

Abstract
Epoxyeicosatrienoic acids (EETs) decrease cardiac ischemia-reperfusion injury; however, the mechanism of their protective effect remains elusive. Here, we investigated the cardioprotective action of a novel EET analog, EET-B, in reperfusion and the role of hypoxia-inducible factor (HIF)-1α in such action of EET-B. Adult male rats were subjected to 30 min of left coronary artery occlusion followed by 2 h of reperfusion. Administration of 14,15-EET (2.5 mg/kg) or EET-B (2.5 mg/kg) 5 min before reperfusion reduced infarct size expressed as a percentage of the area at risk from 64.3 ± 1.3% in control to 42.6 ± 1.9% and 46.0 ± 1.6%, respectively, and their coadministration did not provide any stronger effect. The 14,15-EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (2.5 mg/kg) inhibited the infarct size-limiting effect of EET-B (62.5 ± 1.1%). Similarly, the HIF-1α inhibitors 2-methoxyestradiol (2.5 mg/kg) and acriflavine (2 mg/kg) completely abolished the cardioprotective effect of EET-B. In a separate set of experiments, the immunoreactivity of HIF-1α and its degrading enzyme prolyl hydroxylase domain protein 3 (PHD3) were analyzed in the ischemic areas and nonischemic septa. At the end of ischemia, the HIF-1α immunogenic signal markedly increased in the ischemic area compared with the septum (10.31 ± 0.78% vs. 0.34 ± 0.08%). After 20 min and 2 h of reperfusion, HIF-1α immunoreactivity decreased to 2.40 ± 0.48% and 1.85 ± 0.43%, respectively, in the controls. EET-B blunted the decrease of HIF-1α immunoreactivity (7.80 ± 0.69% and 6.44 ± 1.37%, respectively) and significantly reduced PHD3 immunogenic signal in ischemic tissue after reperfusion. In conclusion, EET-B provides an infarct size-limiting effect at reperfusion that is mediated by HIF-1α and downregulation of its degrading enzyme PHD3.
NEW & NOTEWORTHY The present study shows that EET-B is an effective agonistic 14,15-epoxyeicosatrienoic acid analog, and its administration before reperfusion markedly reduced myocardial infarction in rats. Most importantly, we demonstrate that increased hypoxia-inducible factor-1α levels play a role in cardioprotection mediated by EET-B in reperfusion likely by mechanisms including downregulation of the hypoxia-inducible factor -1α-degrading enzyme prolyl hydroxylase domain protein 3.
Keywords: epoxyeicosatrienoic acid, heart, hypoxia-inducible factor-1α, ischemia-reperfusion, prolyl hydroxylase 3
INTRODUCTION
Epoxyeicosatrienoic acids (EETs) are the cytochrome P-450 monooxidase metabolites of arachidonic acid known to protect mammals against lethal myocardial injury caused by acute ischemia-reperfusion (I/R) insult in various experimental settings (12, 33, 40). It has been shown that the acute elevation of their levels by exogenous administration of EETs or inhibition of the EET-degrading enzyme soluble epoxide hydrolase (sEH) before ischemia as well as before reperfusion reduces myocardial I/R injury (3, 9, 13, 14, 26, 29, 31, 41). Despite strong evidence for cardioprotective effects of EETs, the mechanism(s) by which EETs protect the myocardium against I/R injury is not yet satisfactorily elucidated.
The major limitations of therapeutic application of EETs are their chemical and metabolic instability and very fast conversion (within minutes) to the biologically inactive dihydroxyeicosatrienoic acids by sEH. To circumvent this limitation of endogenous EETs, several synthetic EET analogs with markedly longer half-life (hours) have been developed. EET analogs have biological activity, possess many structural features required for their stability and bioavailability (4, 19), and have demonstrated promising cardioprotective actions (1, 4, 7, 16).
The transcription factor hypoxia-inducible factor (HIF)-1α is a key regulator of physiological and cellular mechanisms of adaptation to oxygen deficiency (38). HIF-1α plays an essential role in triggering cellular protection and metabolic alteration during hypoxic conditions. In hypoxia, HIF-1α levels increase if the HIF-1α-degrading enzymes prolyl-4-hydroxylase domain proteins (PHDs) are inactive because of low oxygen levels. Apart from hypoxia, elevated eicosanoid signaling also can increase HIF-1α levels (35).
It has been suggested that HIF-1α is involved in the protection of the heart against acute I/R injury (34). Mice lacking HIF-1α exhibit an absence of heart protection induced by ischemic preconditioning (11, 37). Moreover, acute administration of the HIF-1α inhibitor acriflavine (Acri) or 2-methoxyestradiol (2-ME) abolished various cardioprotective interventions (37, 42, 50). Finally, ischemic or pharmacological postconditioning markedly increased gene and protein expression of HIF-1α in ischemic tissue after reperfusion (50, 52). Altogether, these findings suggest that activation of HIF-1α plays an important role in cardiac ischemic tolerance.
With respect to HIF-1α and EETs, it is well known that EETs activate HIF-1α-mediated expression of proangiogenic transcription factors (36, 43, 49). Interestingly, Batchu et al. (3) demonstrated that acute administration of sEH inhibitor increased HIF-1α DNA-binding activity in reperfusion and improved resistance of myocytes to acute I/R.
In the present study, we investigated the cardioprotective action of acute administration of EET-B, a novel, stable, and orally active EET analog in reperfusion. Experiments were also carried out to determine the mechanism of cardioprotection afforded by EET-B in relation to a role for HIF-1α and its degrading enzyme PHD3.
METHODS
All experiments were conducted in accordance with the “Position of the American Heart Association on Research and Animal Use” adopted by the American Heart Association and the guidelines of the Biomedical Resource Center and the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. The Medical College of Wisconsin is accredited by the American Association of Laboratory Animal Care.
Materials.
The EET analog N-isopropyl-N-{5-[(2-pivalamidobenzo[d]thiazol-4-yl)oxy]pentyl]heptanamide (EET-B; Fig. 1A) was designed and synthesized together with 14,15-EET and the EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE) in the laboratory of John R. Falck. HIF-1α inhibitors (2-ME and Acri) were obtained from Sigma-Aldrich (St. Louis, MO). EET-B, 14,15-EET, and 14,15-EEZE were dissolved in a vehicle composed of 95% ethanol, polyethylene glycol 200, and 1 N sodium hydroxide (5:5:1). Acri was dissolved in saline, and 2-ME was dissolved in DMSO (0.01% final concentration). All drugs were made up fresh daily.
Fig. 1.

A: structure of the epoxyeicosatrienoic acid (EET) analog N-isopropyl-N-{5-[(2-pivalamidobenzo[d]thiazol-4-yl)oxy]pentyl}heptanamide (EET-B). B: design of the experiments analyzing the cardioprotective action of EET-B in rat hearts. C: design of the experiments analyzing immunoreactivity of hypoxia-inducible factor (HIF)-1α and prolyl hydroxylase domain protein 3 (PHD3) in the ischemic area and nonischemic septum at the end of ischemia and in reperfusion of vehicle- and EET-B-treated rats. See materials and methods for a detailed description. 2-ME, 2-methoxyestradiol; 14,15-EEZE, 14,15-epoxyeicosa-5(Z)-enoic acid; Acri, acriflavine; EET, epoxyeicosatrienoic acid; ISCH, ischemia; REP, reperfusion.
Intact rat preparation protocol.
Adult male Sprague-Dawley rats weighing 250–300 g were fasted overnight and anesthetized with Inactin (100 mg/kg ip, Sigma-Aldrich). Animals were intubated with a cannula connected to a rodent ventilator (Columbus Instruments) and ventilated with room air supplemented with 100% oxygen. Cannulas were placed into the jugular vein and carotid artery to administer drugs and measure peripheral hemodynamics, respectively. Hemodynamics and heart rate were monitored throughout the experiment. Arterial blood pH, Pco2, and Po2 were monitored at selected intervals by an AVL automatic blood gas system and maintained within normal physiological limits (pH 7.35−7.45, Pco2: 30−40 mmHg, and Po2: 85−120 mmHg) by adjusting the respiration rate and oxygen flow or by intravenous administration of 1.5% sodium bicarbonate, if necessary. Body temperature was maintained at 37 ± 1°C with a heating pad.
A left thoracotomy was performed, and the pericardium was removed to reveal the location of the left anterior descending coronary artery (LAD). A polyester ligature (6/0, Ethibond, Ethicon, Scotland) was placed around the LAD ~1–2 mm distal to the origin, and an occlusive snare was placed around it. The ends of the suture were threaded through a polyethylene tube. After the surgical preparation, rats were allowed to stabilize for 10 min before the ischemic interventions. Regional myocardial ischemia was induced by tightening the ligature around the coronary artery. Characteristic changes in myocardial color and a transient decrease in blood pressure verified the complete coronary artery occlusion. After the 30 min of occlusion, the snare was released and reperfusion of the previously ischemic tissue was indicated by transient decrease of blood pressure, change of myocardial color, and appearance of reperfusion arrhythmias.
Experimental treatment protocol.
Rats were assigned to 13 groups (n = 5–9 rats/group) for the different treatments. EET-B (2.5 mg/kg) was administered in 0.15 ml of vehicle by slow (1–2 min) injection into the jugular vein 10 min before the 30-min coronary occlusion or 5 min before reperfusion (Fig. 1B). The dose of EET analog was chosen based on preliminary dose-response experiments with 14,15-EET in the range of 1.0 to 5.0 mg/kg. The results indicated that doses of 14,15-EET above 2.5 mg/kg did not further reduce infarct size (IS). In the HIF-1α inhibitor experiments, the dose of 2-ME (0.5 or 2.5 mg/kg) or Acri (2.0 mg/kg) were chosen from previously published studies (5, 34), and the compounds were administered 10 min before reperfusion (5 min before EET-B; Fig. 1B) by slow (1–2 min) intravenous injection.
IS determination.
At the end of the 2-h reperfusion period, the LAD was reoccluded. To determine the anatomic area at risk (AAR) and the nonischemic area, 1 ml of Patent blue dye was injected into the jugular vein, and the heart was arrested by an intravenous injection of potassium chloride. The heart was then immediately removed, and the left ventricle (LV) was dissected and sliced into serial transverse sections 1–2 mm in thickness. The nonstained ischemic area and the blue-stained nonischemic area were separated and incubated with 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) in 0.1 M phosphate buffer (pH 7.4) at 37°C for 15 min. After an overnight incubation in 10% formaldehyde, the noninfarcted and infarcted tissues within the AAR were separated under a dissecting microscope and determined gravimetrically. IS was expressed as a percentage of the AAR.
Immunohistochemistry.
In separate sets of vehicle- and EET-B-treated rats, immunoreactivity of HIF-1α and its degrading enzyme PHD3 was analyzed in the ischemic area and nonischemic septa at the end of ischemia and in reperfusion. The surgical procedures for the intact rat preparation and EET-B administration before reperfusion have been described above. Hearts were excised at the end of 30-min ischemia (without reperfusion) and after 20 min and 2 h of reperfusion (Fig. 1C). After that, hearts were washed with Tyrode solution, perfusion fixed, and stored in 4% paraformaldehyde for 2 days at 4°C. Hearts were cut perpendicularly to the long axis at the largest circumference and embedded in paraffin. Tissue sections were immunostained with anti-HIF-1α (1:50, Santa Cruz Biotechnology, Dallas, TX) or anti-PHD3 (1:50, Santa Cruz Biotechnologies). Biotinylated donkey anti-mouse secondary antibody (1:200) was used for the development with avidin-biotinylated horseradish peroxidase complex (Vectastain ABC Elite kit, Vector Laboratories, Burlingame, CA) followed by counterstaining with hematoxylin. Stained sections were visualized by light microscopy at ×400 magnification, and digital images of the stained sections were taken for analysis using Nikon NIS Elements Software (Nikon Instruments, Melville, NY). HIF-1α- and PHD3-positive areas, respectively (4 different sections/heart, 6 images in each) were determined by two experienced observers in a blinded fashion. PHD3 nuclear positivity was quantified as the nuclear RGB intensity profile and was normalized to cytosolic values (25 nuclei/section, 4 different sections/heart).
RNA isolation and real-time RT-PCR analysis.
In the last set of experiments, the effect of EET-B treatment on gene expression of HIF-1α (Hif-1α) and PHD3 (Egln3) was analyzed. Rats were anesthetized, a cannula placed into the jugular vein as described above, and vehicle or EET-B (2.5 mg/kg) was administered by slow (1–2 min) injection in 0.15 ml of vehicle. Two hours after drug administration, hearts were excised, washed with cold saline (0°C), and divided to right ventricle, septum, and LV. Total cellular RNA was extracted from each LV sample using Trizol Reagent (Invitrogen) according to the manufacturer’s instruction. The RNA samples were treated by DNase I (Sigma-Aldrich) to avoid contamination of DNA. One microgram of total RNA was converted to cDNA using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA) using random primers according to the manufacturer’s instructions. Real-time PCR was performed on a Light Cycler 480 (Roche Applied Sciences) using the TaqMan Gene Expression Assays (Applied Biosystems). Topoisomerase I (TopI) was used as a reference gene. VIC-labeled probe for TopI was combined with Hif-1a or Egln3 FAM-labeled probes. The appropriate Master kit, 5× HOT FIREPol Probe qPCR Mix Plus (NO ROX) (Solis BioDyne), was used according to the manufacturer’s protocol with the following temperature profile: preincubation (95°C/12 min), amplification 55 cycles (denaturation 95°C/15 s, annealing/elongation 60°C/1 min), and cooling (40°C/30 s). Data were analyzed using GenEx software.
Statistical analysis.
Results are expressed as means ± SE. One-way ANOVA and a subsequent Student-Newman-Keuls test were used for comparison of differences in normally distributed variables between three and more groups, and group-to-group comparisons were done using an unpaired Student’s t-test. Differences were considered statistically significant when P < 0.05.
RESULTS
Hemodynamics.
There were no significant differences in baseline hemodynamic data (Table 1). In ischemia, arterial blood pressure slightly decreased compared with baseline values in all experimental groups. At the end of reperfusion, an additional but mild arterial blood pressure reduction was observed. Any of the pharmacological treatments before reperfusion had no significant effect on blood pressure compared with their corresponding controls. Heart rate did not differ among the groups compared with their corresponding controls in ischemia as well as in reperfusion (Table 1). Altogether, the acute administration of vasoactive 14,15-EET and EET-B and their combinations with other drugs had no hemodynamic effects in rats subjected to acute I/R.
Table 1.
Heart rate, systolic blood pressure, and diastolic blood pressure after stabilization (baseline), at 15 min of coronary artery occlusion, and at the end of 2 h of reperfusion as well as relative sizes of the AAR/LV
| Heart Rate, beats/min |
Systolic Blood Pressure, mmHg |
Diastolic Blood Pressure, mmHg |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 15-min occlusion | 2-h reperfusion | Baseline | 15-min occlusion | 2-h reperfusion | Baseline | 15-min occlusion | 2-h reperfusion | AAR/LV | |
| Vehicle I | 399 ± 18 | 396 ± 17 | 379 ± 14 | 133 ± 5 | 106 ± 4 | 105 ± 5 | 105 ± 4 | 86 ± 5 | 76 ± 5 | 41.0 ± 2.1 |
| Vehicle II | 390 ± 6 | 393 ± 9 | 393 ± 15 | 151 ± 7 | 106 ± 5 | 109 ± 3 | 117 ± 3 | 86 ± 6 | 74 ± 4 | 39.4 ± 2.6 |
| EET-B (Pre) | 408 ± 15 | 422 ± 10 | 392 ± 22 | 134 ± 6 | 124 ± 6 | 108 ± 2 | 107 ± 5 | 103 ± 5 | 80 ± 6 | 43.3 ± 1.8 |
| EET-B | 393 ± 10 | 397 ± 8 | 390 ± 12 | 131 ± 5 | 105 ± 4 | 107 ± 4 | 101 ± 6 | 87 ± 6 | 73 ± 3 | 46.9 ± 1.5 |
| 14,15-EET | 368 ± 7 | 371 ± 8 | 343 ± 6 | 148 ± 3 | 132 ± 8 | 105 ± 5 | 119 ± 3 | 109 ± 7 | 73 ± 6 | 40.4 ± 3.0 |
| EET-B + 14,15-EET | 395 ± 9 | 397 ± 6 | 373 ± 8 | 147 ± 3 | 123 ± 3 | 115 ± 3 | 117 ± 3 | 100 ± 3 | 81 ± 3 | 47.4 ± 1.3 |
| 14,15-EEZE + EET-B | 407 ± 9 | 403 ± 11 | 393 ± 11 | 135 ± 2 | 113 ± 5 | 110 ± 4 | 107 ± 2 | 89 ± 6 | 78 ± 2 | 40.7 ± 2.1 |
| 2-ME (0.5) + EET-B | 372 ± 11 | 368 ± 7 | 370 ± 5 | 126 ± 6 | 125 ± 8 | 103 ± 4 | 96 ± 4 | 104 ± 7 | 68 ± 5 | 39.1 ± 2.0 |
| 2-ME (2.5) + EET-B | 402 ± 10 | 394 ± 11 | 362 ± 9 | 154 ± 9 | 125 ± 6 | 109 ± 4 | 124 ± 3 | 103 ± 7 | 75 ± 4 | 39.9 ± 2.1 |
| 2-ME (2.5) + 14,15-EET | 386 ± 14 | 368 ± 9 | 376 ± 11 | 139 ± 5 | 120 ± 6 | 118 ± 6 | 109 ± 4 | 98 ± 4 | 85 ± 4 | 39.0 ± 1.8 |
| 2-ME (2.5) | 390 ± 7 | 390 ± 9 | 392 ± 8 | 140 ± 6 | 127 ± 4 | 116 ± 2 | 109 ± 5 | 103 ± 6 | 83 ± 3 | 37.7 ± 2.7 |
| Acri + EET-B | 378 ± 11 | 360 ± 11 | 365 ± 11 | 145 ± 4 | 128 ± 8 | 125 ± 3 | 113 ± 4 | 103 ± 8 | 85 ± 4 | 35.3 ± 1.7* |
| Acri | 402 ± 13 | 416 ± 8 | 390 ± 13 | 152 ± 8 | 144 ± 6 | 118 ± 8 | 124 ± 8 | 112 ± 9 | 93 ± 6 | 37.5 ± 1.9 |
All values are expressed as means ± SE of 5−9 rats/group. AAR/LV, area at risk as a percentage of the left ventricle; vehicle I, ethanol + polyethylene glycol + NaOH; vehicle II, DMSO; EET-B, N-isopropyl-N-{5-[(2-pivalamidobenzo[d]thiazol-4-yl)oxy]pentyl}heptanamide; EET, epoxyeicosatrienoic acid; 14,15-EEZE, 14,15-epoxyeicosa-5(Z)-enoic acid; 2-ME, 2-methoxyestradiol; Acri, acriflavine.
P < 0.05 vs. EET-B.
The mean normalized AAR (AAR/LV) was 37.5–47.5% (Table 1) and did not significantly differ among the groups except that the Acri + EET-B-treated group had significantly lower AAR/LV (35.3 ± 1.7%) compared with the EET-B-treated group (46.9 ± 1.5%).
Effect of EET-B on IS.
IS reached 63.6 ± 1.2% of the AAR in vehicle-treated controls. Acute administration of EET-B before ischemia or before reperfusion reduced IS/AAR to 51.3 ± 1.3% and 46.0 ± 1.6%, respectively (Fig. 2A). Figure 2B shows the dependence of IS on the size of AAR, with both normalized to LV size. EET-B decreased the slope of the linear relationship when administrated before ischemia; however, it shifted down the regression line without a change of slope when administered before reperfusion.
Fig. 2.

Myocardial infarct size expressed as a percentage of the area at risk (A) and relationship between area at risk and infarct size, both expressed as a percentage of the left ventricle (B), in rats administrated vehicle or the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg iv) 10 min before ischemia (Pre-Isch) or 5 min before reperfusion (Rep). Values are expressed as means ± SE from 5−9 rats. *P < 0.05 vs. vehicle.
Next, the effect of acute coadministration of EET-B with 14,15-EET or its antagonist 14,15-EEZE before reperfusion was examined. 14,15-EET alone provided cardioprotection comparable to EET-B, and coadministration with EET-B had no additive IS-limiting effect (Fig. 3). Acute administration of 14,15-EEZE before EET-B completely blocked the cardioprotective action of EET-B (62.5 ± 1.1%; Fig. 3A). Neither 14,15-EET nor 14,15-EEZE affected the slope of the linear regression lines (Fig. 3B).
Fig. 3.

Myocardial infarct size expressed as a percentage of the area at risk (A) and relationship between area at risk and infarct size, both expressed as a percentage of the left ventricle (B), in rats administrated vehicle, the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg iv) or 14,15-EET (2.5 mg/kg iv) 5 min before reperfusion, or the EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE; 2.5 mg/kg iv) alone or combined with EET-B 10 min before reperfusion. Values are expressed as means ± SE from 6−9 rats. *P < 0.05 vs. vehicle; †P < 0.05 vs. EET-B.
Effect of HIF-1α inhibitors on cardioprotection conferred by EET-B.
To test whether the cardioprotective effect of EET-B administered before reperfusion is mediated by HIF-1α, its inhibitors (2-ME or Acri) were administered 5 min before EET-B (10 min before reperfusion). 2-ME at low dose (0.5 mg/kg) partially reduced the IS-limiting effect of EET-B from 43.4 ± 1.1% to 52.1 ± 1.3% of the AAR. A five times higher dose of 2-ME (2.5 mg/kg) completely abolished the cardioprotective action of EET-B and 14,15-EET (63.3 ± 1.6% and 61.3 ± 1.1%, respectively; Fig. 4).
Fig. 4.

Myocardial infarct size expressed as a percentage of the area at risk (A) and relationship between area at risk and infarct size, both expressed as a percentage of the left ventricle (B), in rats administrated vehicle, the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg iv) or 14,15-EET (2.5 mg/kg iv) 5 min before reperfusion, or the hypoxia-inducible factor-1α inhibitor 2-methoxyestradiol (2-ME; 0.5 and 2.5 mg/kg iv, respectively) alone or combined with EET-B and 14,15-EET, respectively, 10 min before reperfusion. Values are means ± SE from 5−9 rats. *P < 0.05 vs. vehicle; †P < 0.05 vs. EET-B.
Despite the fact that AAR/LV was significantly lower in the Acri + EET-B-treated group compared with the EET-B-alone group (Table 1), acute administration of Acri before EET-B completely blocked cardioprotection and IS/AAR reached 63.6 ± 1.2% (Fig. 5A). Both Acri and 2-ME at a high dose did not affect IS/AAR when they were administrated alone (Figs. 4A and 5A). Neither 2-ME nor Acri affected the slope of the linear regression lines (Figs. 4B and 5B).
Fig. 5.

Myocardial infarct size expressed as a percentage of the area at risk (A) and relationship between area at risk and infarct size, both expressed as a percentage of the left ventricle (B), in rats administrated vehicle, the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg iv) 5 min before reperfusion, or the hypoxia-inducible factor-1α inhibitor acriflavine (Acri; 2.0 mg/kg iv) alone or combined with EET-B 10 min before reperfusion. Values are means ± SE from 5−9 rats. *P < 0.05 vs. vehicle; †P < 0.05 vs. EET-B.
Effect of EET-B on myocardial HIF-1α and PHD3 expression and distribution.
In a separate set of experiments, the immunoreactivity of HIF-1α and its degrading enzyme PHD3 were analyzed in the ischemic area and nonischemic septum at the end of 30 min of ischemia (without any reperfusion) as well as after 20 min and 2 h of reperfusion. In the nonischemic septum, HIF-1α and PHD3 immunogenic signals were 1.20 ± 0.23% and 2.00 ± 0.16%, respectively (Fig. 6). In the ischemic area, HIF-1α immunoreactivity markedly increased to 10.31 ± 0.78% at the end of 30 min of ischemia (Fig. 6A). Regional LV ischemia without any reperfusion did not change PHD3-positive immunoreactivity (2.21 ± 0.27%); however, a striking change in PHD3 tissue distribution was observed. Although PHD3 immunoreactivity was present mainly in the myocyte nuclei in the nonischemic regions, in the ischemic area this signal was localized mainly to the intercalated disks (Fig. 6B).
Fig. 6.

Representative photomicrographs of immunohistochemical staining showing the expression and localization of hypoxia-inducible factor (HIF)-1α (black arrows; A) and prolyl-4-hydroxylase domain protein 3 (PHD3; black arrows; B) in the nonischemic septum and left ventricle subjected to 30 min of ischemia without reperfusion. Bottom photos show details from the boxed regions shown on the top images. Scale bars = 50 μm.
After 20 min and 2 h of reperfusion, HIF-1α positivity rapidly decreased to 2.40 ± 0.48% and 1.85 ± 0.43%, respectively, in the ischemic area of vehicle-treated controls (Fig. 7, A–C). With respect to PHD3, a slight decrease in immunopositivity to 2.10 ± 0.15% and 1.62 ± 0.27%, respectively, was observed at reperfusion (Fig. 7, D–F). In ischemic tissue after reperfusion, EET-B blunted the fast decrease of HIF-1-positive immunoreactivity (7.80 ± 0.70% and 6.44 ± 1.37%, respectively) and induced more distinct reduction of PHD3 (1.58 ± 0.30% and 0.88 ± 19%, respectively; Fig. 7).
Fig. 7.

Representative photomicrographs of immunohistochemical staining showing the expression and localization of hypoxia-inducible factor (HIF)-1α (black arrows; A and B) and prolyl-4-hydroxylase domain protein 3 (PHD3; black arrows; D and E) in left ventricles subjected to 30 min of ischemia (ISCH) followed by 20 min or 2 h of reperfusion (REP) in rats administrated vehicle (Veh) or the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg) 5 min before reperfusion. Bottom photos show details from the boxed regions shown on the top photos. Scale bars = 50 μm. C and F: quantification of left ventricular myocardial HIF-1α (C)- and PHD3 (F)-positive areas assessed at the end of ischemia and after 20 min and 2 h of reperfusion. Values are means ± SE from 4 different sections (6 images in each) of 3 hearts/group. *P < 0.05 vs. vehicle; †P < 0.05 vs. ischemia.
In the nonischemic septum of vehicle-treated rats, HIF-1α immunopositivity did not change during reperfusion (Fig. 8A). However, EET-B administration significantly increased the HIF-1α immunogenic signal after 20 min of reperfusion (0.98 ± 0.23% vs. 2.25 ± 0.23%). After 2 h of reperfusion, higher HIF-1α immunogenic signal still persisted in EET-B-treated hearts (0.66 ± 0.13% vs. 1.03 ± 0.22%), but the difference did not reach statistical significance (Fig. 8A).
Fig. 8.
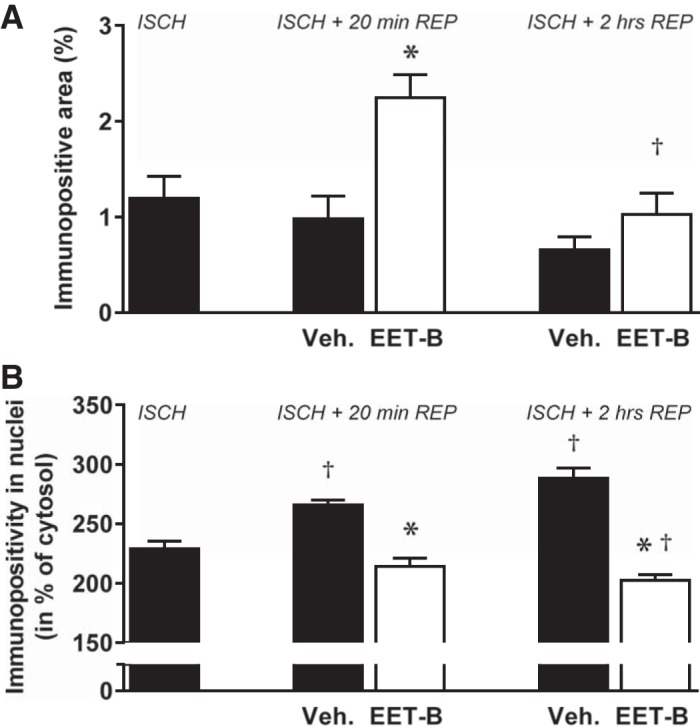
Hypoxia-inducible factor (HIF)-1α tissue immunopositivity (A) and prolyl-4-hydroxylase domain protein 3 (PHD3) nuclear positivity (B) in the nonischemic septum assessed at the end of ischemia (ISCH) and after 20 min and 2 h of reperfusion (REP) in rats administrated vehicle (Veh) or the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg) 5 min before reperfusion. Values are means ± SE. For HIF-1α, 4 different sections (six images in each) of 3 hearts/group were analyzed; for PHD3, 25 nuclei from 4 different sections of 3 hearts/group were analyzed. *P < 0.05 vs. vehicle; †P < 0.05 vs. ischemia.
As the strongest PHD3 signal was observed in myocyte nuclei of the nonischemic septum, we longitudinally analyzed their immunopositivity in detail. PHD3 nuclear positivity progressively increased from 229.1 ± 6.3% in the cytosol at the end of 30 min of ischemia to 265.9 ± 4.0% and 288.3 ± 6.3%, respectively, after 20 min and 2 h of reperfusion in vehicle-treated rats (Fig. 8B). Interestingly, EET-B administration abrogated the increase in PHD3 nuclear positivity after reperfusion in the nonischemic septum. Moreover, significantly lower PHD3 nuclear positivity was observed in EET-B-treated rats after 2 h of reperfusion (202.6 ± 4.0%) compared with values at the end of ischemia (229.1 ± 6.3%; Fig. 8B).
Finally, an additional set of experiments showed that EET-B administration decreased LV mRNA levels of PHD3 by 42% (although the effect did not reach statistical significance) but did not affect HIF-1α gene expression in intact nonischemized rats (Fig. 9).
Fig. 9.
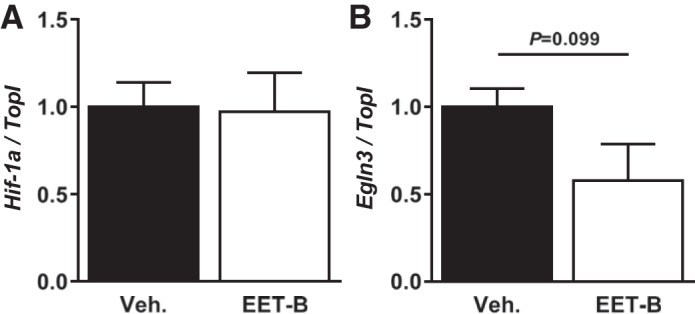
Effect of acute administration of vehicle (Veh) or the epoxyeicosatrienoic acid (EET) analog EET-B (2.5 mg/kg) on mRNA expression of hypoxia-inducible factor-1α (Hif1α; A) and prolyl-4-hydroxylase domain protein 3 (Egln3; B). mRNA transcript levels were assessed 2 h after bolus administration in the left ventricle and normalized to the reference gene topoisomerase I (TopI). Values are means ± SE from six hearts, expressed as a portion of the vehicle-treated group.
DISCUSSION
EET-based therapeutic strategies represent an innovative approach in the prevention of various cardiovascular diseases and provide distinct organ protection (18). Their cardioprotective actions were also demonstrated in hearts subjected to infarction (for a review, see Ref. 40). It has been reported that increased levels of EETs not only before ischemia but also before reperfusion reduce myocardial I/R injury. Indeed, the acute exogenous administration of native 11,12- or 14,15-EET before reperfusion diminished myocardial IS in dogs, rats, and mice (14, 26, 31). Furthermore, inhibition of cytochrome P-450 epoxygenase, the enzyme that metabolizes arachidonic acid to biologically active EETs, completely abolished the IS-limiting effect afforded by ischemic postconditioning (51). Altogether, these findings suggested a potent cardioprotective action of EET-mediated signaling in the clinically relevant experimental setting.
In the present study, a novel, stable, and orally active EET analog, EET-B, administered before ischemia or before reperfusion markedly reduced myocardial IS without affecting arterial blood pressure in normotensive rats. Similarly, acute administration of 14,15-EET or 11,12-EET led to an IS-limiting effect without blood pressure reduction in our previous study (14). Therefore, it seems that the acute effects of EETs on cardiac ischemic tolerance do not depend on arterial blood pressure.
As demonstrated in the present study, the cardioprotective action of EET-B in reperfusion was comparable with that of 14,15-EET. Moreover, coadministration of EET-B with 14,15-EEZE, a 14,15-EET antagonist, completely abolished the IS-limiting effect afforded by EET-B. Therefore, these findings confirmed that EET-B is an effective agonistic 14,15-EET analog, which has been previously suggested (16). Earlier, it has been also documented that chronic EET-B treatment reduced kidney injury in rats; it decreased nephrotoxic injury caused by cisplatin (20) and diminished renal injury in salt- and angiotensin II-dependent forms of hypertension (16, 17). In hypertensive rat hearts, EET-B also reduced myocardial inflammation and fibrosis (16). Finally, EET-B demonstrated therapeutic potential to attenuate the progression of heart failure in spontaneously hypertensive rats after myocardial infarction (J. Neckář, M. A. Hye Khan, G. J. Gross, M. Cyprová, A. Kvasilová, J. R. Falck, W. B. Campbell, V. Olejníčková, M. Gregorovičová, D. Sedmera, F. Kolář, and J. D. Imig, unpublished observations).
EETs are able to activate a number of intracellular and extracellular signaling elements important in increased cardiac ischemic tolerance (33). Some of them represent the well-known mediators involved in cardioprotection induced by various forms of conditioning [for example, ATP-sensitive K+ channels, nitric oxide synthases, the mitochondrial permeability transition pore, etc. (40)]. In the present study, HIF-1α, an original and novel component of intracellular cardioprotective signaling mediated by EET-based therapy, was investigated on the model of improved cardiac ischemic tolerance afforded by EET-B.
HIF-1 is a heterodimeric transcription factor composed of an O2-regulated α-subunit and a constitutively expressed β-subunit (46). HIF-1 regulates the expression of numerous genes through several mechanisms (39). In well-oxygenated cells, the HIF-1α-subunit is ubiquitinated based on the hydroxylation of prolines by PHDs. Hydroxylation of asparagine by a factor inhibiting HIF-1 inactivates the COOH-terminal transactivation domain of HIF-1α. The HIF-1α level is elevated under hypoxia (for example, under high-altitude conditions, in temporary anoxic tissue, etc.) because of low activity of degrading hydroxylases. However, other O2-independent stimuli, including eicosanoids, can also increase HIF-1α availability (35). With respect to HIF-1α and cardiac ischemic tolerance, HIF-1α is involved in the protection of the heart against acute I/R injury (39). Indeed, it has been shown that ischemic preconditioning does not provide cardioprotection in transgenic heterozygous HIF-1α+/− mice as well as in hearts after HIF-1α inhibition by siRNA (8, 11). Until now, it has been found that HIF-1α participates in cardioprotection afforded by various forms of conditioning and modifies different components of protective signaling pathways (for a review, see Ref. 34).
Previously, the relationship between EETs and HIF-1α-mediated signaling has been suggested. Xu et al. (49) reported that acute myocardial infarction reduced gene and protein expression of HIF-1α in human endothelial progenitor cells. Incubation of endothelial progenitor cells with sEH inhibitor markedly elevated the HIF-1α level. Furthermore, Batchu et al. (3) demonstrated that acute administration of sEH inhibitor improved the resistance of H9C2 cells to acute anoxia/reoxygenation injury that was accompanied by increased HIF-1α DNA-binding activity at reoxygenation. Moreover, cotreatment of cells with the 14,15-EET antagonist 14,15-EEZE completely abolished the improved cell viability and decreased sEH inhibitor-mediated elevation of HIF-1α transcription activity.
In our study, two HIF-1α inhibitors with different mechanisms of action were used. Acri binds HIF-1α and inhibits dimerization with HIF-1β (21), whereas 2-ME inhibits HIF-1α nuclear translocation (23). Our aim was to analyze the role of HIF-1α in improved cardiac ischemic tolerance mediated by EET analog administration during reperfusion. As clearly demonstrated, the acute coadministration of EET-B with each HIF-1α inhibitor completely abrogated the IS-limiting effect. Previous studies have shown that acute administration of Acri and 2-ME abolished the cardioprotective effect of ischemic preconditioning (37) and various forms of pharmacological postconditioning (42, 47, 48, 50). It has also been demonstrated that these HIF-1α inhibitors abrogated neuroprotective interventions (22, 54) and deteriorated brain function after traumatic injury and ischemic damage (15, 44, 53). Furthermore, 2-ME abolished the IS-limiting effect of chronic antioxidant treatment in diabetic rat hearts (24). In the study of Dai et al. (10), coculture of neonatal rat cardiomyocytes with bone marrow stem cells reduced H2O2-induced apoptosis that was inhibited by pretreatment of the cells with 2-ME in a concentration-dependent manner. Finally, chronic Acri treatment blunted the cardioprotective effect of long-lasting adaptation to heat stress in mice (2). Altogether, these findings demonstrated that the activation of HIF-1α plays an important role in the susceptibility of organs to acute I/R injury and participates in various protective mechanisms, including those induced by EETs.
In the subsequent part of this study, immunoreactivity of HIF-1α and PHD3 in the myocardium was analyzed at the end of ischemia and during reperfusion. Regional LV ischemia markedly increased immunopositivity of HIF-1α. Because of the restoration of coronary flow and O2 delivery at reperfusion, a quick and substantial decrease in ischemia-induced HIF-1α positivity was observed in vehicle-treated rats. Upregulation of PHD3 gene expression during hypoxia has been implicated in the HIF-1α short half-life at reperfusion (6). Here, we showed that acute administration of EET-B before reperfusion prevented the fast HIF-1α degradation. Similarly, it was demonstrated that ischemia- or sevoflurane-induced postconditioning elevated HIF-1α protein, but not gene, expression levels in the ischemic myocardium at the end of reperfusion (47, 48, 52).
In contrast to HIF-1α, myocardial infarction did not change the PHD3 immunopositivity in the ischemic LV, but it did affect its subcellular distribution. Compared with dominant nuclear staining in nonischemic septa, the PHD3 signal was localized mainly in intercalated disks and in the cytosol, and it did not return to the nuclei during reperfusion. It has been reported that PHDs have a distinct pattern of subcellular localization and that PHD3 occurs preferentially in the nucleus and cytoplasm (25), which agrees with our results. The translocation of PHD3 immunopositivity from nuclei to intercalated disks upon reperfusion is a novel observation, and further investigation is needed to elucidate its physiological significance. Nevertheless, we can speculate that it reflects a dynamic shift between nuclear low-molecular-mass PHD3 species (monomer or dimer) that hydroxylate HIF-1α and larger-molecular-mass PHD3-containing complexes of cytosolic and membrane proteins with low PHD3-specific activity (27).
Compared with HIF-1α, EET-B administration before reperfusion had an opposite effect on PHD3 immunopositivity by potentiating its decrease during reperfusion. EET-B not only eliminated PHD3 in the ischemic area but also decreased its nuclear positivity in the nonischemic septum and tended to suppress its gene expression. Therefore, EET-B changed the balance between HIF-1α and its degrading enzyme PHD3 in favor of the cardioprotective action of HIF-1α. With regard to the role of PHDs in cardiac ischemic tolerance, it has been shown that general PHD inhibitors decreased myocardial IS in mice, rats, and rabbits in preconditioning or postconditioning settings (32, 45, 52, 55). Moreover, Zieseniss et al. (55) showed that cardiomyocyte-specific PHD3 overexpression was associated with lower HIF-1α protein levels and increased myocardial infarction. Finally, transgenic depletion of PHD3 protected mouse hearts against acute I/R injury by decreasing myocyte apoptosis (48). Therefore, downregulation of PHD3 increased cardiac ischemic tolerance. In line with these findings, we demonstrated lower PHD3 immunopositivity at the end of myocardial reperfusion in EET-B-treated rats subjected to I/R. However, we cannot exclude that a reduced activity of other hydroxylases can also participate in the stabilization of HIF-1α during reperfusion and improved cardiac ischemic tolerance, as has been previously suggested (28).
In summary, the data of the present study clearly indicate that EET-B is an effective agonistic 14,15-EET analog, and its administration in reperfusion markedly reduced myocardial IS in rats. We suggest that increased HIF-1α levels play an important role in the cardioprotection afforded by EET-B in reperfusion by a mechanism(s) that decreases HIF-1α degradation.
GRANTS
J. Neckář was supported by Czech Science Foundation Grants 15-07544S and 18-03207S and Institutional Research Projects 67985823 (Institute of Physiology of the Czech Academy of Sciences) and 00023001 (Institute for Clinical and Experimental Medicine). This work was also supported by National Institutes of Health Grants HL-111392 (to G. Gross) and DK-103616 (to J. Imig), a Dr. Ralph and Marian Falk Medical Research Trust Bank of America, North America, Trustee Grant (to J. Imig), and Robert A. Welch Foundation Grant I-0011 (to J. Falck).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.N. and J.D.I. conceived and designed research; J.N., A.H., M.A.H.K., M.C., and D.B. performed experiments; J.N., A.H., M.H., and D.S.-K. analyzed data; J.N., M.A.H.K., G.J.G., K.N., M.H., D.S.-K., J.R.F., D.S., and F.K. interpreted results of experiments; J.N. prepared figures; J.N. drafted manuscript; J.N., M.A.H.K., G.J.G., M.C., D.B., M.H., D.S.-K., J.R.F., F.K., and J.D.I. edited and revised manuscript; J.N., M.A.H.K., G.J.G., M.C., D.B., M.H., D.S.-K., J.R.F., F.K., and J.D.I. approved final version of manuscript.
REFERENCES
- 1.Alánová P, Husková Z, Kopkan L, Sporková A, Jíchová Š, Neckář J, Imig JD, Klevstig M, Kolář F, Rami Reddy N, Falck JR, Sadowski J, Nishiyama A, Kramer HJ, Melenovský V, Červenková L, Kujal P, Vernerová Z, Červenka L. Orally active epoxyeicosatrienoic acid analog does not exhibit antihypertensive and reno- or cardioprotective actions in two-kidney, one-clip Goldblatt hypertensive rats. Vascul Pharmacol 73: 45–56, 2015. doi: 10.1016/j.vph.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 2.Alexander-Shani R, Mreisat A, Smeir E, Gerstenblith G, Stern MD, Horowitz M. Long-term HIF-1α transcriptional activation is essential for heat-acclimation-mediated cross tolerance: mitochondrial target genes. Am J Physiol Regul Integr Comp Physiol 312: R753–R762, 2017. doi: 10.1152/ajpregu.00461.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batchu SN, Lee SB, Samokhvalov V, Chaudhary KR, El-Sikhry H, Weldon SM, Seubert JM. Novel soluble epoxide hydrolase inhibitor protects mitochondrial function following stress. Can J Physiol Pharmacol 90: 811–823, 2012. doi: 10.1139/y2012-082. [DOI] [PubMed] [Google Scholar]
- 4.Batchu SN, Lee SB, Qadhi RS, Chaudhary KR, El-Sikhry H, Kodela R, Falck JR, Seubert JM. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br J Pharmacol 162: 897–907, 2011. doi: 10.1111/j.1476-5381.2010.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belibi F, Zafar I, Ravichandran K, Segvic AB, Jani A, Ljubanovic DG, Edelstein CL. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am J Physiol Renal Physiol 300: F1235–F1243, 2011. doi: 10.1152/ajprenal.00348.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J 22: 4082–4090, 2003. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao J, Tsenovoy PL, Thompson EA, Falck JR, Touchon R, Sodhi K, Rezzani R, Shapiro JI, Abraham NG. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat 116-117: 76–86, 2015. doi: 10.1016/j.prostaglandins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc Res 77: 463–470, 2008. doi: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhary KR, Abukhashim M, Hwang SH, Hammock BD, Seubert JM. Inhibition of soluble epoxide hydrolase by trans-4- [4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is protective against ischemia-reperfusion injury. J Cardiovasc Pharmacol 55: 67–73, 2010. doi: 10.1097/FJC.0b013e3181c37d69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai Y, Xu M, Wang Y, Pasha Z, Li T, Ashraf M. HIF-1α induced-VEGF overexpression in bone marrow stem cells protects cardiomyocytes against ischemia. J Mol Cell Cardiol 42: 1036–1044, 2007. doi: 10.1016/j.yjmcc.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckle T, Köhler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation 118: 166–175, 2008. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 12.Gross GJ, Falck JR, Gross ER, Isbell M, Moore J, Nithipatikom K. Cytochrome P450 and arachidonic acid metabolites: role in myocardial ischemia/reperfusion injury revisited. Cardiovasc Res 68: 18–25, 2005. doi: 10.1016/j.cardiores.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol 42: 687–691, 2007. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross GJ, Hsu A, Pfeiffer AW, Nithipatikom K. Roles of endothelial nitric oxide synthase (eNOS) and mitochondrial permeability transition pore (MPTP) in epoxyeicosatrienoic acid (EET)-induced cardioprotection against infarction in intact rat hearts. J Mol Cell Cardiol 59: 20–29, 2013. doi: 10.1016/j.yjmcc.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang T, Huang W, Zhang Z, Yu L, Xie C, Zhu D, Peng Z, Chen J. Hypoxia-inducible factor-1α upregulation in microglia following hypoxia protects against ischemia-induced cerebral infarction. Neuroreport 25: 1122–1128, 2014. doi: 10.1097/WNR.0000000000000236. [DOI] [PubMed] [Google Scholar]
- 16.Hye Khan MA, Neckář J, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, Falck JR, Imig JD. Orally active epoxyeicosatrienoic acid analog attenuates kidney injury in hypertensive Dahl salt-sensitive rat. Hypertension 62: 905–913, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hye Khan MA, Pavlov TS, Christain SV, Neckář J, Staruschenko A, Gauthier KM, Capdevila JH, Falck JR, Campbell WB, Imig JD. Epoxyeicosatrienoic acid analogue lowers blood pressure through vasodilation and sodium channel inhibition. Clin Sci (Lond) 127: 463–474, 2014. doi: 10.1042/CS20130479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 92: 101–130, 2012. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imig JD, Elmarakby A, Nithipatikom K, Wei S, Capdevila JH, Tuniki VR, Sangras B, Anjaiah S, Manthati VL, Sudarshan Reddy D, Falck JR. Development of epoxyeicosatrienoic acid analogs with in vivo anti-hypertensive actions. Front Physiol 1: 157, 2010. doi: 10.3389/fphys.2010.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J 27: 2946–2956, 2013. doi: 10.1096/fj.12-218040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA 106: 17910–17915, 2009. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Luo C, Ouyang MW, Fang YY, Li SJ, Zhou Q, Fan J, Qin ZS, Tao T. Dexmedetomidine protects mouse brain from ischemia-reperfusion injury via inhibiting neuronal autophagy through up-regulating HIF-1α. Front Cell Neurosci 11: 197, 2017. doi: 10.3389/fncel.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3: 363–375, 2003. doi: 10.1016/S1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 24.Mao X, Wang T, Liu Y, Irwin MG, Ou JS, Liao XL, Gao X, Xu Y, Ng KF, Vanhoutte PM, Xia Z. N-acetylcysteine and allopurinol confer synergy in attenuating myocardial ischemia injury via restoring HIF-1α/HO-1 signaling in diabetic rats. PLoS One 8: e68949, 2013. doi: 10.1371/journal.pone.0068949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Bürgel T, Jelkmann W, Acker H, Fandrey J. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci 116: 1319–1326, 2003. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- 26.Motoki A, Merkel MJ, Packwood WH, Cao Z, Liu L, Iliff J, Alkayed NJ, Van Winkle DM. Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am J Physiol Heart Circ Physiol 295: H2128–H2134, 2008. doi: 10.1152/ajpheart.00428.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakayama K, Gazdoiu S, Abraham R, Pan ZQ, Ronai Z. Hypoxia-induced assembly of prolyl hydroxylase PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem J 401: 217–226, 2007. doi: 10.1042/BJ20061135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natarajan R, Salloum FN, Fisher BJ, Ownby ED, Kukreja RC, Fowler AA III. Activation of hypoxia-inducible factor-1 via prolyl-4 hydoxylase-2 gene silencing attenuates acute inflammatory responses in postischemic myocardium. Am J Physiol Heart Circ Physiol 293: H1571–H1580, 2007. doi: 10.1152/ajpheart.00291.2007. [DOI] [PubMed] [Google Scholar]
- 29.Neckář J, Kopkan L, Husková Z, Kolář F, Papoušek F, Kramer HJ, Hwang SH, Hammock BD, Imig JD, Malý J, Netuka I, Ošťádal B, Červenka L. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-1-ylureido)cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin Sci (Lond) 122: 513–525, 2012. doi: 10.1042/CS20110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am J Physiol Heart Circ Physiol 291: H537–H542, 2006. doi: 10.1152/ajpheart.00071.2006. [DOI] [PubMed] [Google Scholar]
- 32.Ockaili R, Natarajan R, Salloum F, Fisher BJ, Jones D, Fowler AA III, Kukreja RC. HIF-1 activation attenuates postischemic myocardial injury: role for heme oxygenase-1 in modulating microvascular chemokine generation. Am J Physiol Heart Circ Physiol 289: H542–H548, 2005. doi: 10.1152/ajpheart.00089.2005. [DOI] [PubMed] [Google Scholar]
- 33.Oni-Orisan A, Alsaleh N, Lee CR, Seubert JM. Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J Mol Cell Cardiol 74: 199–208, 2014. doi: 10.1016/j.yjmcc.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ong SG, Hausenloy DJ. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol Ther 136: 69–81, 2012. doi: 10.1016/j.pharmthera.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 35.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92: 967–1003, 2012. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rand AA, Barnych B, Morisseau C, Cajka T, Lee KSS, Panigrahy D, Hammock BD. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc Natl Acad Sci USA 114: 4370–4375, 2017. doi: 10.1073/pnas.1616893114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarkar K, Cai Z, Gupta R, Parajuli N, Fox-Talbot K, Darshan MS, Gonzalez FJ, Semenza GL. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc Natl Acad Sci USA 109: 10504–10509, 2012. doi: 10.1073/pnas.1208314109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 1813: 1263–1268, 2011. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat 82: 50–59, 2007. doi: 10.1016/j.prostaglandins.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res 95: 506–514, 2004. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 42.Si J, Wang N, Wang H, Xie J, Yang J, Yi H, Shi Z, Ma J, Wang W, Yang L, Yu S, Li J. HIF-1α signaling activation by post-ischemia treatment with astragaloside IV attenuates myocardial ischemia-reperfusion injury. PLoS One 9: e107832, 2014. doi: 10.1371/journal.pone.0107832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki S, Oguro A, Osada-Oka M, Funae Y, Imaoka S. Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells. J Pharmacol Sci 108: 79–88, 2008. doi: 10.1254/jphs.08122FP. [DOI] [PubMed] [Google Scholar]
- 44.Umschweif G, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E. Hypoxia-inducible factor 1 is essential for spontaneous recovery from traumatic brain injury and is a key mediator of heat acclimation induced neuroprotection. J Cereb Blood Flow Metab 33: 524–531, 2013. doi: 10.1038/jcbfm.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogler M, Zieseniss A, Hesse AR, Levent E, Tiburcy M, Heinze E, Burzlaff N, Schley G, Eckardt KU, Willam C, Katschinski DM. Pre- and post-conditional inhibition of prolyl-4-hydroxylase domain enzymes protects the heart from an ischemic insult. Pflugers Arch 467: 2141–2149, 2015. doi: 10.1007/s00424-014-1667-z. [DOI] [PubMed] [Google Scholar]
- 46.Wang GL, Semenza GL. Purification and characterisation of hypoxia-inducible factor 1. J Biol Chem 270: 1230–1237, 1995. [DOI] [PubMed] [Google Scholar]
- 47.Wu J, Yang L, Xie P, Yu J, Yu T, Wang H, Maimaitili Y, Wang J, Ma H, Yang Y, Zheng H. Cobalt chloride upregulates impaired HIF-1α expression to restore sevoflurane post-conditioning-dependent myocardial protection in diabetic rats. Front Physiol 8: 395, 2017. doi: 10.3389/fphys.2017.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie P, Yang L, Talaiti A, Wu JJ, Yu J, Yu T, Wang HY, Huang B, Wu Q, Maimaitili Y, Wang J, Ma HP, Yang YN, Zheng H. Deferoxamine-activated hypoxia-inducible factor-1 restores cardioprotective effects of sevoflurane postconditioning in diabetic rats. Acta Physiol (Oxf) 221: 98–114, 2017. doi: 10.1111/apha.12874. [DOI] [PubMed] [Google Scholar]
- 49.Xu DY, Davis BB, Wang ZH, Zhao SP, Wasti B, Liu ZL, Li N, Morisseau C, Chiamvimonvat N, Hammock BD. A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARγ to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction. Int J Cardiol 167: 1298–1304, 2013. doi: 10.1016/j.ijcard.2012.03.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Xie P, Wu J, Yu J, Yu T, Wang H, Wang J, Xia Z, Zheng H. Sevoflurane postconditioning improves myocardial mitochondrial respiratory function and reduces myocardial ischemia-reperfusion injury by up-regulating HIF-1. Am J Transl Res 8: 4415–4424, 2016. [PMC free article] [PubMed] [Google Scholar]
- 51.Yu GG, Zeng XJ, Wang HX, Lu LQ, Zheng SP, Ma LQ, Chang J, Wang J, Zhang DM, Du FH, Zhang LK. Cytochrome P450 2J3/epoxyeicosatrienoic acids mediate the cardioprotection induced by ischaemic post-conditioning, but not preconditioning, in the rat. Clin Exp Pharmacol Physiol 38: 63–70, 2011. doi: 10.1111/j.1440-1681.2010.05464.x. [DOI] [PubMed] [Google Scholar]
- 52.Zhao HX, Wang XL, Wang YH, Wu Y, Li XY, Lv XP, Zhao ZQ, Zhao RR, Liu HR. Attenuation of myocardial injury by postconditioning: role of hypoxia inducible factor-1alpha. Basic Res Cardiol 105: 109–118, 2010. doi: 10.1007/s00395-009-0044-0. [DOI] [PubMed] [Google Scholar]
- 53.Zhou D, Matchett GA, Jadhav V, Dach N, Zhang JH. The effect of 2-methoxyestradiol, a HIF-1 alpha inhibitor, in global cerebral ischemia in rats. Neurol Res 30: 268–271, 2008. doi: 10.1179/016164107X229920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu T, Zhan L, Liang D, Hu J, Lu Z, Zhu X, Sun W, Liu L, Xu E. Hypoxia-inducible factor 1α mediates neuroprotection of hypoxic postconditioning against global cerebral ischemia. J Neuropathol Exp Neurol 73: 975–986, 2014. doi: 10.1097/NEN.0000000000000118. [DOI] [PubMed] [Google Scholar]
- 55.Zieseniss A, Hesse AR, Jatho A, Krull S, Hölscher M, Vogel S, Katschinski DM. Cardiomyocyte-specific transgenic expression of prolyl-4-hydroxylase domain 3 impairs the myocardial response to ischemia. Cell Physiol Biochem 36: 843–851, 2015. doi: 10.1159/000430260. [DOI] [PubMed] [Google Scholar]