serum activity of XOD.
Collected blood was centrifuged at 2,500 rpm/min for 5 min, and 100 μl serum were harvested to detect XOD content. Serum activity of XOD was determined according to the protocol provided by the manufacturer.
Assessment of tubular injury.
The degree of tubular injury was determined using a semiquantitative grade scale that tubular injury was scored on a scale from 0 to 3, where 0 = normal, 1 = injury <30%, 2 = injury 30–60%, and 3 = injury >60%. Two sections were randomly selected from each sample of at least three for every group, and 10 fields were randomly selected at a magnification of ×200 from each section in periodic acid Schiff staining. Last, an average score was calculated and then made into column diagram.
TUNEL staining.
To determine cell apoptosis, TdT-mediated dUTP nick-end labeling (TUNEL) staining was performed according to the protocol provided by a Roche Molecular System (Branchburg, NJ). For quantitative assessment of TUNEL, the TUNEL-positive renal tubular cells were counted, and the average ratio to each microscopic field (original magnification, × 200) was calculated and graphed.
Immunoblot analysis.
Immunoblot analysis for tissue samples and NRK-49F cells was performed according to our previous protocol (66). Densitometry analysis of immunoblot results was conducted with ImageJ software (National Institutes of Health, Bethesda, MD).
Immunofluorescent and immunohistochemical staining.
Immunohistochemical staining was conducted on the basis of the procedure described in our previous studies (66). The quantification of TNF-α and MCP-1 was accounted by percentage of positive areas and reported as the mean of 20 random high-power (×200) fields in six mice per group. Immuofluorescent staining was carried out according to the procedure described previously (66). Slides were viewed with a Nikon Eclipse 80i microscope equipped with a digital camera (DS-Ri1; Nikon, Shanghai, China).
ELISA analysis.
ELISA detection of TNF-α was performed in accordance with the manufacturer’s instructions.
CCK-8 proliferation assay.
The CCK-8 proliferation kit was used according to the manufacturer’s instructions. NRK-49F cells were starved for 24 h with DMEM containing 0.5% FBS and then exposed to uric acid (800 μM) in the presence or absence of 3-DZNeP (1, 5, and 10 μM). After 36 h, the original culture medium was removed, and 100 μl new DMEM/F12 medium containing 10 μl CCK-8 were added to each well in a 96-well plate for 37 °C incubation for an additional 4 h. The final optical density values were read at 450 nm.
Statistical analysis.
All the experiments were conducted at least three times. Data depicted in graphs represent the means ± SD for each group. Intergroup comparison was made using one-way ANOVA. Multiple means were compared using Turkey’s test. The differences between two groups were determined by Student’s t-test. Statistical significant difference between mean values was marked in each graph. P < 0.05 is considered significant. The statistical analyses were conducted by using IBM SPSS Statistics 20.0 (Beijing, China).
RESULTS
3-DZNeP blocks uric acid-induced activation and proliferation of rat renal interstitial fibroblasts.
Activation and proliferation of renal interstitial fibroblasts have been identified as pivotal mechanisms in renal fibrosis (13). Thus we examined the effect of 3-DZNeP, a selective inhibitor of EZH2, on the expression of collagen I, an ECM protein, and α-SMA, a hallmark of fibroblast activation in vitro. As shown in Fig. 1A, a small amount of α-SMA and collagen I was detected in the starved rat renal interstitial fibroblast (NRK-49F) cells, while addition of uric acid to the culture increased expression of these proteins. Treatment with 3-DZNeP inhibited these responses in a dose-dependent manner with the maximum effect at 10 μM (Fig. 1B). Exposure of NRK-49F cells to uric acid resulted in increased expression of EZH2 and H3K27me3, which were dose dependently inhibited by 3-DZNeP (Fig. 1, C and D). Moreover, inhibition of EZH2 with 3-DZNeP prominently suppressed cell proliferation in cultured rat renal interstitial fibroblast cells (Fig. 1, E and F).
Fig. 1.

3-DZNeP blocks uric acid (UA)-induced activation and proliferation of rat renal interstitial fibroblasts. Serum-starved NRK-49F cells were pretreated with various concentrations of 3-DZNeP (0, 1, 5, and 10 μM) for 1 h and then exposed to uric acid (800 μM) for an additional 36 h. Cell lysates were subjected to immunoblot analysis with antibodies against various molecules as indicated (A and C). Expression levels of α-smooth muscle actin (α-SMA; B), collagen I (B), enhancer of zeste homolog 2 (EZH2; D), or trimethylation of histone H3 at lysine27 (H3K27me3; D) were quantified by densitometry and normalized with β-actin. Photomicrographs (original magnification: ×100) illustrated the characterization of the NRK-49F phenotype (E). Cell proliferation was assessed by the CCK-8 assay (F). Values are means ± SD of at least 3 independent experiments. Means with different letters (a–e) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 suppresses phosphorylation of Smad3, EGFR, and ERK1/2 in NRK-49F.
Our previous studies have shown that activation of TGF-β/Smad3 and EGFR/ERK pathways is involved in the differentiation of renal interstitial fibroblasts to myofibroblasts (32, 72). To demonstrate whether EZH2 plays a role in regulating activation of these two pathways, we examined the effect of 3-DZNeP on the activation of Smad3, EGFR, and ERK1/2 in NRK-49F. As shown in Fig. 2, A and B, expression of expression of phosphorylated Smad3 (p-Smad3) was detected in the starved NRK-49F cells, and exposure to uric acid increased expression levels of p-Smad3 but had no effect on total Smad3. Inhibition of EZH2 with 3-DZNeP suppressed activation of Smad3 in a dose-dependent manner, with a maximum inhibition at 10 μM. Similarly, uric acid exposure induced phosphorylation of EGFR and ERK1/2 in NRK-49F, and 3-DZNeP treatment dose dependently inhibited their phosphorylation (Fig. 2, C and E) without affecting the expression levels of total EGFR and ERK1/2 (Fig. 2, C–F). These data suggest that EZH2 mediates activation of Smad3, EGFR, and ERK1/2 in cultured renal interstitial fibroblasts.
Fig. 2.

Inhibition of enhancer of zeste homolog 2 (EZH2) suppresses phosphorylation of Smad3, epidermal growth factor receptor (EGFR), and ERK1/2 in NRK-49F. Serum-starved NRK-49F cells were pretreated with various concentration of 3-DZNeP (0, 1, 5, and 10 μM) for 1 h and then exposed to uric acid (UA; 800 μM) for an additional 36 h. Cell lysates were subjected to immunoblot analysis with antibodies against various molecules as indicated (A, C, and E). Expression of p-Smad3 was quantified by densitometry and normalized with Smad3 (B). Expression of p-EGFR was quantified by densitometry and normalized with EGFR (D). Expression of p-ERK1/2 was quantified by densitometry and normalized with ERK1/2 (F). Values are means ± SD of at least 3 independent experiments. Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
SiRNA-mediated silencing of EZH2 inhibits activation of rat renal interstitial fibroblasts as well as phosphorylation of Smad3, EGFR, and ERK1/2.
To confirm the above observations, we further examined the effect of siRNA-mediated EZH2 silencing on the renal interstitial fibroblast activation in cultured NRK-49F. As shown in Fig. 3, A and B, there were very low expression levels of α-SMA and collagen I in both control and EZH2 silencing groups without uric acid exposure. Uric acid stimulation resulted in increased expression of α-SMA and collagen I, while treatment with EZH2 siRNA remarkably inhibited their expression. The levels of EZH2 and H3K27me3 in EZH2 silencing cells were >50% lower than control cells after uric acid exposure (Fig. 3, C and D).
Fig. 3.

Small interfering RNA (siRNA)-mediated silencing of enhancer of zeste homolog 2 (EZH2) inhibits the activation of rat renal interstitial fibroblasts as well as the phosphorylation of Smad3, epidermal growth factor receptor (EGFR), and ERK1/2 in NRK-49F. Serum-starved NRK-49F cells were transferred with siRNA targeting EZH2 or scrambled siRNA and then incubated with uric acid (UA; 800 μM) for an additional 36 h. Cell lysates were subjected to immunoblot analysis with antibodies against α-smooth muscle actin (α-SMA), collagen I, EZH2, trimethylation of histone H3 at lysine27 (H3K27me3), p-Smad3, Smad3, p-EGFR, EGFR, p-ERK1/2, ERK1/2, or β-actin (A, C, E, and G). Expression levels of α-SMA, collagen I (B), and EZH2, H3K27me3 (D) were quantified by densitometry and normalized with β-actin. Expression of p-Smad3 was quantified by densitometry and normalized with Smad3 (F). Expression of p-EGFR was quantified by densitometry and normalized with EGFR (H). Expression of p-ERK1/2 was quantified by densitometry and normalized with ERK1/2 (H). Values are means ± SD of at least 3 independent experiments. Means with different letters (a–d) for each molecule are significantly different from one another (P < 0.05).
We also examined whether silencing of EZH2 would have an inhibitory effect on the phosphorylation of Smad3, EGFR and ERK1/2 in cultured NRK-49F. As shown in Fig. 3, E–H, low phosphorylation levels of Smad3, EGFR, and ERK1/2 were observed in both control and EZH2 silencing cells without uric acid treatment. Transfection of siRNA specifically targeting EZH2 reduced phosphorylation of these three molecules in NRK-49F exposed to uric acid. There were no significant changes in the expression levels of total proteins for each molecule. These data further confirm the importance of EZH2 in activating renal interstitial fibroblasts induced by uric acid.
3-DZNeP blocks uric acid-induced EMT in cultured human tubular epithelial cells.
Renal tubular epithelial-mesenchymal transition (EMT) promotes the development and progression of renal fibrogenesis (16). Thus we further verified the effect of 3-DZNeP on EMT in human tubular epithelial (HK2) cells. As shown in Fig. 4, A–C, uric acid stimulation significantly upregulated the expression of α-SMA and collagen I, compared with starved HK2 cells alone. Administration of 3-DZNeP downregulated levels of α-SMA and collagen I in a dose-dependent manner. Similarly, exposure to uric acid led to elevated expression of EZH2 and H3K27me3, which was blocked by 3-DZNeP treatment (Fig. 4, D–F). These results, together with Figs. 1–3, demonstrate that EZH2 plays an important role in regulating uric acid-stimulated activation and proliferation of renal interstitial fibroblasts and EMT of renal tubular epithelial cells.
Fig. 4.

3-DZNeP blocks uric acid (UA)-induced epithelial-to-mesenchymal transition (EMT) in cultured human tubular epithelial cells. Serum-starved human tubular epithelial (HK2) cells were pretreated with various concentration of 3-DZNeP (0, 1, 5, and 10 μM) for 1 h and then exposed to UA (800 μM) for an additional 36 h. Cell lysates were subjected to immunoblot analysis with specific antibodies against α-smooth muscle actin (α-SMA), collagen I, enhancer of zeste homolog 2 (EZH2), trimethylation of histone H3 at lysine27 (H3K27me3), or GAPDH (A and D). Expression levels of α-SMA (B), collagen I (C), EZH2 (E), or H3K27me3 (F) were quantified by densitometry and normalized with GAPDH. Values are means ± SD of at least 3 independent experiments. Means with different letters (a–d) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 reduces histone methylation in the kidney of hyperuricemic mice.
To determine the role of EZH2 in renal fibrosis in vivo, we first examined the effect of 3-DZNeP on the expression levels of EZH2 and H3K27me3 in the kidney of hyperuricemic mice. The basal expression levels of EZH2 and H3K27me3 were detected in the sham kidney, injection of uric acid for 21 days resulted in a significant increase of them. Administration of 3-DZNeP reduced the levels of EZH2 and H3K27me3 to ~90 and 50%, respectively, in the kidney tissues of hyperuricemia mice (Fig. 5, A–C). Immunofluorescence staining showed that EZH2 is expressed in both renal tubules and myofibroblasts as evidenced by its abundant expression in tubules and colocalization with α-SMA in the interstitium (Fig. 5D). Collectively, these data indicate that EZH2 and H3K27me3 are highly expressed in the kidney of hyperuricemic mice and 3-DZNeP is a potent EZH2 inhibitor.
Fig. 5.

Inhibition of enhancer of zeste homolog 2 (EZH2) reduces expression of trimethylation of histone H3 at lysine27 (H3K27me3) in the kidney of hyperuricemic mice. A mouse model of hyperuricemia was established by intraperitoneal injection with uric acid (UA: 250 mg/kg) daily. In some mice, 3-DZNeP were simultaneously administrated intraperitoneally. After 3 wk, the kidneys were taken for immunoblot analysis of EZH2, H3K27me3, or β-actin (A). Expression levels of EZH2 or H3K27me3 were quantified by densitometry and normalized with β-actin (B and C). Photomicrographs illustrated costaining of EZH2 and α-smooth muscle actin (α-SMA) in the kidney collected 21 days after UA injection (original magnification: × 200) (D). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 reduces the level of serum uric acid and blocks serum XOD activity and protects against renal tubular damage in hyperuricemic mice.
To determine the efficiency of EZH2 inhibition in mice with hyperuricemia, we initially examined the effect of 3-DZNeP on serum level of uric acid in mice. As shown in Fig. 6A, the level of serum uric acid was relatively low and no different in the sham groups with/without 3-DZNeP treatment. Hyperuricemic mice exhibited a high level of uric acid, which was reduced to the basal level following administration of 3-DZNeP. As XOD is the primary enzyme that contributes to synthesis of uric acid (2), we further examined the effect of EZH2 inhibition on serum XOD activity. As indicated in Fig. 6B, the increased XOD activity was detected in the kidney tissue of hyperuricemic mice, and 3-DZNeP administration reduced its activity.
Fig. 6.

Inhibition of enhancer of zeste homolog 2 (EZH2) reduces the level of serum uric acid (UA) and blocks serum xanthine oxidase (XOD) activity and protects renal histopathologic structure in hyperuricemia mice. A mouse model of hyperuricemia was established by intraperitoneal injection with UA (250 mg/kg) daily. Expression level of serum UA was examined using automatic biochemistry assay (P800; Modular) (A). Serum XOD activity was examined by XOD kit (B). Photomicrographs (original magnification: ×200) illustrated periodic acid-Schiff (PAS) staining of the kidney tissues in control or hyperuricemia mice with or without 3-DZNeP (C). Morphologic changes were scored on the basis of the scale described in the concise materials and methods (D). Serum creatinine was measured in each group (E). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
Furthermore, we examined the effect of EZH2 inhibition on renal histopathologic changes in hyperuricemic mice. As shown in Fig. 6C, hyperuricemic mice displayed mild glomerulosclerosis and severe tubulointerstitial damages, such as tubular dilatation and tubular atrophy. Administration of 3-DZNeP also relieved glomerulosclerosis and tubulointerstitial damages and largely preserved renal structure. Seminal scoring analysis indicated that EZH2 inhibition improved tubular injury by 50% (Fig. 6D). However, either uric acid injection or 3-DZNeP treatment had no effect on the level of serum creatinine in mice (Fig. 6E). Taken together, inhibition of EZH2 can decrease the level of serum uric acid and alleviate renal injury in hyperuricemic mice.
Inhibition of EZH2 alleviates uric acid-induced renal tubular injury.
Lipocalin-2 (Lcn2) is a biomarker of renal tubule injury (34, 63). We thus assessed the effect of EZH2 inhibition on its expression in the kidney of hyperuricemic mice. Immunofluorescence analysis indicated that Lcn2 expression is not observed in the normal kidney either subjected to DMSO or 3-DZNeP, but it markedly increased in renal tubular cells of hyperuricemic mice, 3-DZNeP treatment significantly reduced its expression (Fig. 7A). Immunoblot analysis also showed that Lcn2 was highly expressed in the kidney of hyperuricemic mice, which was inhibited by 3-DZNeP administration (Fig. 7, B and C). Taken together, we suggest that EZH2 activation is associated with renal tubular injury of hyperuricemic mice.
Fig. 7.

Inhibition of enhancer of zeste homolog 2 (EZH2) alleviates uric acid (UA)-induced renal tubular injury. A mouse model of hyperuricemia was established by intraperitoneal injection with UA (250 mg/kg) daily. Photomicrographs (original magnification: × 200) illustrated staining of Lipocalin-2 (Lcn2) in the kidney collected 21 days after UA injection with or without 3-DZNeP (A). The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against Lcn2 or β-actin (B). Expression level of Lcn2 was quantified by densitometry and normalized with β-actin (C). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 attenuates renal tubular cell apoptosis in hyperuricemic mice.
It has been reported that renal tubular cell apoptosis is involved in the progression of renal interstitial fibrosis (52, 71). Thus we examined the effect of EZH2 on tubular cell apoptosis by TUNEL staining (Fig. 8, A and B). There were no TUNEL-positive renal tubular cells in the sham kidney tissues either subjected to DMSO or 3-DZNeP, while uric acid injection for 21 days increased the number of TUNEL-positive renal tubular cells. 3-DZNeP treatment reduced this count by ~70% in hyperuricemia-injured kidney compared with the group injected with uric acid alone. Meanwhile, we also examined expression of cleaved caspase-3, a critical mediator in apoptosis, in the kidney of hyperuricemic mice with or without 3-DZNeP treatment by immunoblot analysis. As shown in Fig. 8, C and D, there was little cleaved caspase-3 expression in sham group with or without 3-DZNeP treatment, but an abundant expression of cleaved caspase-3 was observed in the kidney after uric acid injection. 3-DZNeP administration suppressed the expression of cleaved caspase-3 by ~50% in the kidney of hyperuricemic mice compared with those injected with uric acid alone. Taken together, our data suggest that EZH2 is involved in the initiation of renal tubular cell apoptosis in uric acid-induced renal injury and interstitial fibrosis.
Fig. 8.

Inhibition of enhancer of zeste homolog 2 (EZH2) attenuates renal tubular cell apoptosis in hyperuricemia mice. A mouse model of hyperuricemia was established by intraperitoneal injection with uric acid (UA; 250 mg/kg) daily. Photomicrographs (original magnification: ×200) illustrated TdT-mediated dUTP nick-end labeling (TUNEL)-positive tubular cells in the kidney tissue (A). The number of TUNEL tubular cells from 10 random cortical fields was counted and statistical analysis was conducted (B). The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against cleaved caspase-3 or β-actin (C). Expression level of cleaved caspase-3 was quantified by densitometry and normalized with β-actin (D). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 suppresses renal interstitial fibroblasts activation and ECM deposition in hyperuricemic mice.
A high level of uric acid plays an important role in mediating renal interstitial fibroblast activation and ECM deposition (34, 48, 53). As shown in Masson trichrome staining, there were more positive areas of interstitial fibrosis in hyperuricemic mice than sham mice with/without 3-DZNeP treatment. 3-DZNeP administration reduced the increased Masson trichrome-positive areas within the tubular interstitium in hyperuricemic mice (Fig. 9A). Further semiquantitative analysis of Masson trichrome-positive areas showed an approximate eightfold increase in positive areas of interstitial fibrosis from hyperuricemia-injured kidney compared with control kidneys, while 3-DZNeP treatment significantly reduced ECM deposition by 60% (Fig. 9B). To investigate the effect of EZH2 on myofibroblast activation and ECM deposition after uric acid exposure, we examined the expression of α-SMA and collagen I by immunoblot analysis. As indicated in Fig. 9, C and D, basal levels of α-SMA and collagen I were observed in the sham group, and their expression levels were dramatically upregulated after exposure to high concentration of uric acid for 21 days. Administration of 3-DZNeP largely reduced expression of α-SMA and collagen I. Collectively, these results suggest that EZH2 is involved in the development of renal interstitial fibrosis.
Fig. 9.

Inhibition of enhancer of zeste homolog 2 (EZH2) suppresses renal interstitial fibroblasts activation and extracellular matrix (ECM) deposition in hyperuricemia mice. A mouse model of hyperuricemia was established by intraperitoneal injection with uric acid (UA; 250 mg/kg) daily. Photomicrographs illustrated Masson trichrome staining of kidney tissues collected at day 21 after injecting with UA with or without 3-DZNeP (A). The graph shows the percentage of Masson-positive tubulointerstitial area (blue) relative to the whole area from 10 random cortical fields (original magnification: ×200) (B). The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against α-smooth muscle actin (α-SMA), collagen I, or β-actin (C). Expression level of α-SMA or collagen I was quantified by densitometry and normalized with β-actin (D). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 blocks activation of TGF-β1/Smad3 signaling pathway in the kidney of hyperuricemic mice.
Because TGF-β1/Smad3 signaling plays a vital role in renal interstitial fibrosis, we examined the effect of EZH2 on the activation of this signaling pathway in the kidney of hyperuricemic mice. Figure 10, A and B, shows that a small amount of TGF-βRI was expressed in sham kidney with/without 3-DZNeP treatment, and its expression levels was upregulated in the kidney administrated with a high dose of uric acid; 3-DZNeP treatment reduced TGF-βRI expression. We also examined the phosphorylation of Smad3, a key molecule of the TGF-β signaling pathway. There was a low expression level of phosphorylated Smad3 in the normal kidney tissues, but the degree of Smad3 phosphorylation doubled in the hyperuricemic mice. 3-DZNeP treatment suppressed phosphorylation of Smad3 to the basal level. Furthermore, either uric acid injection or 3-DZNeP administration had no effect on the expression level of total Smad3 (Fig. 10, C and D). These data suggest that EZH2 is implicated in the activation of the TGF-β signaling pathway.
Fig. 10.
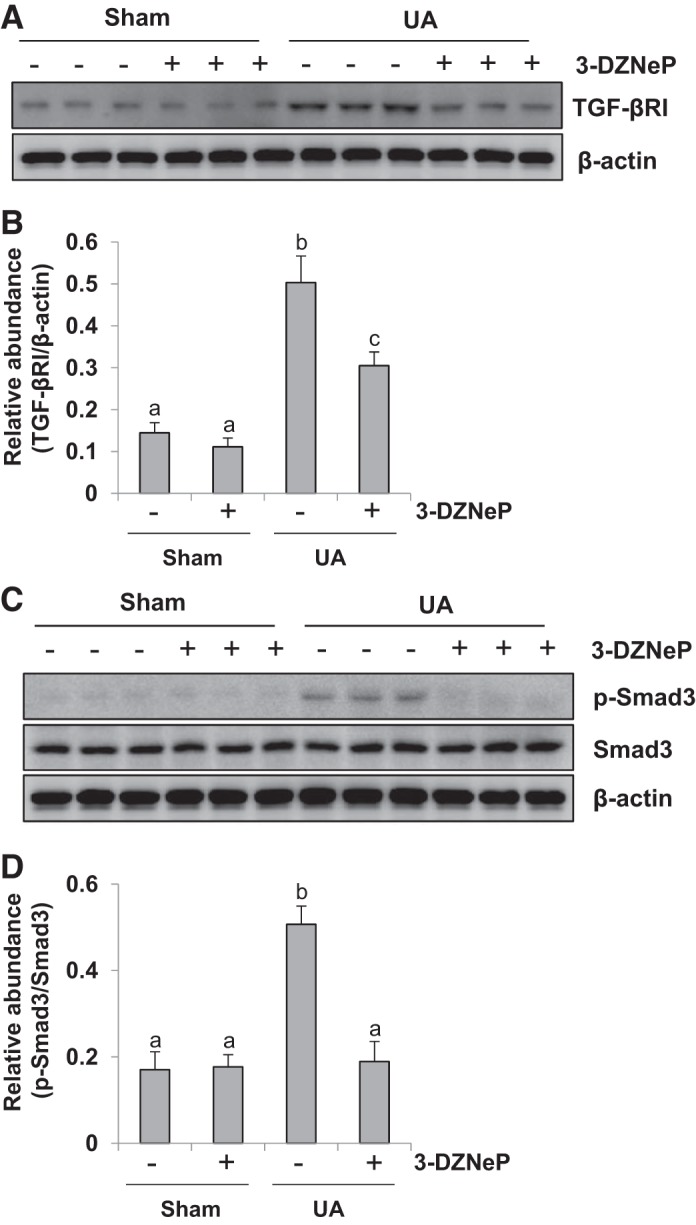
Inhibition of enhancer of zeste homolog 2 (EZH2) blocks activation of transforming growth factor-β1 (TGF-β1)/Smad3 signaling pathway in the kidney of hyperuricemia mice. A mouse model of hyperuricemia was established by intraperitoneal injection with uric acid (UA; 250 mg/kg) daily. The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against TGF-βRI or β-actin (A). Expression level of TGF-βRI was quantified by densitometry and normalized with β-actin (B). The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against Smad3, p-Smad3, or β-actin (C). Expression level of p-Smad3 was quantified by densitometry and normalized with Smad3 (D). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 abrogates activation of EGFR/ERK1/2 signaling in the kidney of hyperuricemic mice.
Studies from our group and others have illustrated that activation of the EGFR/ERK1/2 signaling pathway is critically involved in the progression of renal interstitial fibrosis (64, 72). Thus we ascertained whether EZH2 plays a role in the phosphorylation of EGFR and ERK1/2. As shown in Fig. 11, A–C, phosphorylated EGFR was rarely detected in the sham kidney while injection of uric acid within 21 days remarkably increased the levels of phosphorylated EGFR as well as total EGFR. 3-DZNeP treatment resulted in a complete inhibition of EGFR phosphorylation but did not alter the level of total EGFR. Figure 11, D–F, indicates that the similar inhibitory effect of 3-DZNeP on the phosphorylation ERK1/2, but did not affect the expression of total ERK1/2. These results indicate that EZH2 inhibition suppressed activation of the EGFR/ERK1/2 pathway.
Fig. 11.

Inhibition of enhancer of zeste homolog 2 (EZH2) abrogates activation of epidermal growth factor receptor (EGFR)/ERK1/2 signaling in the kidney of hyperuricemia mice. A mouse model of hyperuricemia was established by intraperitoneal injection with uric acid (UA; 250 mg/kg) daily. The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against EGFR, p-EGFR, or β-actin (A). Expression level of p-EGFR was quantified by densitometry and normalized with EGFR (B). Expression level of EGFR was quantified by densitometry and normalized with β-actin (C). The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against ERK1/2, p-ERK1/2, or β-actin (D). Expression level of p-ERK1/2 was quantified by densitometry and normalized with ERK1/2 (E). Expression level of ERK1/2 was quantified by densitometry and normalized with β-actin (F). Data are represented as the means ± SD (n = 6). Means with different letters (a, b) for each molecule are significantly different from one another (P < 0.05).
Inhibition of EZH2 blocks NF-κB phosphorylation and attenuates release of inflammatory cytokines and chemokines in the kidney of hyperuricemic mice.
As a transcriptional factor associated with inflammation, NF-κB phosphorylation sets off mononuclear cells infiltration (33, 58). We thus assessed the role of EZH2 in the activation of NF-κB signaling and the infiltration of mononuclear cells. As shown in Fig. 12, A and B, a large dose of uric acid led to a notable phosphorylation of NF-κB, which was inhibited by 3-DZNeP completely. The basal expression of p-NF-κB was detected in sham groups with/without 3-DZNeP. Moreover, 3-DZNeP treatment did not affect expression levels of total NF-κB in each group. Increased inflammatory cells in the renal interstitium contribute to renal interstitial fibrosis (6, 14, 31). To determine whether EZH2 was involved in this response, immunohistochemistry staining was used to assess the expression level of monocyte chemoattractant protein 1 (MCP-1) in the hyperuricemic kidney with or without 3-DZNeP administration. MCP-1 level was increased markedly in the kidney of hyperuricemic mice but reduced to the basal level with 3-DZNeP treatment (Fig. 12, C and D).
Fig. 12.

Inhibition of enhancer of zeste homolog 2 (EZH2) blocks phosphorylation of NF-κB signaling and attenuates release of inflammatory cytokines and chemokines in the kidney in hyperuricemia mice. A mouse model of hyperuricemia was established by intraperitoneal injection with uric acid (UA; 250 mg/kg) daily. The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against NF-κB, p-NF-κB, or β-actin (A). Expression level of p-NF-κB was quantified by densitometry and normalized with NF-κB (B). Photomicrographs (original magnification: × 200) illustrated monocyte chemoattractant protein-1 (MCP-1) immunohistochemistry staining of kidney tissues collected at day 21 after UA injection with or without 3-DZNeP (C). The percentage of MCP-1-positive area was calculated from ten random fields of six mice kidneys (D). Photomicrographs (original magnification: ×200) illustrated TNF-α immunochemistry staining of kidney tissues collected at day 21 after UA injection with or without 3-DZNeP (E). The percentage of TNF-α-positive area was calculated from 10 random fields of 6 mice kidneys (F). Graphs showed the expression level of TNF-α by ELISA (G). Data are represented as the means ± SD (n = 6). Means with different letters (a–c) for each molecule are significantly different from one another (P < 0.05).
As increased expression of inflammatory cytokines and chemokines contributes to renal interstitial fibrosis (13), we further evaluated the effect of EZH2 inhibition on the expression of TNF-α, one of key inflammatory cytokines in the kidney. TNF-α was mainly localized in renal tubules as indicated by immunohistrochemistry staining. There was a basal level of TNF-α in the sham group with or without 3-DZNeP treatment. Its expression level was markedly upregulated in the kidney of hyperuricemic mice and downregulated following 3-DZNeP administration (Fig. 12, E and F). By using the ELISA assay, we also revealed an increase of TNF-α in the kidney of hyperuricemic mice, whereas 3-DZNeP treatment reduced its expression levels (Fig. 12G). Thus these results indicate that EZH2 activity is required for the production and release of inflammatory cytokines in the renal interstitial fibrosis induced by hyperuricemia. Collectively, these data suggest that activation of EZH2 is required for the activation of NF-κB signaling as well as infiltration of inflammatory cells in the kidney of hyperuricemic mice.
DISCUSSION
Hyperuricemia has been considered as an independent risk for chronic kidney disease (CKD) and promotes the development and progression of CKD (12). In this study, we demonstrated that blockade of EZH2 with 3-DZNeP or EZH2 siRNA inhibited activation of renal interstitial fibroblasts, ECM deposition, and EMT. Inhibition of EZH2 also attenuated renal tubule injury, apoptosis, and inflammation in the kidney of hyperuricemic mice. These data demonstrated that EZH2 is a critical mediator in chronic kidney injury induced by hyperuricemia and suggested that EZH2 is a novel therapeutic target for CKD associated with hyperuricemia.
Although elevated serum urate has been considered to be associated with progression of kidney disease, it has been debated whether uric acid plays a causal role in human CKD. CKD is characterized by activation of renal interstitial fibroblasts and excessive production of ECM proteins. Our investigations demonstrated that uric acid can directly induce activation of renal interstitial fibroblasts in culture. On the other hand, uric acid may also indirectly induce activation of renal interstitial fibroblasts through induction of EMT. This is because that partial EMT is the prerequisite for epithelial cells arrested at the G2/M arrest of cell cycle, a state that produces an excessive amount of profibrotic cytokines/growth factors, promoting renal interstitial fibroblast activation and fibrosis (37). In this study, we found that uric acid stimulates EMT in cultured HK2 cells. We thus provide the essential evidence that uric acid may be a causative factor for the development of renal fibrosis and CKD. In addition, we observed that EZH2 and H3K27me3 are highly expressed not only in uric acid-treated rat renal interstitial fibroblasts and tubular epithelial cells in culture but also in the kidney of mice with hyperuricemia. In addition, inhibition of EZH2 blocked all these pathological responses. This suggests the importance of EZH2 in mediating development of hyperuricemia associated CKD.
In vivo, we observed that the EZH2 inhibitor lowered serum uric acid to the base level. This suggests that the decrease of serum uric acid levels may also contribute to EZH2 inhibition elicited attenuation of kidney injury in vivo, in addition to its direct renal protective effect. Previous studies have shown that uric acid-lowering therapy can effectively ameliorate renal function and protect the kidney from acute and chronic renal injury (18, 20). Allopurinol and febuxostat, two inhibitors of xanthine oxidase, can normalize serum uric acid levels and postpone the deterioration of renal function in hyperuricemic patients (10, 55, 57). Furthermore, they alleviated oxonic acid-induced renal injury in rat hyperuricemic models (51) and inhibited EMT in cultured rat renal tubular epithelial cells (NRK-52E cells) (49). In conclusion, our in vitro and in vivo data validated two aspects in renal protection of 3-DZNeP, downregulated serum uric acid and antifibrosis.
Numerous studies have demonstrated that overexpression of EZH2 is associated with various forms of cancers, involving breast, liver, and bladder cancer (27, 61, 69). Considering several preclinical studies suggest that blocking EZH2 activity has inhibitory effects on tumor growth, increased expression of EZH2 in the kidney may be correlated with pathogenesis of renal injury induced by high level of serum uric acid. In support of this hypothesis, we detected a high expression level of EZH2 in the injured kidney induced by hyperuricemia. EZH2 inhibition also prevented activation of several signaling pathways, such as TGF-β1/Smad3 signaling and EGFR/ERK1/2 signaling, and multiple proinflammation chemokines/cytokines, which contribute to renal interstitial fibrosis (4, 30, 34, 46). Moreover, 3-DZNeP attenuated apoptosis in renal tubular cells in the kidney of hyperuricemia mice, indicating that 3-DZNeP protects the kidney at least partly by reducing renal tubular injury.
The maintenance of normal serum uric acid levels is through balancing production, excretion and reabsorption of uric acid. The production of uric acid largely depends on the activity of XOD. XOD is an enzyme which has a catalyst function, speeding up hypoxanthine oxidation, and then accelerating uric acid production (42). Our results showed that 3-DZNeP treatment decreases the level of uric acid and XOD activity, indicating that EZH2-mediated XOD activation increases uric acid production and suggesting that inhibition of XOD activity may be one of mechanism by which 3-DZNeP reduces renal injury. Meanwhile, we noticed that despite injection of large amounts of uric acid (250 mg/kg daily), the serum levels of urate did not elevate to a very high (~1.7× baseline or ~3 mg/dl). This may be attributed to a feedback response of activated urate excretion transporters to the instant uric acid stimulus in vivo. It may also be due to relatively preserved glomerular filtration function in our mice model, as indicated by no significant fluctuation of serum creatinine in each group. However, despite the small increase of serum uric acid, the clear kidney injury was observed. Currently, the detailed mechanism by which EZH2 regulates the level of serum uric acid and XOD activity remains obscure and needs further investigations.
Our data demonstrated that EZH2 inhibition attenuates renal interstitial fibrosis by blocking the TGF-β1/Smad3 and EGFR/ERK1/2 signaling pathways activation. This is evidenced by our results that inhibiting EZH2 with 3-DZNeP markedly suppresses the phosphorylation of Smad3 in cultured renal interstitial fibroblasts and has the same effects with EZH2-specific siRNA. In vivo, administration of 3-DZNeP also blocked the activation of TGF-β1/Smad3 signaling in the injured kidney. Previous studies suggested that activating TGF-βRI, a vital receptor in mediating renal interstitial fibrosis, can recruit Smad3 from microtubules and then phosphorylate it (8, 74). In addition, it has been reported that trimethylation of H3K27 suppresses the expression the antifibrogenic transcriptional factor, peroxisome proliferator-activated receptor-γ, and then increases transcription of profibrogenic TGF-β1 (45). Given the role of EGFR/ERK1/2 activation in mediating TGF-β1 production during the renal interstitial fibrosis (33, 34), EZH2-dependent H3K27 trimethylation may alter TGF-β1 transcription and affect the phosphorylation of Smad3 by activating EGFR/ERK1/2 signaling pathways. Furthermore, it was documented that injury to the kidney led to downregulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a protein phosphatase activity with the ability to dephosphorylate some tyrosine kinases, including EGFR (1, 5) whereas EZH2 inhibition retained PTEN expression. Thus the 3-DZNeP elicited inhibition of Smad-3 may be associated with preservation of PTEN and subsequent inactivation of the EGFR/ERK1/2 signaling pathway.
EZH2 inhibition-elicited suppression of inflammation may attenuate renal injury induced by hyperuricemia. It is well known that release of various chemokines/cytokines and infiltration of macrophages are two main features of inflammation (11). In this regard, we observed the expression of inflammatory factors such as TNF-α was markedly increased in the kidney of hyperuricemia mice and suppressed by 3-DZNeP administration. EZH2 may control the release of inflammatory factors through suppressing transcriptional factors. In this context, we have previously shown that blockage of EZH2 with 3-DZNeP inhibits the phosphorylation of NF-κB(p65), a key transcriptional factor in multiply chemokines/cytokines expression (9).
Unlike with other mammals, uric acid is the final product of purine in human due to a lack of uricase. Uric acid is mostly excreted in urine through proximal tubular lumen. In this study, we found that a high level of uric acid can trigger the apoptosis of renal tubular cells. Inhibition of EZH2 attenuates the tubular cells apoptosis and protects the kidney against the injury. EZH2 reduces the apoptosis of tubular cells via suppressing Lcn2 expression, a biomarker for chronic renal injury (68).
Renal tubular epithelial cells are rich in uric acid transporters, which are important target cells to ensure the transport of uric acid. When they are damaged, the number of uric acid transporters is reduced. Verzola et al. (60) have shown that high uric acid levels induce apoptosis of renal proximal tubule cells by triggering the NADPH oxidase signal and the pathway of urate transporter 1 (URAT1). Uric acid promotes the interaction between Bax protein and mitochondrial membrane, which opens the mitochondrial voltage dependent anion channel. As a result, cytochrome c and other apoptotic factors are released from mitochondria, leading to activation of caspase-9 and apoptosis. In addition, Sánchez-Lozada et al. (50) suggested that uric acid-induced cell apoptosis is associated with abnormal mitochondrial respiratory chain and the key injury factor is the exceptional surplus of reactive oxygen species. These results underline the relevance of apoptosis and oxidative stress. Hence, our next work will focus on oxidative stress and its markers and confirm the inhibition effect of 3-DZNeP on oxidative stress in hyperuricemia-induced renal injury.
In conclusion, our data suggest that EZH2 inhibition significantly decreases serum uric acid levels by lowering serum XOD activity and alleviates renal pathological damages induced by hyperuricemia through various mechanisms, involving suppressing of TGF-β1/Smad3 and EGFR/ERK1/2 signaling, downregulating the level of proinflammation chemokines/cytokines, and attenuating apoptosis of renal tubular cells. Thus EZH2 may represent a viable target for attenuating hyperuricemia-induced renal injury and delaying the development of CKD.
GRANTS
This study was supported by the National Nature Science Foundation of China Grants 81670690, 81470991, and 81200492 (to N. Liu) and 81270778, 81470920, 81670623, and 81830021 (to S. Zhuang), Key Discipline Construction Project of Pudong Health Bureau of Shanghai Grant PWZxk2017-05 (to N. Liu), Branch Grant of National Key Grants of Ministry of Science and Technology 2018YFA0108802 (to S. Zhuang), National Institute of Diabetes and Digestive and Kidney Diseases Grant 2R01-DK-08506505A1 (to S. Zhuang), Shanghai Scientific Committee of China Grant 13PJ1406900 (to N. Liu), and Science Technology Grant of Jiangxi Province Municipal Health Commision 20184077 (to L. Fang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.L. conceived and designed research; Y.S., L.X., M.T., L.F., J.L., H.G., S.M., T.L., Y.W., W.B., and N.L. performed experiments; Y.S., L.X., M.T., W.B., and N.L. analyzed data; Y.S. and N.L. interpreted results of experiments; Y.S. and N.L. prepared figures; Y.S., L.X., S.Z., and N.L. drafted manuscript; Y.S., A.Q., S.Z., and N.L. edited and revised manuscript; Y.S., L.X., M.T., L.F., J.L., H.G., S.M., T.L., Y.W., W.B., A.Q., S.Z., and N.L. approved final version of manuscript.
REFERENCES
- 1.Abouantoun TJ, Castellino RC, MacDonald TJ. Sunitinib induces PTEN expression and inhibits PDGFR signaling and migration of medulloblastoma cells. J Neurooncol 101: 215–226, 2011. doi: 10.1007/s11060-010-0259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzai N, Kanai Y, Endou H. New insights into renal transport of urate. Curr Opin Rheumatol 19: 151–157, 2007. doi: 10.1097/BOR.0b013e328032781a. [DOI] [PubMed] [Google Scholar]
- 3.Bae JM. Interpretation of the hygiene and microflora hypothesis for allergic diseases through epigenetic epidemiology. Epidemiol Health 40: e2018006, 2018. doi: 10.4178/epih.e2018006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrera-Chimal J, Estrela GR, Lechner SM, Giraud S, El Moghrabi S, Kaaki S, Kolkhof P, Hauet T, Jaisser F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int 93: 1344–1355, 2018. doi: 10.1016/j.kint.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 5.Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beauséjour M, Beaulieu JF, Lipkin SM, Gudas LJ. Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res 319: 1463–1470, 2013. doi: 10.1016/j.yexcr.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bideak A, Blaut A, Hoppe JM, Müller MB, Federico G, Eltrich N, Gröne HJ, Locati M, Vielhauer V. The atypical chemokine receptor 2 limits renal inflammation and fibrosis in murine progressive immune complex glomerulonephritis. Kidney Int 93: 826–841, 2018. doi: 10.1016/j.kint.2017.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Bontha SV, Maluf DG, Archer KJ, Dumur CI, Dozmorov MG, King AL, Akalin E, Mueller TF, Gallon L, Mas VR. Effects of DNA methylation on progression to interstitial fibrosis and tubular atrophy in renal allograft biopsies: a multi-omics approach. Am J Transplant 17: 3060–3075, 2017. doi: 10.1111/ajt.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Böttinger EP. TGF-beta in renal injury and disease. Semin Nephrol 27: 309–320, 2007. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Zhouhua W, Jie Z, Xinlu F, Jinqiang L, Yuwen Q, Zhiying H. Renal interstitial fibrosis induced by high-dose mesoporous silica nanoparticles via the NF-κB signaling pathway. Int J Nanomedicine 10: 1–22, 2014. doi: 10.2147/ijn.s73538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou HW, Chiu HT, Tsai CW, Ting IW, Yeh HC, Huang HC, Kuo CC; CMUH Kidney Research Group . Comparative effectiveness of allopurinol, febuxostat and benzbromarone on renal function in chronic kidney disease patients with hyperuricemia: a 13-year inception cohort study. Nephrol Dial Transplant 33: 1620–1627, 2017. doi: 10.1093/ndt/gfx313. [DOI] [PubMed] [Google Scholar]
- 11.Gao L, Liu MM, Zang HM, Ma QY, Yang Q, Jiang L, Ren GL, Li HD, Wu WF, Wang JN, Wei B, Liu XQ, Jiang C, Huang C, Li J, Meng XM. Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab Invest 98: 911–923, 2018. doi: 10.1038/s41374-018-0052-5. [DOI] [PubMed] [Google Scholar]
- 12.Garofalo C, De Stefano T, Vita C, Vinci G, Balia F, Nettuno F, Scarpati L, Sguazzo A, Sagliocchi A, Pacilio M, Minutolo R, De Nicola L, Borrelli S. [Hyperuricaemia and chronic kidney disease] G Ital Nefrol 35: 2018-vol1, 2018. [PubMed] [Google Scholar]
- 13.Gewin LS. Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68-69: 248–262, 2018. doi: 10.1016/j.matbio.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh M, Thangada S, Dasgupta O, Khanna KM, Yamase HT, Kashgarian M, Hla T, Shapiro LH, Ferrer FA. Cell-intrinsic sphingosine kinase 2 promotes macrophage polarization and renal inflammation in response to unilateral ureteral obstruction. PLoS One 13: e0194053, 2018. doi: 10.1371/journal.pone.0194053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, Ljungman M, Merajver SD, Kleer CG. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 28: 843–853, 2009. doi: 10.1038/onc.2008.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan M, Li W, Xu L, Zeng Y, Wang D, Zheng Z, Lyv F, Xue Y. Metformin improves epithelial-to-mesenchymal transition induced by TGF-β1 in renal tubular epithelial NRK-52E cells via inhibiting Egr-1. J Diabetes Res 2018: 1031367, 2018. doi: 10.1155/2018/1031367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gude D, Chennamsetty S, Jha R. Fathoming uric acid nephropathy. Saudi J Kidney Dis Transpl 24: 1259–1261, 2013. doi: 10.4103/1319-2442.121307. [DOI] [PubMed] [Google Scholar]
- 18.Hahn K, Kanbay M, Lanaspa MA, Johnson RJ, Ejaz AA. Serum uric acid and acute kidney injury: a mini review. J Adv Res 8: 529–536, 2017. doi: 10.1016/j.jare.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hibino S, Saito Y, Muramatsu T, Otani A, Kasai Y, Kimura M, Saito H. Inhibitors of enhancer of zeste homolog 2 (EZH2) activate tumor-suppressor microRNAs in human cancer cells. Oncogenesis 3: e104, 2014. doi: 10.1038/oncsis.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalal DI, Chonchol M, Chen W, Targher G. Uric acid as a target of therapy in CKD. Am J Kidney Dis 61: 134–146, 2013. doi: 10.1053/j.ajkd.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson RJ, Bakris GL, Borghi C, Chonchol MB, Feldman D, Lanaspa MA, Merriman TR, Moe OW, Mount DB, Sanchez Lozada LG, Stahl E, Weiner DE, Chertow GM. Hyperuricemia, acute and chronic kidney disease, hypertension, and cardiovascular disease: report of a scientific workshop organized by the National Kidney Foundation. Am J Kidney Dis 71: 851–865, 2018. doi: 10.1053/j.ajkd.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson RJ, Nakagawa T, Jalal D, Sánchez-Lozada LG, Kang DH, Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrol Dial Transplant 28: 2221–2228, 2013. doi: 10.1093/ndt/gft029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joo CK, Kim HS, Park JY, Seomun Y, Son MJ, Kim JT. Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-beta involves multiple signaling pathways. Oncogene 27: 614–628, 2008. doi: 10.1038/sj.onc.1210649. [DOI] [PubMed] [Google Scholar]
- 24.Kanellis J, Watanabe S, Li JH, Kang DH, Li P, Nakagawa T, Wamsley A, Sheikh-Hamad D, Lan HY, Feng L, Johnson RJ. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 41: 1287–1293, 2003. doi: 10.1161/01.HYP.0000072820.07472.3B. [DOI] [PubMed] [Google Scholar]
- 25.Keen HI, Davis WA, Latkovic E, Drinkwater JJ, Nossent J, Davis TM. Ultrasonographic assessment of joint pathology in type 2 diabetes and hyperuricemia: The Fremantle Diabetes Study Phase II. J Diabetes Complications 32: 400–405, 2018. doi: 10.1016/j.jdiacomp.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Kim IY, Lee DW, Lee SB, Kwak IS. The role of uric acid in kidney fibrosis: experimental evidences for the causal relationship. BioMed Res Int 2014: 638732, 2014. doi: 10.1155/2014/638732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondo Y. Targeting histone methyltransferase EZH2 as cancer treatment. J Biochem 156: 249–257, 2014. doi: 10.1093/jb/mvu054. [DOI] [PubMed] [Google Scholar]
- 28.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905, 2002. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee E, Yi JY, Chung E, Son Y. Transforming growth factorbeta(1) transactivates EGFR via an H2O2-dependent mechanism in squamous carcinoma cell line. Cancer Lett 290: 43–48, 2010. doi: 10.1016/j.canlet.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Hong Z, Peng Z, Zhao Y, Shao R. Acetylshikonin from Zicao ameliorates renal dysfunction and fibrosis in diabetic mice by inhibiting TGF-β1/Smad pathway. Hum Cell 31: 199–209, 2018. doi: 10.1007/s13577-017-0192-8. [DOI] [PubMed] [Google Scholar]
- 31.Liu B, Ding F, Hu D, Zhou Y, Long C, Shen L, Zhang Y, Zhang D, Wei G. Human umbilical cord mesenchymal stem cell conditioned medium attenuates renal fibrosis by reducing inflammation and epithelial-to-mesenchymal transition via the TLR4/NF-κB signaling pathway in vivo and in vitro. Stem Cell Res Ther 9: 7, 2018. [Erratum in Stem Cell Res Ther 9: 76, 2018]. doi: 10.1186/s13287-017-0760-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu N, Guo JK, Pang M, Tolbert E, Ponnusamy M, Gong R, Bayliss G, Dworkin LD, Yan H, Zhuang S. Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J Am Soc Nephrol 23: 854–867, 2012. doi: 10.1681/ASN.2011050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu N, Wang L, Yang T, Xiong C, Xu L, Shi Y, Bao W, Chin YE, Cheng SB, Yan H, Qiu A, Zhuang S. EGF receptor inhibition alleviates hyperuricemic nephropathy. J Am Soc Nephrol 26: 2716–2729, 2015. doi: 10.1681/ASN.2014080793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu N, Xu L, Shi Y, Fang L, Gu H, Wang H, Ding X, Zhuang S. Pharmacologic targeting ERK1/2 attenuates the development and progression of hyperuricemic nephropathy in rats. Oncotarget 8: 33807–33826, 2017. doi: 10.18632/oncotarget.16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 69: 213–217, 2006. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- 36.Long CL, Qin XC, Pan ZY, Chen K, Zhang YF, Cui WY, Liu GS, Wang H. Activation of ATP-sensitive potassium channels protects vascular endothelial cells from hypertension and renal injury induced by hyperuricemia. J Hypertens 26: 2326–2338, 2008. doi: 10.1097/HJH.0b013e328312c8c1. [DOI] [PubMed] [Google Scholar]
- 37.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21: 998–1009, 2015. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, Armaiz-Pena GN, Hu W, Stone RL, Munkarah A, Ravoori MK, Shahzad MM, Lee JW, Mora E, Langley RR, Carroll AR, Matsuo K, Spannuth WA, Schmandt R, Jennings NB, Goodman BW, Jaffe RB, Nick AM, Kim HS, Guven EO, Chen YH, Li LY, Hsu MC, Coleman RL, Calin GA, Denkbas EB, Lim JY, Lee JS, Kundra V, Birrer MJ, Hung MC, Lopez-Berestein G, Sood AK. Regulation of tumor angiogenesis by EZH2. Cancer Cell 18: 185–197, 2010. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulay SR, Evan A, Anders HJ. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant 29: 507–514, 2014. doi: 10.1093/ndt/gft248. [DOI] [PubMed] [Google Scholar]
- 40.Mulè G, Nardi E, Lattuca L, Cottone S. Hyperuricemia and high blood pressure at rest and during exercise: guilty or innocent? The jury is still out. J Clin Hypertens (Greenwich) 20: 557–559, 2018. doi: 10.1111/jch.13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyễn-Thanh T, Kim D, Lee S, Kim W, Park SK, Kang KP. Inhibition of histone deacetylase 1 ameliorates renal tubulointerstitial fibrosis via modulation of inflammation and extracellular matrix gene transcription in mice. Int J Mol Med 41: 95–106, 2018. doi: 10.3892/ijmm.2017.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niu Y, Li H, Gao L, Lin H, Kung H, Lin MC, Leung KS, Wong MH, Xiong W, Li L. Old drug, new indication: olsalazine sodium reduced serum uric acid levels in mice via inhibiting xanthine oxidoreductase activity. J Pharmacol Sci 135: 114–120, 2017. doi: 10.1016/j.jphs.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol 19: 2407–2413, 2008. doi: 10.1681/ASN.2008010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 297: F996–F1005, 2009. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, White SA, Burt AD, Oakley F, Tsukamoto H, Mann DA, Mann J. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 56: 1129–1139, 2012. doi: 10.1002/hep.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rayego-Mateos S, Morgado-Pascual JL, Rodrigues-Diez RR, Rodrigues-Diez R, Falke LL, Mezzano S, Ortiz A, Egido J, Goldschmeding R, Ruiz-Ortega M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J Pathol 244: 227–241, 2018. doi: 10.1002/path.5007. [DOI] [PubMed] [Google Scholar]
- 47.Rock KL, Kataoka H, Lai JJ. Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol 9: 13–23, 2013. doi: 10.1038/nrrheum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romi MM, Arfian N, Tranggono U, Setyaningsih WA, Sari DC. Uric acid causes kidney injury through inducing fibroblast expansion, Endothelin-1 expression, and inflammation. BMC Nephrol 18: 326, 2017. doi: 10.1186/s12882-017-0736-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryu ES, Kim MJ, Shin HS, Jang YH, Choi HS, Jo I, Johnson RJ, Kang DH. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol 304: F471–F480, 2013. doi: 10.1152/ajprenal.00560.2012. [DOI] [PubMed] [Google Scholar]
- 50.Sánchez-Lozada LG, Lanaspa MA, Cristóbal-García M, García-Arroyo F, Soto V, Cruz-Robles D, Nakagawa T, Yu MA, Kang DH, Johnson RJ. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron, Exp Nephrol 121: e71–e78, 2013. doi: 10.1159/000345509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sánchez-Lozada LG, Tapia E, Soto V, Avila-Casado C, Franco M, Zhao L, Johnson RJ. Treatment with the xanthine oxidase inhibitor febuxostat lowers uric acid and alleviates systemic and glomerular hypertension in experimental hyperuricaemia. Nephrol Dial Transplant 23: 1179–1185, 2008. doi: 10.1093/ndt/gfm783. [DOI] [PubMed] [Google Scholar]
- 52.Savitskaya MA, Onishchenko GE. Mechanisms of apoptosis. Biochemistry (Mosc) 80: 1393–1405, 2015. doi: 10.1134/S0006297915110012. [DOI] [PubMed] [Google Scholar]
- 53.Sibunruang C, Ingsathit A, Kantachuvesiri P, Radinahamed P, Rattanasiri S, Pootracool P, Kijvikai K, Sumethkul V, Kantachuvesiri S. Increased urine transforming growth factor β1 (TGF-β1) and serum uric acid are associated with an early decline of glomerular filtration rate in kidney transplant recipients. Transplant Proc 47: 304–308, 2015. doi: 10.1016/j.transproceed.2014.11.037. [DOI] [PubMed] [Google Scholar]
- 54.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 647: 21–29, 2008. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 47: 51–59, 2006. doi: 10.1053/j.ajkd.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 56.Tampe B, Zeisberg M. Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol Dial Transplant 29, Suppl 4: iv72–iv79, 2014. doi: 10.1093/ndt/gft025. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka K, Nakayama M, Kanno M, Kimura H, Watanabe K, Tani Y, Hayashi Y, Asahi K, Terawaki H, Watanabe T. Renoprotective effects of febuxostat in hyperuricemic patients with chronic kidney disease: a parallel-group, randomized, controlled trial. Clin Exp Nephrol 19: 1044–1053, 2015. doi: 10.1007/s10157-015-1095-1. [DOI] [PubMed] [Google Scholar]
- 58.Tang W, Ma J, Gu R, Ding X, Lei B, Wang X, Zhuang H, Xu G. Lipocalin 2 suppresses ocular inflammation by inhibiting the activation of NF-κβ pathway in endotoxin-induced uveitis. Cell Physiol Biochem 46: 375–388, 2018. doi: 10.1159/000488472. [DOI] [PubMed] [Google Scholar]
- 59.Umekawa T, Chegini N, Khan SR. Increased expression of monocyte chemoattractant protein-1 (MCP-1) by renal epithelial cells in culture on exposure to calcium oxalate, phosphate and uric acid crystals. Nephrol Dial Transplant 18: 664–669, 2003. doi: 10.1093/ndt/gfg140. [DOI] [PubMed] [Google Scholar]
- 60.Verzola D, Ratto E, Villaggio B, Parodi EL, Pontremoli R, Garibotto G, Viazzi F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS One 9: e115210, 2014. doi: 10.1371/journal.pone.0115210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Völkel P, Dupret B, Le Bourhis X, Angrand PO. Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res 7: 175–193, 2015. [PMC free article] [PubMed] [Google Scholar]
- 63.Woitas RP, Scharnagl H, Kleber ME, Delgado GE, Grammer TB, Pichler M, Krämer BK, März W, Stojakovic T. Neutrophil gelatinase-associated lipocalin levels are U-shaped in the Ludwigshafen Risk and Cardiovascular Health (LURIC) study–Impact for mortality. PLoS One 12: e0171574, 2017. doi: 10.1371/journal.pone.0171574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y, Wang L, Deng D, Zhang Q, Liu W. Renalase protects against renal fibrosis by inhibiting the activation of the ERK signaling pathways. Int J Mol Sci 18: 855, 2017. doi: 10.3390/ijms18050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao X, Senavirathna LK, Gou X, Huang C, Liang Y, Liu L. EZH2 enhances the differentiation of fibroblasts into myofibroblasts in idiopathic pulmonary fibrosis. Physiol Rep 4: e12915, 2016. doi: 10.14814/phy2.12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiong C, Liu N, Fang L, Zhuang S, Yan H. Suramin inhibits the development and progression of peritoneal fibrosis. J Pharmacol Exp Ther 351: 373–382, 2014. doi: 10.1124/jpet.114.215228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan M, Chen K, He L, Li S, Huang D, Li J. Uric acid induces cardiomyocyte apoptosis via activation of calpain-1 and endoplasmic reticulum stress. Cell Physiol Biochem 45: 2122–2135, 2018. doi: 10.1159/000488048. [DOI] [PubMed] [Google Scholar]
- 68.Yang HT, Yim H, Cho YS, Kym D, Hur J, Kim JH, Chun W, Kim HS. Assessment of biochemical markers in the early post-burn period for predicting acute kidney injury and mortality in patients with major burn injury: comparison of serum creatinine, serum cystatin-C, plasma and urine neutrophil gelatinase-associated lipocalin. Crit Care 18: R151, 2014. doi: 10.1186/cc13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang J, Zhang N, Gao R, Zhu Y, Zhang Z, Xu X, Wang J, Li Z, Liu X, Li Z, Li J, Bi J, Kong C. TGF-β1 induced fascin1 expression facilitates the migration and invasion of kidney carcinoma cells through ERK and JNK signaling pathways. Biochem Biophys Res Commun 501: 913–919, 2018. doi: 10.1016/j.bbrc.2018.05.081. [DOI] [PubMed] [Google Scholar]
- 70.Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 4: 331–341, 2013. doi: 10.1007/s13238-013-2093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan J, Shen Y, Yang X, Xie Y, Lin X, Zeng W, Zhao Y, Tian M, Zha Y. Thymosin β4 alleviates renal fibrosis and tubular cell apoptosis through TGF-β pathway inhibition in UUO rat models. BMC Nephrol 18: 314, 2017. doi: 10.1186/s12882-017-0708-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou X, Zang X, Ponnusamy M, Masucci MV, Tolbert E, Gong R, Zhao TC, Liu N, Bayliss G, Dworkin LD, Zhuang S. Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol 27: 2092–2108, 2016. doi: 10.1681/ASN.2015040457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Y, Fang L, Jiang L, Wen P, Cao H, He W, Dai C, Yang J. Uric acid induces renal inflammation via activating tubular NF-κB signaling pathway. PLoS One 7: e39738, 2012. doi: 10.1371/journal.pone.0039738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu S, Goldschmidt-Clermont PJ, Dong C. Transforming growth factor-beta-induced inhibition of myogenesis is mediated through Smad pathway and is modulated by microtubule dynamic stability. Circ Res 94: 617–625, 2004. doi: 10.1161/01.RES.0000118599.25944.D5. [DOI] [PubMed] [Google Scholar]