

Abstract
A deficit or loss in the number of nephrons, the functional unit of the kidney, can induce compensatory growth and hyperfunction of remaining nephrons. An increase in single nephron glomerular filtration rate (SNGFR) aims to compensate but may be deleterious in the long term. The increase in SNGFR is determined by the dynamics of nephron loss, total remaining GFR, the body’s excretory demand, and the functional capacity to sustain single nephron hyperfunction.
Introduction
Nephrons are the functional units of the kidney. Together, the kidneys of a healthy young adult have on average ~1.8–2 million nephrons, but there is a wide variability (8). Nephron loss is critical to the pathology of diseases such as acute kidney injury (AKI), non-dialysis-dependent chronic kidney disease (CKD), and end-stage renal disease (ESRD), all of which are associated with significant morbidity and mortality. The degree of nephron loss that corresponds to ESRD is difficult to establish since the degree of glomerular sclerosis, tubulointerstitial scaring and atrophy, and nephron dysfunction all affect net kidney function (48). Other than nephron loss, a low nephron population may also arise from differences in nephrogenesis during fetal kidney development, with nephron numbers reported as low as 0.2 million in humans (8). How the kidneys respond to the deficit or loss of nephrons, in terms of structural and functional adaptations, is still incompletely understood. The current belief is that the initial responses are compensatory and beneficial, since they presumably aim to normalize total kidney function. However, in the long term, these adaptive changes may become deleterious if they overwhelm the physiological capacity of the remaining nephrons. This review outlines the determinants of nephron number during nephrogenesis, briefly discusses clinical consequences of the deficit or loss of nephrons, which may drive structural and functional adaptations in the remaining nephrons in the first place, and describes in more detail these glomerular and tubular adaptations, including insights from experimental, human, and theoretical studies.
Determinants of Nephron Endowment at Birth and Birth Weight as a Surrogate for Nephron Number
In humans, nephrogenesis ends shortly before term birth (35). As a result, any deficit in the number of nephrons at birth (nephron endowment) is permanent. Maternal exposures to specific diets or toxins, genetic predisposition, and low birth weight can all be associated with decreased nephrogenesis. For example, maternal diets low in vitamin A, iron, and protein, and high in alcohol, hyperglycemia, fetal exposure to steroids, gentamicin, cyclosporine, β-lactam antibiotics, and non-steroidal anti-inflammatory drugs have all been linked to low nephron endowment in preclinical studies (35). Mutations in genes involved in nephrogenesis can reduce nephron numbers at birth. Examples include the AAA variant of the PAX2 gene and the RET1476A variant of the RET gene, which cause ~10% reduction in kidney volume, a surrogate for nephron number (41, 53). Low nephron numbers have also been associated with a family history of ESRD as well as with short height (8). Finally, for most racial groups, birth weight positively correlates with nephron number; thus birth weight can be used to estimate nephron number at birth (20, 36). A positive correlation, however, has not been detected in all studies; e.g., one study reported a positive correlation between birth weight and nephron number in southeastern U.S. white subjects but not in African-American subjects (22).
Why Should the Kidneys Aim for Compensation When Nephron Number is Reduced?
The kidneys play a key role in fluid and electrolyte homeostasis, acid base balance, calcium and phosphate homeostasis, and the renal excretion of endogenous and exogenous compounds, to name but a few of its tasks. As nephron number and GFR decrease, the ability of the kidney to fulfill these tasks becomes impaired (FIGURE 1), and compensatory or alternative mechanisms are induced to partially normalize kidney function and the body’s excretory capacity. The compensation may affect both the glomeruli and the tubular system, and these responses need to be coordinated. Although it is easy to imagine how a massive deficit or loss of nephrons may impair kidney function and require compensation, it is less clear how the kidneys respond to a subtle lowering in nephron number or GFR. Moreover, the responses may differ when nephron number is reduced due to impaired nephrogenesis, kidney donation, aging, or acute or chronic kidney injury (FIGURE 2).
FIGURE 1.
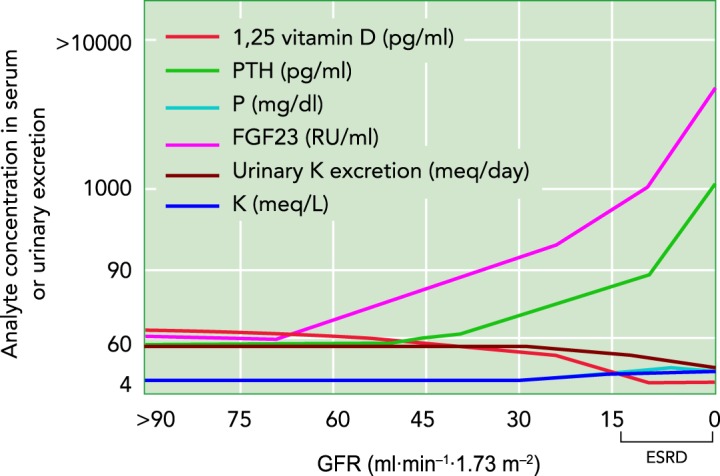
Impairment of the kidneys’ ability to maintain body homeostasis when GFR, and potentially nephron number, is declining
CKD stages 2, 3, 4, and 5 correspond to GFR values (in ml·min−1·1.73 m−2) of 60-89, 30-59, 15-29, and 0-15 (=ESRD), respectively. The blood levels of phosphaturic fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH) rise with decreasing GFR, and thus filtered phosphate, to maintain renal phosphate excretion and plasma phosphate concentrations. Hyperphosphatemia occurs in CKD stages 4 and 5, primarily as a result of the reduction in GFR. FGF23 inhibits renal conversion of inactive vitamin D to active 1,25 vitamin D, whereas PTH increases this conversion. Blood levels of FGF23 rise in CKD stage 2 and before PTH, the latter increasing in CKD stage 3, thus causing a decrease in active 1,25 vitamin D. Urinary potassium excretion decreases starting in CKD stage 4; serum potassium concentrations increase but remain within normal range through adaptive renal and non-renal mechanisms. Of note, serum potassium and phosphorous graphically overlap to a large extent. Adapted from Ref. 47, with permission from Kidney Research and Clinical Practice, and Ref. 52, with permission from the Journal of the American Society of Nephrology.
FIGURE 2.

The causes, adaptations, and consequences of nephron deficit or loss
Green arrows indicate an enhancing effect. Red lines indicate an inhibitory effect. A nephron deficit or loss reduces the ability of the kidney to maintain body homeostasis. This triggers adaptations that can include an increase in mean SNGFR and tubular transport machinery. With acute nephron loss, as in nephrectomy, there is a rise in mean SNGFR. With a chronic deficit or loss of nephrons, mean SNGFR only rises when total GFR or nephron number decrease below a certain threshold, with the latter increasing when the body’s excretory demand on the kidney rises. The increase in mean SNGFR may also depend on the kidney’s ability to sustain single nephron hyperfunction. High individual SNGFR can induce deleterious consequences and lead to further nephron loss, when the physiological capacity of the given nephron is not able to sustain hyperfunction. Even though mean SNGFR may not increase, a low nephron number may enhance the internephron variation in SNGFR with some nephrons hyperfiltering. See text for additional details. HTN, hypertension; NE, nephron endowment; ROS, reactive oxygen species; SNO2, single nephron oxygen consumption.
Much has been learned about compensation from observations in patients with CKD. Urinary phosphate excretion and plasma levels are maintained close to normal levels until later CKD stages, which are defined by the level of estimated GFR (eGFR) (FIGURE 1). The hormones fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH) regulate phosphate homeostasis by inhibiting proximal tubular phosphate reabsorption and thereby enhancing urinary phosphate excretion to match intestinal uptake (23). The blood levels of both hormones rise with decreasing GFR, and thus filtered phosphate, to maintain renal phosphate excretion and plasma phosphate concentrations. Hyperphosphatemia occurs in stages 4 and 5, primarily as a result of the severe reduction in GFR (23).
Similarly, urinary potassium excretion is typically not altered in individuals with earlier CKD stages 1–3 (FIGURE 1). However, in advanced CKD stages 4–5, urinary potassium excretion decreases, potassium is retained, and serum potassium levels increase (47). Serum potassium levels increase only modestly, in part due to the aldosterone-driven compensatory upregulation of the potassium excretion machinery in the digestive system (44). Computational modeling studies in the rat kidney indicated that an increase in single nephron GFR (SNGFR) would help to facilitate potassium excretion in response to nephron loss, in part by filtering more potassium and in part by delivering more sodium to the connecting tubule, where potassium secretion is linked to sodium reabsorption. Moreover, model simulations predicted that a high potassium intake raised potassium secretion along the individual connecting tubule in sham rats, and even more when nephron number was reduced by half or 5/6. However, the increased potassium secretion per tubule failed to completely compensate for the reduction in nephron number, such that the ability to excrete an acute or chronic potassium load would become impaired (31).
Thus the organism and the remaining nephrons need to compensate for reduced nephron numbers to maintain body homeostasis. The examples in CKD illustrate that adaptations in SNGFR and in hormone-regulated tubular transport can help to preserve kidney function, but the extent of compensation becomes limited when total GFR and potentially nephron number drop excessively.
Renal Adaptations to Deficit or Loss of Nephrons
What is the Relationship Between Nephron Number and SNGFR?
Due to the inherent challenges in counting the number of nephrons in human subjects, it is difficult to establish the relationship between nephron loss and the change in whole kidney GFR and SNGFR. The current gold-standard method is the stereological dissector/fractionator method (19), which is labor intensive and requires the whole kidney ex vivo. A new method using MRI and cationized ferritin contrast agent, which labels glomeruli, holds promise for a noninvasive measurement of nephron number (1). No studies have been published counting glomeruli in living humans other than in living kidney donors (8).
Denic and colleagues determined in living kidney donors total GFR (via urinary iothalamate clearance before kidney removal) and nephron number [by the stereological Weibel-Gomez (49) method for counting nephrons on kidney biopsies]. They reported that GFR and nephron number positively correlated and that both parameters were inversely associated with age (8, 9). Glomerular number also positively correlated with GFR in a study that used MRI to measure glomeruli number in stable renal transplants (15) and in autopsies of patients suffering from CKD (24). These data indicated in a number of clinical scenarios that a lower total GFR correlated with a lower nephron number.
Whether due to low nephron endowment at birth, kidney donation, or progressive loss of nephrons with aging, AKI, or CKD, it might make sense for the remaining nephrons to elevate mean SNGFR to increase their functional capacity. The discussion below, however, indicates that the relationship between nephron number and mean SNGFR is likely more complex and is potentially determined by the dynamics of nephron loss, the total remaining GFR and nephron number, and the excretory demand on the kidney.
SNGFR response to acute nephron loss.
A compensatory rise in SNGFR, along with glomerular hypertrophy, in response to nephron loss was initially described by Brenner and colleagues in the 1980s using rat models. Two groups of rats underwent uninephrectomy (UNX) and 5/6th ablation of the remaining kidney; one group received a normal diet, whereas the other received a low protein diet. A third group underwent sham surgery and received a normal diet (21). A high protein intake can increase GFR and glomerular hypertrophy in rodents, potentially to facilitate the excretion of the increased urea load (14). Seven days after surgery, single nephron hyperfiltration was indeed seen primarily in the first group, as were changes in glomerular morphology such as mesangial cell expansion and podocyte foot process effacement. These changes were attenuated by a low protein diet. Moreover, micropuncture studies indicated that increased glomerular capillary pressure and single nephron plasma flow were responsible for single nephron hyperfiltration in response to acute nephron loss (21).
Tan and colleagues showed that single kidney GFR increased quickly after kidney donation (47 to 64 ml·min–1·1.73 m–2), and remained stable from the early (median 0.8 yr) to the late (median 6.3 yr) post-donation period (34). Mean SNGFR must also have increased since nephron number in the remaining kidney should be unchanged. Single kidney renal plasma flow (RPF) showed a similar pattern, indicating that renal blood flow is likely the driver of the rise in single kidney GFR and SNGFR. Thus acute nephron loss, due to kidney resection or donation, induces a rapid and sustained increase in mean SNGFR in rodents and humans (FIGURE 2).
SNGFR response to gradual nephron loss.
The response in mean SNGFR may be different when nephrons are lost gradually. Despite 30% fewer nephrons and 30% lower total GFR in 65- to 69-yr-old vs. 18- to 29-yr-old individuals, Denic and colleagues (study introduced above) found no significant difference in mean SNGFR among individuals 18–69 yr of age (9) (FIGURE 3). These findings indicated that mean SNGFR does not necessarily increase following a slow nephron loss in these age groups. The slow nephron loss typical of aging may be compensated by subtler or alternative compensation mechanisms than an SNGFR increase. This could also reflect the aging-associated decrease in the body’s metabolic and excretory demand due to decreased body mass and physical activity, which requires less kidney function to maintain body homeostasis (FIGURE 2). Of note, in the same study, there was a further reduction in nephron number in individuals 70–75 yr of age (~50% fewer nephrons) that was associated with a 40% rise in SNGFR (FIGURE 3), which would support the hypothesis that, in the healthy aging kidney, mean SNGFR only increases when total nephron number or GFR reaches a certain threshold. A limitation of the study was that only 13 individuals were in this oldest age group out of a total number of 1,388 individuals (9). Moreover, there was an inherent selection bias in this oldest age group since the donor selection process might favor those with the best preserved total GFR. In the kidney donor selection process, a patient with an eGFR of <70 ml·min–1·1.73 m–2 is typically not considered for donation. However, with normal aging, the mean GFR is ~70 and 66 ml·min–1·1.73 m–2 in the 70- to 75-yr age group for healthy males and females, respectively (50). Mean GFR in the 70- to 75-yr age group was indeed 96 ml·min–1·1.73 m–2 (9). Considering the further reduced nephron number in this age group, the preserved total GFR required single nephron hyperfiltration (FIGURE 3). Thus one may speculate that aging individuals with “better kidneys” or “better preserved GFR” have an enhanced functional capacity to sustain single nephron hyperfunction, including a better preserved tubular transport capacity and integrity.
FIGURE 3.
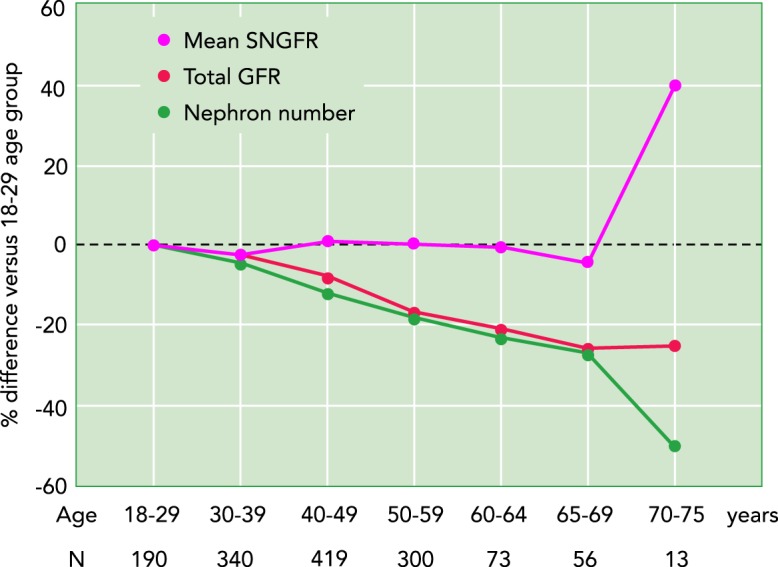
Effect of aging on total GFR, nephron number, and mean SNGFR
Data are presented as percent difference compared with the youngest age group (18–29 yr). For age ranges up to 69 yr, total GFR and nephron number decrease in parallel with age, whereas mean SNGFR does not change. In the 70–75 age range, a further reduction in nephron number is associated with an increase in mean SNGFR while total GFR was maintained. Based on data from Ref. 9.
In a recent study on autopsy kidneys of Japanese normotensive, hypertensive, and CKD patients (n = 9/group), kidneys of CKD subjects had fewer non-sclerosed glomeruli (mean: 268,043; range: 65,086–402,102) than hypertensives (mean: 392,108; range: 264,770–507,077) or normal subjects (mean: 640,399; range: 390,352–938,659). Patients with CKD had an average eGFR of 34.8 vs. 88.1 ml·min–1·1.73 m–2 in the normotensive group, but estimated mean SNGFR was similar in both groups. Glomerular volume, however, was significantly higher in CKD patients compared with the other two (24). In the study by Denic et al. (9), larger glomerular and tubular volume were independently and positively associated with SNGFR. Additionally, the correlation with other risk factors associated with higher SNGFR, such as obesity and family history of ESRD, was lessened when corrected for glomerular size. This suggests that larger nephrons are the main reason SNGFR is associated with these risk factors (9). Based on these two studies, one may speculate that glomerular hypertrophy is a more common adaptation to nephron loss than single nephron glomerular hyperfiltration. It is also possible that the glomeruli in the CKD group were in a post-hyperfiltration state. Moreover, the internephron variability in SNGFR may increase with the reduction in total GFR and nephron number, i.e., mean SNGFR could be maintained because the reduced SNGFR of more damaged nephrons would be compensated by more intact and hyperfiltering nephrons (FIGURE 2).
Mechanisms Inducing Glomerular Hyperfiltration and Hypertrophy Following a Deficit or Loss of Nephrons
The above studies indicated that an increase in RPF drives the increase in SNGFR in response to acute kidney mass resection in rodents and humans. Moreover, Brenner and colleagues argued that the primary increase in SNGFR causes secondary glomerular changes like hypertrophy and potentially sclerosis (21) (FIGURE 2). Kaufman and colleagues demonstrated in rats that the remaining renal mass increased proportionally to the amount of renal ablation. At 4 wk after surgery, total mass of the remaining renal tissue had increased by 50% and 140% in rats subjected to 50% ( = UNX) and 70–75% renal ablation, respectively; this was primarily due to tubular hypertrophy (see below) but was also associated with glomerular enlargement and a twofold and a three- to fourfold rise in estimated mean SNGFR, respectively (25). In a 5/6 nephrectomy (NX) model in mice, in which one kidney and 2/3 of the remaining kidney were removed, a 50% increase in the individual glomerular area was reported, together with a near twofold increase in mesangial expansion and a 30% increase in collagen deposition vs. sham mice (18).
Various growth signals have been implicated in the increase in SNGFR and glomerular hypertrophy, including growth hormone, epidermal growth factor, interleukins-1 and -6, tumor necrosis factor-α (TNF-α), angiotensin II, hepatocyte growth factor, and endothelin (13). The genetic background of the animal or human also contributes. As an example, mice carrying the Os mutation, which is associated with 50% reduction in nephron mass, have a higher predisposition to glomerular hypertrophy in the background of the ROP (sclerosis prone) strain compared with the C57 strain in response to UNX (11). Of note, glomerular hypertrophy due to UNX in rats induces an increase in glomerular volume but not necessarily in glomerular surface area. The latter, measured by binding of antiGBM (glomerular basement membrane) antibodies to the glomerular surface, did not change 22 days after UNX compared with pre-surgery, whereas a rise in glomerular surface area during early normal growth was demonstrated (29).
Larger glomerular size has been implicated as a poor prognostic factor in patients suffering from focal segmental glomerulosclerosis (FSGS) (16). The renin angiotensin aldosterone system (RAAS) plays a critical role in the interplay between glomerular growth and glomerulosclerosis, including the stimulation of multiple pathways linked to hypertrophy and the production of reactive oxygen species (ROS) and cytokines that mediate fibrosis (46). An enhanced mechanical strain on the glomerular capillaries, due to an increase in nephron plasma flow or glomerular capillary hypertension, can increase the production of extracellular matrix (ECM), thereby linking glomerular hyperperfusion and hyperfiltration to secondary glomerular hypertrophy and potentially sclerosis (6) (FIGURE 2). By suppressing the RAAS, reducing blood pressure, and, via dilation of the efferent arteriole, lowering glomerular capillary pressure, angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are a mainstay of proteinuria treatment and kidney protection, including in CKD (16).
Adaptive Tubular Changes Following Deficit or Loss of Nephrons
Studies by Denic and colleagues suggested that, although glomeruli size appeared not to change, tubular size increased with age-related nephron loss in humans (7, 10). Using a computational model of the rat nephrons (33), Layton and colleagues simulated epithelial transport from the proximal tubule to the cortical collecting duct following nephron loss (30). They adjusted the model parameters including higher SNGFR and larger tubular dimensions based on values observed at 4 wk after UNX or 5/6-NX in rats, respectively (26, 28). The increased tubular load was predicted to enhance apical cell membrane recruitment and expression of tubular transport proteins and induce tubular hypertrophy. The changes in SNGFR and tubular growth to a large extent normalized fluid and electrolyte excretion (FIGURE 2) but were not enough to fully restore salt balance (30). Additional adaptive increases in protein density of various sodium transporters, including Na-K-ATPase, Na-K-2Cl cotransporter (NKCC2), Na-Cl cotransporter (NCC), and epithelial sodium channel (ENaC), were needed to fully match urine flow, and potassium and sodium excretion in UNX and 5/6-NX models to sham levels, assuming similar fluid and food intake (30). The model predicted that fractional water and salt reabsorption is similar to sham from the proximal tubule to the distal convoluted tubule, but there is a need to further reduce sodium reabsorption and increase potassium secretion along the connecting tubules and collecting ducts. Additionally, the simulations indicated that oxygen consumption per individual nephron segment (SNO2) increases substantially due to the increased SNGFR, and thus tubular transport load, in the remaining nephrons (FIGURE 2). However, due to the lower nephron number, total renal oxygen use was reduced (30).
Layton and Vallon also used the rat model to predict the effects of new anti-hyperglycemic drugs, sodium glucose cotransporter 2 (SGLT2) inhibitors, on solute transport in diabetic and non-diabetic kidneys with normal or reduced nephron numbers (32). SGLT2 reabsorbs ~97% of the filtered glucose in the early proximal tubule, and SGLT2 inhibitors are used as anti-hyperglycemic drugs in diabetic patients (12). As expected, SGLT2 inhibition led to glucosuria, diuresis, natriuresis, and kaliuresis in non-diabetic kidneys. These effects were reduced in kidneys with low nephron numbers due to lesser overall filtration of glucose and thus lesser SGLT2-dependent glucose reabsorption. In diabetic kidneys with reduced nephron numbers, however, the diuretic, natriuretic, and kaliuretic effect of chronic SGLT2 inhibition was largely preserved due to an increased delivery of glucose to the remaining hyperfiltering individual nephrons causing proximal tubular paracellular sodium secretion (32). These model predictions could explain why the blood pressure-lowering effect of SGLT2 inhibitors is preserved in diabetic patients with CKD despite a blunted effect on blood glucose control.
Transition From Adaptation to Disease
A low nephron number and GFR can lead to hypertension as a mechanism to partly restore salt homeostasis through pressure natriuresis. Brenner and colleagues hypothesized, based on observations in rat studies, that lower nephron numbers might explain why some patients are more prone to CKD and hypertension than others (3). Keller and colleagues measured the number of nephrons in 10 patients who had suffered from hypertension and in 10 control patients. All patients had passed away due to accidents, and the two groups were age (median age of 46 yr) and sex matched. The patients with a history of hypertension had only about half the number of nephrons than the control group: means for two kidneys were 702,379 vs. 1,429,200. The lower nephron number was associated with more than a doubling in glomerular volume (27). An increased rate of hypertension has also been observed in the post-kidney donation period in previous studies (17, 34, 51).
The exact etiology of the development of hypertension with low nephron endowment is unclear. In two mouse models of renal agenesis, there was no difference in mean arterial pressure among the mice with 25% or 65% lower nephron numbers compared with wild-type when fed a normal diet. However, after exposure to a high salt diet, the mean arterial pressure increased significantly in the low nephron-numbered mice and no change was seen in wild-type mice (43). This supports the theory that low nephron endowment alone may not lead to the development of hypertension, but it sensitizes blood pressure to salt intake.
A number of studies have been conducted in human adults regarding the relationship between low birth weight (as a surrogate for low nephron number in most situations) and its renal consequences. Among the Australian aboriginal population, Hoy and colleagues observed an increased urinary albumin-to-creatinine ratio in patients with low vs. normal birth weight (21a). Interestingly, infants with high birth weight also had an increased risk of albuminuria, possibly due to the high prevalence of maternal diabetes in this population and the subsequent development of hyperglycemia during gestation (37, 38). The odds of developing hypertension, diabetic nephropathy, IgA nephropathy, membranous nephropathy, nephrotic syndrome, and ESRD are all increased in patients with low birth weight (4).
Robson and colleagues showed that, in a rat model of pyelonephritis, unilateral nephrectomy of the control kidney led to increased proteinuria in the diseased kidney (42). The authors further observed that glomerular selectivity measured by clearance of polydisperse polyvinal pyrrolidone (PVP) was significantly reduced in rats undergoing 50% or greater reduction in renal mass (42). These findings support the theory that tubular injury combined with hyperfiltration in remaining nephrons can enhance proteinuria as a consequence of both tubular and glomerular changes.
If the initial injury occurs in the glomerulus, tubular damage can be induced through increased delivery of protein to the tubules, which can cause inflammation, oxidative stress, and eventual injury (46) (FIGURE 2). As mentioned in the computational modeling studies above (30), whole kidney oxygen use is lower with nephron loss, but oxygen demand is increased on the level of the remaining nephron. The outer medulla is particularly susceptible to hypoxic damage due to low blood supply to this segment despite high transport activity (5). The increased oxygen demand in remaining tubules will lead to more anaerobic metabolism and local acidosis (46). Hypoxia can enhance production of reactive oxygen species, which leads to further inflammation possibly through activation of angiotensin AT1 receptors and CD36 receptors (39). The combination of local hypoxia, inflammation, and acidosis promotes tubular and kidney damage (FIGURE 2).
At the cellular level, partial epithelial-to-mesenchymal transition (EMT) of proximal tubular cells may promote cell proliferation, cytokine production, and cell dedifferentiation in remaining nephrons (40). This dedifferentiation may be triggered by persistent tubular growth signals and can lead to apoptosis of the cells or eventual inflammation and fibrosis with scar formation. The mechanism behind this fibrosis is not completely understood but likely includes excess production of cytokines such as TGF-β, fibroblast growth factor-2 (FGF), platelet-derived growth factor (PDGF), and endothelin. The partial EMT of proximal tubular cells may primarily recruit fibroblasts, which secrete scar forming proteins such as extracellular matrix proteins, rather than undergoing transformation into myofibroblasts (45, 46).
Conclusion and Perspectives
Nephron deficit or loss plays an important role in the pathogenesis of kidney disease and hypertension. Adaptation of the remaining nephrons is needed for compensation when nephron function and capacity is lost relatively rapidly (e.g., after kidney donation or potentially after recovering from episodes of AKI). These adaptations include increases in mean single nephron plasma flow and mean SNGFR along with glomerular hypertrophy. The increased tubular load enhances the membrane recruitment and expression of tubular transport proteins, and induces tubular hypertrophy. This enhances the tubular transport capacity but increases consumption of oxygen at the single nephron level (FIGURE 2). Eventually, if the nephron loss is sufficiently severe or long lasting, these adaptations may overwhelm the physiological capacities and lead to hypertension or CKD.
What determines whether a certain acute or chronic degree of nephron loss increases SNGFR or leads to glomerular hypertrophy and eventually glomerular sclerosis and tubulointerstitial fibrosis, the prerequisite for CKD? The recent study by Denic and colleagues provided new insights by showing that, with an age-dependent 30% nephron loss, there appeared to be no increase in mean SNGFR in the remaining nephrons (9). This may be due to the relative abundance of nephron endowment to meet the body's demands on the kidneys. In addition, an age-dependent decrease in total body metabolism or reduced dietary intake (including protein, salt, and potentially potassium and phosphate) may lower the excretory burden on the kidneys. Are the adaptations to nephron loss different among those born with a low nephron number vs. those who lose nephrons due to aging? One may hypothesize that, in persons born with a modestly reduced number of nephrons, mean SNGFR may also not adapt or increase. Nevertheless, there is likely a threshold that requires a rise in SNGFR when the nephron endowment is sufficiently low. This threshold concept for the SNGFR response is supported by the findings by Denic and colleagues, which reveal a significant increase in mean SNGFR in a small group of even older individuals (70–75 yr old) with an even lesser nephron number (~50% loss) (9).
The study by Kanzaki et al. indicated that mean SNGFR was unchanged in CKD patients vs. normal controls, whereas glomerular volume was greater in the CKD group (24). Thus the glomerular volume increase may dominate the glomerular adaptation to nephron loss, or SNGFR may initially rise in the course of the disease and eventually normalize to reduce the metabolic load on the remnant nephrons. It is noteworthy that the sample sizes of some of these studies are small: only 27 patients were included in this study (24), and 13 patients were included in the 70- to 75-yr-old group in the Denic et al. study (9). Thus larger and further clinical studies are warranted. There is a need to better define the relationship between nephron number, SNGFR, and glomerular volume in patients with CKD following kidney donation or in response to aging or low nephron endowment at birth. New MRI methods (2) may help determine the adaptive mechanisms to nephron loss in humans, provide a better understanding of the clinical consequences, and lead to improved therapeutic interventions. There is also a need to further investigate the hypothesis that aging individuals with better preserved GFR have an enhanced functional capacity to sustain single nephron hyperfunction, including a better preserved tubular transport capacity and integrity. Assessment of SNGFR in rodent models of more gradual nephron loss may also provide new insights, including the occurence and relevance of greater internephron variability in SNGFR.
Acknowledgments
The authors were supported by National Institute of Health (NIH) Grants R01 DK-112042, R01 DK-106102, the UAB/UCSD O’Brien Center of Acute Kidney Injury NIH-P30 DK-079337, NIH Training Grant T32DK104717, the Department of Veterans Affairs, and the Canada 150 Research Chair program.
Over the past 36 mo, V.V. has served as a consultant and received honoraria from Bayer, Boehringer Ingelheim, Eli Lilly, Janssen Pharmaceutical, and Merck, and received grant support for investigator-initiated research from Astra-Zeneca, Bayer, Boehringer Ingelheim, Fresenius, and Janssen Pharmaceutical. H.F. and A.L. have no conflicting interests to disclose.
H.F., A.T.L., and V.V. interpreted results of experiments; H.F. and V.V. prepared figures; H.F., A.T.L., and V.V. drafted manuscript; H.F., A.T.L., and V.V. edited and revised manuscript; H.F., A.T.L., and V.V. approved final version of manuscript.
References
- 1.Bennett KM, Bertram JF, Beeman SC, Gretz N. The emerging role of MRI in quantitative renal glomerular morphology. Am J Physiol Renal Physiol 304: F1252–F1257, 2013. doi: 10.1152/ajprenal.00714.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertram JF, Cullen-McEwen LA, Egan GF, Gretz N, Baldelomar E, Beeman SC, Bennett KM. Why and how we determine nephron number. Pediatr Nephrol 29: 575–580, 2014. doi: 10.1007/s00467-013-2600-y. [DOI] [PubMed] [Google Scholar]
- 3.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens 1: 335–347, 1988. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 4.Brenner BM, Rector FC. Brenner and Rector’s the Kidney. Philadelphia, PA: Saunders Elsevier, 2008. [Google Scholar]
- 5.Brezis M, Rosen S, Silva P, Epstein FH. Renal ischemia: a new perspective. Kidney Int 26: 375–383, 1984. doi: 10.1038/ki.1984.185. [DOI] [PubMed] [Google Scholar]
- 6.Cortes P, Zhao X, Riser BL, Narins RG. Role of glomerular mechanical strain in the pathogenesis of diabetic nephropathy. Kidney Int 51: 57–68, 1997. doi: 10.1038/ki.1997.8. [DOI] [PubMed] [Google Scholar]
- 7.Denic A, Alexander MP, Kaushik V, Lerman LO, Lieske JC, Stegall MD, Larson JJ, Kremers WK, Vrtiska TJ, Chakkera HA, Poggio ED, Rule AD. Detection and clinical patterns of nephron hypertrophy and nephrosclerosis among apparently healthy adults. Am J Kidney Dis 68: 58–67, 2016. doi: 10.1053/j.ajkd.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denic A, Lieske JC, Chakkera HA, Poggio ED, Alexander MP, Singh P, Kremers WK, Lerman LO, Rule AD. The substantial loss of nephrons in healthy human kidneys with aging. J Am Soc Nephrol 28: 313–320, 2017. doi: 10.1681/ASN.2016020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denic A, Mathew J, Lerman LO, Lieske JC, Larson JJ, Alexander MP, Poggio E, Glassock RJ, Rule AD. Single-nephron glomerular filtration rate in healthy adults. N Engl J Med 376: 2349–2357, 2017. doi: 10.1056/NEJMoa1614329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denic A, Mathew J, Nagineni VV, Thompson RH, Leibovich BC, Lerman LO, Lieske JC, Alexander MP, Augustine JJ, Kremers WK, Rule AD. Clinical and pathology findings associate consistently with larger glomerular volume. J Am Soc Nephrol 29: 1960–1969, 2018. doi: 10.1681/ASN.2017121305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esposito C, He C-J, Striker GE, Zalups RK, Striker LJ. Nature and severity of the glomerular response to nephron reduction is strain-dependent in mice. Am J Pathol 154: 891–897, 1999. doi: 10.1016/S0002-9440(10)65336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fattah H, Vallon V. The potential role of SGLT2 inhibitors in the treatment of Type 1 diabetes mellitus. Drugs 78: 717–726, 2018. doi: 10.1007/s40265-018-0901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fogo AB. Glomerular hypertension, abnormal glomerular growth, and progression of renal diseases. Kidney Int Suppl 75: S15–S21, 2000. doi: 10.1046/j.1523-1755.2000.07505.x. [DOI] [PubMed] [Google Scholar]
- 14.Friedman AN. High-protein diets: potential effects on the kidney in renal health and disease. Am J Kidney Dis 44: 950–962, 2004. doi: 10.1053/j.ajkd.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 15.Fulladosa X, Moreso F, Narváez JA, Grinyó JM, Serón D. Estimation of total glomerular number in stable renal transplants. J Am Soc Nephrol 14: 2662–2668, 2003. doi: 10.1097/01.ASN.0000088025.33462.B0. [DOI] [PubMed] [Google Scholar]
- 16.Gansevoort RT, de Zeeuw D, de Jong PE. ACE inhibitors and proteinuria. Pharm World Sci 18: 204–210, 1996. doi: 10.1007/BF00735961. [DOI] [PubMed] [Google Scholar]
- 17.Garg AX, Prasad GVR, Thiessen-Philbrook HR, Ping L, Melo M, Gibney EM, Knoll G, Karpinski M, Parikh CR, Gill J, Storsley L, Vlasschaert M, Mamdani M; Donor Nephrectomy Outcomes Research (DONOR) Network . Cardiovascular disease and hypertension risk in living kidney donors: an analysis of health administrative data in Ontario, Canada. Transplantation 86: 399–406, 2008. doi: 10.1097/TP.0b013e31817ba9e3. [DOI] [PubMed] [Google Scholar]
- 18.Gava AL, Freitas FP, Balarini CM, Vasquez EC, Meyrelles SS. Effects of 5/6 nephrectomy on renal function and blood pressure in mice. Int J Physiol Pathophysiol Pharmacol 4: 167–173, 2012. [PMC free article] [PubMed] [Google Scholar]
- 19.Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Møller A, Nielsen K, Nyengaard JR, Pakkenberg B, SØRensen FB, Vesterby A, West MJ. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS 96: 857–881, 1988. doi: 10.1111/j.1699-0463.1988.tb00954.x.http://www.ncbi.nlm.nih.gov/pubmed/3056461 [DOI] [PubMed] [Google Scholar]
- 20.Hinchliffe SA, Lynch MR, Sargent PH, Howard CV, Van Velzen D. The effect of intrauterine growth retardation on the development of renal nephrons. Br J Obstet Gynaecol 99: 296–301, 1992. doi: 10.1111/j.1471-0528.1992.tb13726.x. [DOI] [PubMed] [Google Scholar]
- 21.Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol Renal Physiol 241: F85–F93, 1981. [DOI] [PubMed] [Google Scholar]
- 21a.Hoy W, Rees M, Kile E, Mathews J, McCredie D, Pugsley D, Wang Z. Low birthweight and renal disease in Australian aborigines. Lancet 352: 1826–1827, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Hughson MD, Douglas-Denton R, Bertram JF, Hoy WE. Hypertension, glomerular number, and birth weight in African Americans and white subjects in the southeastern United States. Kidney Int 69: 671–678, 2006. doi: 10.1038/sj.ki.5000041. [DOI] [PubMed] [Google Scholar]
- 23.Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CAM, Lash JP, Hsu C-Y, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79: 1370–1378, 2011. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanzaki G, Puelles VG, Cullen-McEwen LA, Hoy WE, Okabayashi Y, Tsuboi N, Shimizu A, Denton KM, Hughson MD, Yokoo T, Bertram JF. New insights on glomerular hyperfiltration: a Japanese autopsy study. JCI Insight 2: 94334, 2017. doi: 10.1172/jci.insight.94334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufman JM, DiMeola HJ, Siegel NJ, Lytton B, Kashgarian M, Hayslett JP. Compensatory adaptation of structure and function following progressive renal ablation. Kidney Int 6: 10–17, 1974. doi: 10.1038/ki.1974.72. [DOI] [PubMed] [Google Scholar]
- 26.Kaufman JM, Siegel NJ, Hayslett JP. Functional and hemodynamic adaptation to progressive renal ablation. Circ Res 36: 286–293, 1975. doi: 10.1161/01.RES.36.2.286. [DOI] [PubMed] [Google Scholar]
- 27.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med 348: 101–108, 2003. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, Heo NJ, Jung JY, Son M-J, Jang HR, Lee JW, Oh YK, Na KY, Joo KW, Han JS. Changes in the sodium and potassium transporters in the course of chronic renal failure. Nephron, Physiol 115: 31–41, 2010. doi: 10.1159/000314542. [DOI] [PubMed] [Google Scholar]
- 29.Knutson DW, Chieu F, Bennett CM, Glassock RJ. Estimation of relative glomerular capillary surface area in normal and hypertrophic rat kidneys. Kidney Int 14: 437–443, 1978. doi: 10.1038/ki.1978.148. [DOI] [PubMed] [Google Scholar]
- 30.Layton AT, Edwards A, Vallon V. Adaptive changes in GFR, tubular morphology, and transport in subtotal nephrectomized kidneys: modeling and analysis. Am J Physiol Renal Physiol 313: F199–F209, 2017. doi: 10.1152/ajprenal.00018.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Layton AT, Edwards A, Vallon V. Renal potassium handling in rats with subtotal nephrectomy: modeling and analysis. Am J Physiol Renal Physiol 314: F643–F657, 2018. doi: 10.1152/ajprenal.00460.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Layton AT, Vallon V. SGLT2 inhibition in a kidney with reduced nephron number: modeling and analysis of solute transport and metabolism. Am J Physiol Renal Physiol 314: F969–F984, 2018. doi: 10.1152/ajprenal.00551.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Layton AT, Vallon V, Edwards A. A computational model for simulating solute transport and oxygen consumption along the nephrons. Am J Physiol Renal Physiol 311: F1378–F1390, 2016. doi: 10.1152/ajprenal.00293.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenihan CR, Busque S, Derby G, Blouch K, Myers BD, Tan JC. Longitudinal study of living kidney donor glomerular dynamics after nephrectomy. J Clin Invest 125: 1311–1318, 2015. doi: 10.1172/JCI78885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luyckx VA, Shukha K, Brenner BM. Low nephron number and its clinical consequences. Rambam Maimonides Med J 2: e0061, 2011. doi: 10.5041/RMMJ.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mañalich R, Reyes L, Herrera M, Melendi C, Fundora I. Relationship between weight at birth and the number and size of renal glomeruli in humans: a histomorphometric study. Kidney Int 58: 770–773, 2000. doi: 10.1046/j.1523-1755.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- 37.Nelson RG, Morgenstern H, Bennett PH. Birth weight and renal disease in Pima Indians with type 2 diabetes mellitus. Am J Epidemiol 148: 650–656, 1998. doi: 10.1093/aje/148.7.650. [DOI] [PubMed] [Google Scholar]
- 38.Nelson RG, Morgenstern H, Bennett PH. Intrauterine diabetes exposure and the risk of renal disease in diabetic Pima Indians. Diabetes 47: 1489–1493, 1998. doi: 10.2337/diabetes.47.9.1489. [DOI] [PubMed] [Google Scholar]
- 39.Okamura DM, Himmelfarb J. Tipping the redox balance of oxidative stress in fibrogenic pathways in chronic kidney disease. Pediatr Nephrol 24: 2309–2319, 2009. doi: 10.1007/s00467-009-1199-5. [DOI] [PubMed] [Google Scholar]
- 40.Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int 80: 41–50, 2011. doi: 10.1038/ki.2011.77. [DOI] [PubMed] [Google Scholar]
- 41.Quinlan J, Lemire M, Hudson T, Qu H, Benjamin A, Roy A, Pascuet E, Goodyer M, Raju C, Zhang Z, Houghton F, Goodyer P. A common variant of the PAX2 gene is associated with reduced newborn kidney size. J Am Soc Nephrol 18: 1915–1921, 2007. doi: 10.1681/ASN.2006101107. [DOI] [PubMed] [Google Scholar]
- 42.Robson AM, Mor J, Root ER, Jager BV, Shankel SW, Ingelfinger JR, Kienstra RA, Bricker NS. Mechanism of proteinuria in nonglomerular renal disease. Kidney Int 16: 416–429, 1979. doi: 10.1038/ki.1979.145. [DOI] [PubMed] [Google Scholar]
- 43.Ruta L-AM, Dickinson H, Thomas MC, Denton KM, Anderson WP, Kett MM. High-salt diet reveals the hypertensive and renal effects of reduced nephron endowment. Am J Physiol Renal Physiol 298: F1384–F1392, 2010. doi: 10.1152/ajprenal.00049.2010. [DOI] [PubMed] [Google Scholar]
- 44.Sandle GI, Gaiger E, Tapster S, Goodship TH. Enhanced rectal potassium secretion in chronic renal insufficiency: evidence for large intestinal potassium adaptation in man. Clin Sci (Lond) 71: 393–401, 1986. doi: 10.1042/cs0710393. [DOI] [PubMed] [Google Scholar]
- 45.Sato Y, Yanagita M. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm Regen 37: 17, 2017. doi: 10.1186/s41232-017-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnaper HW. Remnant nephon physiology and the progression of chronic kidney disease. Pediatr Nephrol 29: 193–202, 2015. doi: 10.1007/s00467-013-2494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueda Y, Ookawara S, Ito K, Miyazawa H, Kaku Y, Hoshino T, Tabei K, Morishita Y. Changes in urinary potassium excretion in patients with chronic kidney disease. Kidney Res Clin Pract 35: 78–83, 2016. doi: 10.1016/j.krcp.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.University of Utah Renal pathology for medical education - WebPath [Online]. https://library.med.utah.edu/WebPath/RENAHTML/RENALIDX.html [7 Nov. 2018].
- 49.Weibel ER, Gomez DM. A principle for counting tissue structures on random sections. J Appl Physiol (1985) 17: 343–348, 1962. doi: 10.1152/jappl.1962.17.2.343. [DOI] [PubMed] [Google Scholar]
- 50.Wetzels JFM, Kiemeney LALM, Swinkels DW, Willems HL, den Heijer M. Age- and gender-specific reference values of estimated GFR in Caucasians: the Nijmegen Biomedical Study. Kidney Int 72: 632–637, 2007. doi: 10.1038/sj.ki.5002374. [DOI] [PubMed] [Google Scholar]
- 51.Wills AK, Lawlor DA, Matthews FE, Sayer AA, Bakra E, Ben-Shlomo Y, Benzeval M, Brunner E, Cooper R, Kivimaki M, Kuh D, Muniz-Terrera G, Hardy R. Life course trajectories of systolic blood pressure using longitudinal data from eight UK cohorts. PLoS Med 8: e1000440, 2011. doi: 10.1371/journal.pmed.1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolf M. Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol 21: 1427–1435, 2010. doi: 10.1681/ASN.2009121293. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Z, Quinlan J, Hoy W, Hughson MD, Lemire M, Hudson T, Hueber P-A, Benjamin A, Roy A, Pascuet E, Goodyer M, Raju C, Houghton F, Bertram J, Goodyer P. A common RET variant is associated with reduced newborn kidney size and function. J Am Soc Nephrol 19: 2027–2034, 2008. doi: 10.1681/ASN.2007101098. [DOI] [PMC free article] [PubMed] [Google Scholar]