

Abstract
Synthesis and remodeling of the lung matrix is necessary for primary and compensatory lung growth. Because cyclic negative force is applied to developing lung tissue during the respiratory cycle, we hypothesized that stretch is a critical regulator of lung matrix remodeling. By using quantitative image analysis of whole-lung and whole-lobe elastin in situ zymography images, we demonstrated that elastase activity increased twofold during the alveolar stage of postnatal lung morphogenesis in the mouse. Remodeling was restricted to alveolar walls and ducts and was nearly absent in dense elastin band structures. In the mouse pneumonectomy model of compensatory lung growth, elastase activity increased threefold, peaking at 14 days postpneumonectomy and was higher in the accessory lobe compared with other lobes. Remodeling during normal development and during compensatory lung growth was different with increased major airway and pulmonary arterial remodeling during development but not regeneration, and with homogenous remodeling throughout the parenchyma during development, but increased remodeling only in subpleural regions during compensatory lung growth. Left lung wax plombage prevented increased lung elastin during compensatory lung growth. To test whether the adult lung retains an innate capacity to remodel elastin, we developed a confocal microscope-compatible stretching device. In ex vivo adult mouse lung sections, lung elastase activity increased exponentially with strain and in peripheral regions of lung more than in central regions. Our study demonstrates that lung elastase activity is stretch-dependent and supports a model in which externally applied forces influence the composition, structure, and function of the matrix during periods of alveolar septation.
Keywords: elastin, lung development, lung regeneration, protease, stretch
unlike other organs, the postnatal lung develops and functions in a cyclic vacuum as negative pressure expands alveoli with subsequent passive exhalation due to the elastic lung recoil. Although lung specification and branching morphogenesis can occur ex vivo (4), only the most rudimentary of alveolar septae can form outside of the lung's native environment (14). If we are to more fully understand the mechanisms controlling alveolarization, the models we use to study lung morphogenesis must faithfully reproduce the conditions in which the postnatal lung grows and develops.
Lung stretch and strain are critical regulators of alveolar size. Three observations support this assertion. First, alveolar size is remarkably consistent within animals of the same species and between apical and basal areas of the lung (23). Second, in the mouse, right lung compensatory growth after left pneumonectomy completely restores lung volume, alveolar number, and alveolar size by 4 wk (22). Third, thoracic plombage (5, 7) and ipsilateral phrenic nerve transection (24) inhibit postpneumonectomy compensatory lung growth. At the other end of the spectrum an imbalance of applied forces likely potentiates the progressive airspace simplification of chronic obstructive pulmonary disease (19). From a therapeutic perspective, compensatory lung growth with reseptation was recently demonstrated in an adult human (3). Because stretch is a critical determinant of lung size and is also an important force underlying septal elongation (1), we sought to understand how stretch regulates macroscopic and microscopic distal lung remodeling during development and compensatory lung growth.
To answer this question, we focused experiments to understand how lung stretch regulates elastin remodeling. We chose this focus for four reasons. First, elastin is both highly expressed in the lung and critical for hysteresis during the respiratory cycle (23). Second, the formation of a collagen/elastin bundle is a critical early step in septal development (18). Third, after lung formation, there is little if any elastin turnover (17). Fourth, the failure to initiate elastase activity following premature birth has been associated with the development of bronchopulmonary dysplasia (2), and the lungs of infants with bronchopulmonary dysplasia have excessive and dysplastic elastin structures within them (12). To study elastase activity, we developed a novel elastin in situ zymography technique to quantify and normalize the endogenous elastase activity of an entire lung section or postpneumonectomy lung lobe section with masking and quantification of activity within different structures.
In this study, we utilized this technique to demonstrate that stretch induces lung elastase activity. We demonstrate a regional remodeling response during compensatory lung growth and show a stoichiometric dyssynchrony of this response with increased remodeling of the distal lung parenchyma but not of central conducting structures. At the acinar level, we show that elastin of dense elastin bands is not remodeled during septation but rather that this remodeling is confined to alveolar walls and ducts. Finally, we demonstrate that the adult mouse lung retains some level of innate remodeling capacity, which is regulated by stretch. Our novel finding, that stretch regulates lung elastase activity, provides insight into how the thoracic vacuum regulates distal lung morphogenesis and informs future investigations in lung regenerative medicine.
METHODS
Animal use and housing.
All animal use was approved by the animal use and care committee at Cincinnati Children's Hospital Medical Center in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Bethesda, MD). C57BL/6J mice from the Jackson Laboratory (Bar Harbor, ME) were maintained in a pathogen-free barrier facility with 12:12-h light-dark cycles and with filtered water and autoclaved chow provided ad libitum.
Animal death and tissue preparation.
Animals were killed by intraperitoneal injection of ketamine, xylazine, and acepromazine (100, 6, and 2 mg/kg, respectively) and severing of the left renal artery. Lungs were inflated with a 1:1 solution of 50% sucrose and optimal cutting temperature (OCT) compound, snap-frozen, and embedded in OCT.
Mouse surgical procedures.
Left pneumonectomy or sham thoracotomy was performed in 8- to 10-wk-old male C57BL/6J mice as previously described (11). For the pneumonectomy plus wax lung plombage, we created lung molds by using the fixed, inflated left lung of weight-matched mice and plaster of Paris. Sterile dental wax was melted and instilled to make sterile wax lung molds.
Lung magnetic resonance imaging.
Mice were imaged on a Bruker (Madison, WI) 7T scanner during free breathing with isoflurane. A standard two-dimensional multislice gradient-echo sequence with an appropriate radio frequency pulse (flip angle = 30°) was used for proton magnetic resonance (MR) imaging. Respiratory-gated 1H MR images were acquired at end expiration for all mice. Twenty-nine transverse slices (thickness = 0.587 mm, gap = 0.2 mm) were acquired to cover the entire mouse lung. The lung was semimanually segmented from the chest with commercial graphics processing software (Amira; FEI, Hillsboro, OR); the major bronchi and blood vessels were excluded. Lung volumes were determined by measuring the sum of all segmented lung voxels (with known voxel size); these data were compared with previously acquired and published data using the same surgical technique and mouse (22).
Fluorescent plate-based elastase assay.
After death, mouse lungs were collected and snap-frozen in liquid nitrogen. RIPA buffer was used to make lung homogenates of equal protein concentration, and 10 mg of lung homogenate protein was used to quantify lung elastase activity at 30 min using the Enzcheck elastase assay (Life Technologies, Carlsbad, CA) per manufacturer specifications.
Desmosine ELISA.
Urine was collected from adult mice after death by bladder puncture, and desmosine ELISA was performed per manufacturer specifications (CUSABIO, Wuhan, China).
Elastin in situ zymography.
Ten micrometer frozen sections were allowed to air dry and then briefly fixed in 2% paraformaldehyde in PBS for 5 min to adhere them to polysine slides. Sections were incubated overnight in DMEM with 10% fetal bovine serum and 1 μg/ml BODIPY-conjugated soluble elastin (Enzcheck Elastase Assay; Life Biosciences), and stained with antitropoelastin antibody (1:1,000, AbCam) and Alexa Fluor 594 secondary antibody. For each in situ zymography experiment except one, sections were stained and imaged together. Lungs and lung lobes were imaged using a Nikon 90i scanning wide-field microscope with motorized stage using a ×10 objective. Images were tile-scanned with 15% overlap and 4×4 binning to reduce the file size. The one experiment in which lung sections were not imaged contiguously was the postpneumonectomy accessory lobe time course. In this experiment, control through 4-wk specimens were imaged together, but the 8-wk section was imaged at a later time with the values normalized to concurrently analyze control and 14-day specimens. For each experiment, three to four lungs were analyzed from mice of at least two different litters.
Quantitative histological image analysis.
Lung and lung lobe images were analyzed using Imaris (version 7.7.2; Bitplane). An elastase surface was generated by first using a negative control lung (no substrate) to set a threshold FITC signal. An elastin surface was created in a similar fashion using the tetramethylrhodamine isothiocyanate (TRITC) signal. Secondary FITC and TRITC channels were generated on the basis of these masks. Next, major arteries and airways (defined as being greater than 200 μm in diameter) were manually masked. Debris and folded tissue were excluded by a manual mask. Elastase and elastin signal were defined as the masked FITC and TRITC signal within the elastase surface. Elastin bands were defined as any elastin object with a sphericity greater than 0.8, and nonband elastin was defined as all other elastin objects. The subserosal surface was defined as tissue within 200 μm of the serosal surface.
Confocal-compatible lung stretching device.
A confocal microscope-compatible stretching device was created that used a threaded shaft and computer-controlled motor to move an open stage. Below this stage on either side were two nylon mounts to which a silastic membrane (Flexcell International) was mounted using nylon strings. A distensible perfusion well was attached (Electron Microsciences) to contain the substrate solution, and a 200-μm-thick lung section was secured with fibrin glue (Abbott). To make the sections, we inflated lungs of mice that were 2 to 3 mo old with 1.2% low-melting-point agarose and embedded the lungs in 4% low melting point agarose. Sections (200 μm) were cut and kept in PBS at 4°C until they adhered to the membrane.
The lung sections were stretched at room temperature in increments of 679 μm every 1 min with elastin in situ zymography substrate at 1:100 dilution in PBS. Strain was measured as the percent change in length. As a control, lung sections were stretched in PBS or incubated in substrate without stretch. Imaging was performed using a Nikon A1Rsi inverted confocal microscope. Blue autofluorescence was used as a loading control. Major lung structures (>200 μm) were manually excluded and elastase and tissue surfaces were created and applied identically to each image. Lung section elastase activity was quantified as the masked elastase signal divided by the masked tissue signal. For whole-lobe stretch experiments, apical and basal segments of the lung section were adhered to two coverslips and suspended in the substrate solution on a glass slide. The lung section was imaged in stretched and unstretched states at ×4 magnification.
Statistical analysis.
SigmaPlot (Systat) was used for statistical analysis. One-way ANOVA was used to compare multiple data sets with pairwise comparisons by the Holm-Sidak method. Longitudinal data sets were analyzed by two methods. First, they were analyzed by one-way ANOVA to answer the question of whether a statistically significant difference existed between the groups. Second, consecutive time points were analyzed by Student's t-test to determine whether a statistically significant difference existed from one time point to the next. All error bars indicate measurements of standard error.
RESULTS
During postnatal lung development, elastase activity increases during the alveolar stage and is restricted to alveolar walls and ducts.
To answer the question of whether lung elastin fibers are selectively degraded during lung development, we quantified lung elastase activity by both fluorometric elastase assay and by elastin in situ zymography. By fluorometric elastase assay, elastase activity was 1.5 to 2.7 times higher from embryonic day 15.5 (E15.5) to postnatal day 14 (PND14) lung homogenates compared with postnatal week 8 lung homogenates, but none of these differences were statistically significant by ANOVA. To more precisely measure lung elastase activity, we performed elastin in situ zymography (Fig. 1A). An elastin surface was created from the TRITC channel, and a manual surface was created to exclude folded tissue, debris, and nonpulmonary organs from analysis (Fig. 1B). The elastase signal was defined as the FITC signal within the elastin mask and outside the exclusion mask (Fig. 1C) and was then normalized to the TRITC signal within the elastin mask. More than 99% of nonexcluded elastase signal was within the elastin mask. This technique yielded much higher intersample agreement than the lung homogenate elastase assay and demonstrated that lung elastase activity increased during the alveolar stage of lung development (between PND3 and PND7), peaked at PND10, and gradually decreased to baseline by postnatal week 8 (Fig. 1D). To quantify the remodeling activity of different lung structures, artery and airway masks were drawn (Fig. 2A). Lung parenchymal elastase activity constituted more than 95% of remodeling activity in all sections. To determine whether elastase activity synchronously increased and decreased in arteries, airways, and lung parenchyma, the elastase/elastin ratios for each structure were normalized to the postanatal week 8 value. The lung parenchyma experienced the greatest increase in elastase activity (Fig. 2B) but both arterial (Fig. 2C) and major airway (Fig. 2D) elastase activity increased in parallel with the parenchyma. Lung sections were not analyzed at E15.5 because structures could not be reliably identified in these sections.
Fig. 1.

Elastin remodeling over the course of lung development. A: to quantify elastin remodeling during development, we performed elastin in situ zymography (green) and normalized the signal to tropoelastin (red). A lung image at postnatal day 10 (PND10) is shown. Scale bar = 1,000 μm. B: an example of the elastin surface created based on the tetramethylrhodamine isothiocyanate channel (red) and the manual exclusion mask for debris, nonpulmonary organs, and folded tissue (gray). C: within this elastin surface, the elastase (green) and elastin (not shown) signals were quantified. D: the lungs of three different animals per time point were imaged. Lung elastin remodeling began increasing with the onset of alveolarization [between PND3 and PND7, peaked at PND10, and then decreased to embryonic levels by postnatal week 8 (PN 8wk)]. This level of remodeling (normalized to 1) represents the baseline signal of the assay (dashed line). Only differences by t-test are shown. Elastase activity values were significantly different by ANOVA (P < 0.001).
Fig. 2.

Parenchymal, arterial, and major airway elastase activity increase and decrease in parallel during the alveolar stage of lung development. A: to differentiate the elastase activity of different lung structures, we applied major artery (red) and major airway (blue) masks with parenchymal elastase activity defined as the unmasked signal. B: parenchymal lung elastase activity peaked at PND10 as did arterial elastase activity (C) and major airway elastase activity (D). Only differences by t-test are shown. Parenchymal (P < 0.001), arterial (P = 0.004), and major airway (P < 0.001) elastase activity were all significantly different by ANOVA.
Creation of a subepithelial elastin and collagen band is critical for the formation of a secondary alveolar septum (18). Elastin bands are also present at the alveolar duct mouth. To determine in which distal lung structures elastin is cleaved, we quantified elastase activity in elastin bands and elastin exclusive of these bands (Fig. 3, A–D). To validate this technique, we quantified the number of defined bands per square millimeter of lung tissue. As expected, the number of elastin bands gradually increased from 7 per mm2 at PND3 to 30 per mm2 at PND14 (Fig. 3E). Despite the increased number of elastin bands, the elastase activity in these bands did not increase between saccular and alveolar stages of lung development. Rather, increases in elastase activity were in nonband elastin structures (Fig. 3F). Increased lung elastase activity during the alveolar stage of lung development was due to activity in the walls of alveolar ducts and terminal respiratory units and not the elastin bands present in alveolar septal tips and the mouths of alveolar ducts.
Fig. 3.
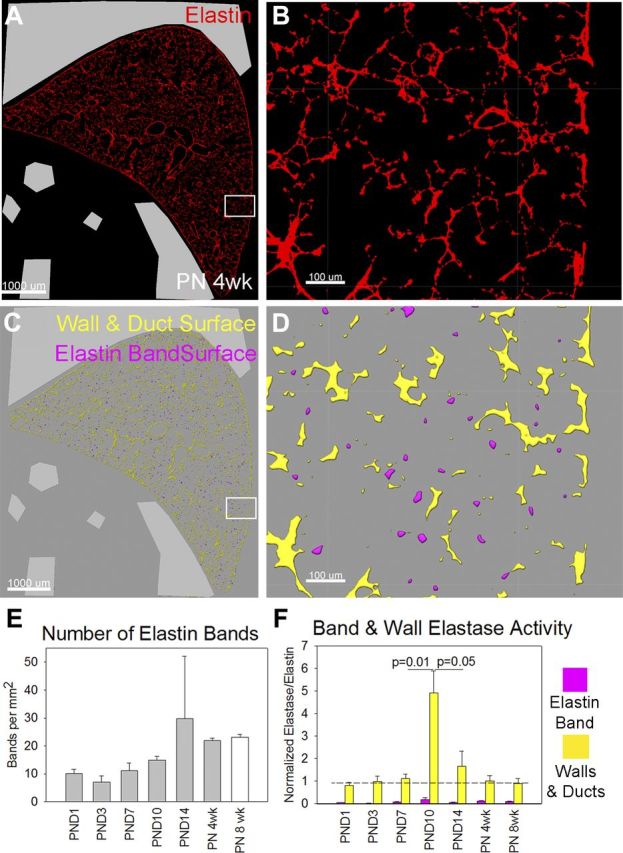
During alveolarization, elastase activity increases in acinar and alveolar duct walls. A: to differentiate elastase activity localized to elastin bands (septal tips and the openings of alveolar ducts) vs. acinar and alveolar duct walls, we used the tropoelastin portion of our in situ zymography images. Scale bar = 1,000 μm. B: at higher magnification, elastin structures could be differentiated into more linear components (nonband elastin) and more circular ones (band elastin). Scale bar = 100 μm. C: after creation of an elastase surface, we segregated the components of that surface into circular (purple) and noncircular (yellow) components. D: at higher magnification, the circular components were defined as elastin bands and the noncircular components were defined as walls and ducts. E: to validate our method, we quantified the number of defined tips per square millimeter of lung tissue. The number of defined tips tripled between the saccular stage of lung development and the completion of alveolarization. F: despite the increased number of elastin band structures, elastin band (purple) elastase activity did not increase during postnatal lung development, but the elastase activity in walls and ducts (yellow) increased fivefold. Only differences by t-test are shown. Wall and duct normalized elastase activity was significantly different by ANOVA (P < 0.001), but differences in elastin band activity were not.
These three sets of data demonstrate that elastase activity increases during the alveolar stage of lung development; that during postnatal lung development parenchymal, arterial, and major airway elastase activity increases and decreases in concert; and that in the lung parenchyma elastase activity is largely restricted to alveolar walls and ducts.
Reinduction of elastase activity during compensatory lung growth requires stretch.
The mouse pneumonectomy model of compensatory lung growth can be used to elucidate critical mechanisms controlling alveolar septation and regeneration. Left pneumonectomy results in expansion of the right lung, principally the accessory lobe, with enlargement of existing airspaces followed by reseptation. By 4 wk postpneumonectomy in the mouse, there is complete recovery of lung volume and normalization of alveolar size (22). Because elastase activity increased during primary alveolar septation, we sought to determine whether this process was reinduced during compensatory lung growth.
We first quantified elastase activity in the accessory lobe postpneumonectomy. Compared with control mice, lung elastase activity gradually increased threefold by postpneumonectomy day 14 and gradually decreased to near-control levels by 8 wk postpneumonectomy (Fig. 4A). To control for surgery-related inflammation, we quantified elastase activity in sham-operated animals and found no difference at 1 or 14 days postprocedure (Fig. 4B). Postpneumonectomy compensatory lung growth occurs in the upper, middle, and lower lung lobes but to a lesser degree than in the accessory lobe. We quantified the level of elastase activity in all four right lung lobes 7 days postpneumonectomy. The upper and middle lobes did not demonstrate an appreciable increase in lung elastase activity, but activity of the lower lobe was increased to a lesser degree than in the accessory lobe (Fig. 4C). To validate these findings, we performed desmosine ELISA on the urine of control, sham-operated, and 7-day postpneumonectomy mice. Urinary desmosine, a soluble byproduct of elastin breakdown, was 9% higher in postpneumonectomy mouse urine than control mice and 6% higher than sham-operated mice. These data demonstrate that elastase activity is induced during compensatory lung growth and decreases to control levels once compensatory growth is complete and that this increased activity is greatest in the lung lobe, which undergoes the most compensatory growth.
Fig. 4.

Elastase activity over the course of compensatory lung growth. A: during postpneumonectomy compensatory lung growth, accessory lobe elastase activity increased threefold compared with control accessory lobe activity (white bar). Elastase activity peaked at 14 days and returned to baseline by 56 days postpneumonectomy (post-PNX). B: in contrast to increased postpneumonectomy elastase activity (black bars), sham-operated animals (gray bars) had levels of lung elastase activity comparable to those of control animals. C: because upper, middle, and lower lobes also experience postpneumonectomy compensatory lung growth but less than the accessory lobe, we quantified elastin remodeling in all lobes at 7 days postpneumonectomy. The elastase activity of the upper (Up) and middle (Mid) lobes was not increased. Elastase activity of the lower (Low) lobe was increased, but not as much as the accessory (Ac) lobe. D: accessory lobes structures were segregated as before. Postpneumonectomy, there was an increase in remodeling of the lung parenchyma (green), but not of the arterial (red) or major airway (blue) components of the lung.
During postpneumonectomy lung growth, there is evidence that central conducting lung structures may lengthen but may not otherwise enlarge or remodel (20). We applied airway and artery masks to determine whether or not arterial and airway elastase activity changed postpneumonectomy. In contrast to the increases in arterial and major airway elastase activity observed during normal lung development, arterial and large airway elastase activity did not increase during compensatory lung growth (Fig. 4D). Our data support the concept that postpneumonectomy lung regeneration occurs in distal lung structures to a greater extent than in large central structures such as arteries and major airways.
Postpneumonectomy accessory lobe in situ zymography demonstrated nonuniform elastase activity with increased activity in subpleural regions (Fig. 5, A and B) By applying a subpleural 200-μm mask to our postpneumonectomy sections, elastase activity was increased only in these subpleural regions during compensatory lung growth (Fig. 5C). We similarly analyzed subpleural and central elastase activity during the alveolar stage of normal lung development (Fig. 5D). In contrast to the elastase activity changes occurring during compensatory lung growth, the increase in lung elastase activity during normal lung development was evenly distributed between the subpleural and nonsubpleural parenchyma (Fig. 5E). Because the subpleural region of the accessory lobe experiences the most strain and deformation during compensatory lung growth (6), we considered whether this increase in elastase activity was caused by a regional increase in lung stretch and that the homogenous remodeling response during normal lung development was due to a more even distribution of stretch.
Fig. 5.

Elastase activity is confined to subpleural regions during compensatory lung growth but not during development. A: postpneumonectomy (post-PNX), the elastase activity of subpleural regions appeared increased compared with more central regions. Scale bar = 1,000 μm. B: when viewed as a heat map of the FITC channel (red = high pixel intensity, blue = low pixel intensity) elastase activity was nearly absent in central regions and increased in subpleural regions. C: to quantify this increased remodeling, we created a 200-μm subpleural mask to quantify subpleural elastase activity and compare it with central elastase activity. The increase in lung elastase activity postpneumonectomy was entirely due to increased activity in subpleural regions (black box with hash marks) with little increase in central regions (black box with lines). D: during development, this subpleural predominance of elastase activity was not observed. Scale bar = 1,000 μm. E: quantification of subpleural and central tissue elastase activity in our lung development time course demonstrated that lung elastase activity was roughly equivalent in subpleural and central lung.
Stretch induces elastase activity during compensatory lung growth.
To test whether lung stretch triggers elastase activity during compensatory lung growth and to isolate stretch-mediated changes from blood flow and inflammation-related changes, we inserted weight-matched wax lungs into the thoracic cavity following left pneumonectomy (Fig. 6A). At 4 to 5 wk postsurgery, wax plombage completely prevented postpneumonectomy right lung reexpansion and growth (Fig. 6, B–E). Because pneumonectomy induces an inflammatory response, we quantified tumor necrosis factor alpha (TNF-α), macrophage chemoattractant protein 1 (MCP1), and neutrophil elastase (ELANE) mRNA of accessory lobes 24 h postsurgery. Levels of MCP1 and ELANE were higher in pneumonectomy with and without plombage compared with those of sham-operated mice, and levels of TNF-α were similar (Fig. 6F). Wax plombage completely prevented the induction of accessory lobe elastase activity (Fig. 6G). These data demonstrate that stretch is necessary for induction elastase activity during compensatory lung growth.
Fig. 6.

Lung stretch is required for induction of lung elastin remodeling. A: the anterior and posterior surfaces of the wax lung used as plombage are demonstrated. B: we used magnetic resonance imaging 4 wk after sham thoracotomy to quantify right and left lung volumes. C: four wk after pneumonectomy, the right lung completely filled the left thorax. D: five wk after pneumonectomy with placement of plombage, the wax lung prevented left lung reexpansion. E: quantification of lung volumes showed that the wax plombage completely prevented compensatory lung growth after pneumonectomy. The white box denotes the left lung volume, the black box denotes the right lung volume, and the white dotted box denotes the wax lung volume. F: we quantified mRNA levels of inflammatory cytokine levels at 24 h postsurgery in pneumonectomy and pneumonectomy + wax plombage. There was no statistical difference in cytokine levels [tumor necrosis factor-α (TNF-α), macrophage chemoattractant protein-1 (MCP1), and neutrophil elastase (ELANE)] between sham-operated mice (white box), or mice that underwent pneumonectomy (black box) or pneumonectomy + wax plombage (black box with lines), although there tended to be more ELANE and MCP1 in mice that underwent pneumonectomy (P = 0.06 by ANOVA). G: wax plombage prevented the induction of lung elastin remodeling at 7 days postpneumonectomy.
The adult mouse lung retains an innate capacity to remodel elastin in response to stretch.
To determine the rapidity with which lung elastase activity can be induced in the adult mouse lung, we developed a confocal-microscope compatible lung stretching device (Fig. 7A). After using fibrin glue to adhere 200-μm subpleural left lung sections to the silastic membrane (Fig. 7B) we obtained sequential elastin in situ zymography images stretching the membrane by 650 μm every minute. As a control, we imaged an unstretched lung section before and after 10 min of incubation with substrate and stretched lung without substrate. Incubating the unstretched lung in substrate for 10 min did not result in increased elastase signal nor did stretching lung sections in the absence of substrate. When lung sections were stretched in the presence of substrate, elastase activity increased exponentially with strain (Fig. 7, C–E). These data indicate that the adult mouse lung retains an innate capacity to remodel elastin in response to strain and that this increased elastase activity develops in a time frame inconsistent with de novo gene transcription and translation.
Fig. 7.

Stretch rapidly induces lung elastase activity. A: to quantify the direct influence of stretch on inducing lung elastin remodeling, we developed an inverted confocal microscope-compatible stretching device that stretches a silastic membrane at 32-μm increments. A schematic of the device is shown. B: subpleural left lung sections (200 μm) were glued to the membrane using fibrin glue and a silastic well was placed around the lung. The membrane was then attached to the underside of the stretching device, substrate solution was applied, and elastin in situ zymography was performed with serial stretches every minute. An image of the underside of the stretching device with secured membrane and glued lung is shown. C: at zero stretch, very little elastase activity (green) was apparent. Autofluorescence in the blue channel was used as a loading control for analysis. Large conducting structures were excluded from analysis (magenta). To measure strain, the distance between two of these major structures (white line) was measured. D: increasing degrees of stretch increased elastase signal and strain. E: when plotted against strain, lung elastase activity increased in an exponential manner (black circles). Control sections stretched with PBS alone (white circles) did not demonstrate increased elastase signal.
To test whether the application of strain to ex vivo lung sections increased lung elastase activity in a pattern similar to that observed postpneumonectomy, we manually stretched sections in the presence of elastase substrate by imaging the entire section at ×4 magnification with confocal microscopy. Prior to stretching, there was little elastase activity (Fig. 8A), but afterward, there was increased activity (Fig. 8B). To quantify this, we applied 11 cylindrical masks to the unstretched image and corresponding masks to the stretched image (Fig. 8C). After measuring the strain for each region, this strain was plotted against change in normalized elastase activity. Elastase activity increased exponentially with strain (Fig. 8D).
Fig. 8.

Stretch increases the lung elastase activity of subpleural regions more than central regions. A: the apical and basal regions of 200-μm mouse left lung sections were glued to coverslips and the central region imaged by confocal microscopy. B: stretching the lung section increased lung elastase activity. C: cylindrical masks were applied to 11 regions of the unstretched image (not shown) and then corresponding masks were applied to the stretched image (teal). Strain and the change in normalized elastase activity were measured for each region. D: an exponential relationship between strain and change in elastase activity was observed.
DISCUSSION
In this study, we demonstrated that lung elastase activity is maximal during the alveolar stage of lung development and that elastase activity is reinduced during compensatory lung growth. Increased lung elastase activity was restricted to alveolar walls and ducts and largely absent from the elastin bands of alveolar septal tips and alveolar duct openings. The lung elastase activity of larger structures and lung parenchyma increased proportionately during normal lung development, but elastin remodeling of major arteries and airways did not increase during compensatory lung growth. The increase in elastase activity during compensatory lung growth was restricted to subpleural regions, which contrasts with more uniform elastase activity during normal development. Preventing postpneumonectomy lung expansion with wax plombage prevented the induction of lung elastase activity. Finally, in ex vivo lung stretch experiments, we demonstrated the endogenous remodeling capacity of the adult lung and demonstrated a direct relationship between strain and the induction of elastase activity.
Our study is consistent with findings that elastin is remodeled in the developing lung, but not in the mature adult lung. In 1991, using the carbon-14 content of lung specimens obtained at autopsy, Shapiro et al. estimated a tropoelastin carbon residence time of 74 yr in adult humans (17). This is not the case in younger humans. In 1992, Bruce et al. quantified urinary desmosine levels in ventilated premature infants, and found that infants with higher desmosine levels after the first postnatal week were less likely to develop bronchopulmonary dysplasia (2). Although lung elastin content increases with gestational age, infants with bronchopulmonary dysplasia have significantly more total lung elastin (21) and the architecture of these fibers is distorted (2). These studies indicate that the quantity, localization, and structure of lung elastin fibers are important for proper septation. Our data demonstrate that stretch is an important regulator of lung elastin remodeling during septation. On the other end of the spectrum, chronic obstructive pulmonary disease is associated with an increase in lung protease activity and elastolysis (16). Although neutrophil elastase is a key player in this disease, the stretch dependent-binding of other elastases to lung elastin fibers (8) and our findings of an innate, stretch-activated remodeling response suggest that stretch itself may play a role in potentiating distal airspace destruction.
We acknowledge two limitations of our study. First, although our assay allows for localization and normalization of lung elastase activity and is thus an improvement on homogenate-based assays, our assay does not directly measure elastin remodeling. This being said, our assay results do correlate with urinary desmosine measurements in postpneumonectomy mice, which is a direct measure of elastin fiber degradation. Second, our method for defining elastin band structures (septal tips and opening of alveolar ducts) vs. nonband elastin structures (acinar and alveolar duct walls) likely misclassifies some structures, but any bias would tend toward classifying elastin band structures as nonband structures. With regards to the elastase activity in these structures, although differences in matrix composition could alter the accessibility of the soluble elastin substrate to local proteases, the activity in band structures did not increase despite a tripling of band density. The bias toward misclassifying band structures as nonband structures and lack of increased elastase activity in elastin bands despite increased density further supports our conclusion that the increases in lung elastase activity during alveolar septation is largely due to increased activity in acinar and alveolar duct walls.
We have demonstrated similar patterns of elastase activity during lung development and compensatory lung growth. In 2012, Kho et al. demonstrated that during postpneumonectomy lung growth, the regenerating lung undergoes limited dedifferentiation and then adopts a gene expression profile reminiscent of the alveolar stage of lung development (9). The observed increase and decrease in lung elastase activity during alveolarization and regeneration is consistent with those findings. However, whereas during primary lung septation remodeling occurred in all structures, during compensatory lung growth elastase activity was not increased in major arteries and airways. Furthermore, whereas in lung development remodeling is homogenously distributed throughout the lung lobe, in compensatory lung growth, remodeling is largely restricted to the subpleural region—the region that experiences the most deformation postpneumonectomy (6). As the regenerative medicine research community develops strategies to develop lung tissue on extracellular matrix scaffolds, we need to remain cognizant of the physiological and pathological roles that stretch plays in the different lung structures and regions.
Although it may seem paradoxical, our finding that lung strain increases lung elastase activity within minutes of application is consistent with a sustained, stretch-regulated elastin remodeling response over the course of compensatory lung growth. After left pneumonectomy in adult mice, right lung expansion occurs through at least the first 21 days (22). We propose that during postnatal lung development and during compensatory lung growth, the strain applied to the walls of acinar units and alveolar ducts defines the level of elastase activity in those structures and that expansion relieves strain, which subsequently attenuates the remodeling response. When considered in the context of tropoelastin and collagen type Iα mRNA increasing in the ends, bends, and junctions of alveolar walls during compensatory lung growth (10), we propose that at the acinar level, an as yet undefined mechanotransductive signal may initiate the formation of a new septal band, and that this new band and stretch-regulated elastase activity act to minimize and equally distribute wall strain in enlarging distal airspaces. Because the subpleural regions of the accessory lobe experience the most postpneumonectomy deformation (6), the sum of these acinar changes explains why elastase activity is restricted to subpleural regions during compensatory lung growth but not during development.
Our finding that during compensatory lung growth elastase activity is induced in the subpleural parenchyma but not arteries or major airways raises a fundamental biological question: is the distal lung morphogenic program in continuity with branching morphogenesis or are the programs discrete? Previous gene expression studies demonstrated that the sacculation/alveolarization program antagonizes the lung epithelial cell branching morphogenesis program (4). In further support of distinct alveolarization and branching morphogenesis programs, the Xenopus lung undergoes sacculation and septation without any discernible branching despite the preservation of lung specification (15). Furthermore, four evolutionarily divergent classes of fish (bichir and ropefish, gar, ray-finned fish, and lungfish) have developed varying degrees of swim bladder septation to augment gas exchange capacity in low oxygen environments (13, 25). Together, these findings suggest that whereas the more evolutionarily conserved distal lung morphogenic program may be relatively easy to reinitiate via stretch, the more highly evolved branching morphogenic program is not as easily manipulated and may represent a more difficult hurdle to overcome in utilizing compensatory lung growth as a therapeutic strategy.
In summary, we have demonstrated the stretch-dependency of lung elastase activity and put forth a model of stretch-regulated distal alveolar morphogenesis that explains the localization of remodeling at both lobar and acinar levels. Understanding the regulatory molecules and downstream signaling mechanisms mediating the stretch-regulated remodeling response will be necessary to leverage our findings into translational therapies.
GRANTS
Support for this study was provided by the Proctor Scholar Program at Cincinnati Children's Hospital Research Foundation and the Parker B. Francis Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.M.V. conception and design of research; S.M.Y., S.L., R.J., M.R.B., J.M.K., J.G., and J.C.W. performed experiments; S.M.Y., S.L., R.J., J.M.K., J.C.W., and B.M.V. analyzed data; S.M.Y., S.L., R.J., J.M.K., and B.M.V. interpreted results of experiments; B.M.V. prepared figures; S.M.Y. and B.M.V. drafted manuscript; S.M.Y., M.R.B., J.C.W., and B.M.V. edited and revised manuscript; S.M.Y., S.L., R.J., M.R.B., J.M.K., J.C.W., and B.M.V. approved final version of manuscript.
REFERENCES
- 1.Ad hoc Statement Committee, American Thoracic Society. Mechanisms and limits of induced postnatal lung growth. Am J Respir Crit Care Med : 319–343, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Bruce MC, Schuyler M, Martin RJ, Starcher BC, Tomashefski JF Jr, Wedig KE. Risk factors for the degradation of lung elastic fibers in the ventilated neonate. Implications for impaired lung development in bronchopulmonary dysplasia. Am Rev Respir Dis : 204–212, 1992. [DOI] [PubMed] [Google Scholar]
- 3.Butler JP, Loring SH, Patz S, Tsuda A, Yablonskiy DA, Mentzer SJ. Evidence for adult lung growth in humans. N Engl J Med : 244–247, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang DR, Martinez Alanis D, Miller RK, Ji H, Akiyama H, McCrea PD, Chen J. Lung epithelial branching program antagonizes alveolar differentiation. Proc Natl Acad Sci USA : 18042–18051, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dane DM, Yilmaz C, Estrera AS, Hsia CC. Separating in vivo mechanical stimuli for postpneumonectomy compensation: physiological assessment. J Appl Physiol : 99–106, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibney BC, Houdek JP, Chamoto K, Lee GS, Ackermann M, Lin M, Collings-Simpson D, Konerding MA, Tsuda A, Mentzer SJ. Mechanostructural adaptations preceding postpneumonectomy lung growth. Exp Lung Res : 396–405, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffman AM, Shifren A, Mazan MR, Gruntman AM, Lascola KM, Nolen-Walston RD, Kim CF, Tsai L, Pierce RA, Mecham RP, Ingenito EP. Matrix modulation of compensatory lung regrowth and progenitor cell proliferation in mice. Am J Physiol Lung Cell Mol Physiol : L158–L168, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jesudason R, Sato S, Parameswaran H, Araujo AD, Majumdar A, Allen PG, Bartolak-Suki E, Suki B. Mechanical forces regulate elastase activity and binding site availability in lung elastin. Biophys J : 3076–3083, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kho AT, Liu K, Visner G, Martin T, Boudreault F. Identification of dedifferentiation and redevelopment phases during postpneumonectomy lung growth. Am J Physiol Lung Cell Mol Physiol : L542–L554, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koh DW, Roby JD, Starcher B, Senior RM, Pierce RA. Postpneumonectomy lung growth: a model of reinitiation of tropoelastin and type I collagen production in a normal pattern in adult rat lung. Am J Respir Cell Mol Biol : 611–623, 1996. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Cimprich J, Varisco BM. Mouse pneumonectomy model of compensatory lung growth. J Vis Exp. First published December 17, 2014; doi: 10.3791/52294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margraf LR, Tomashefski JF Jr, Bruce MC, Dahms BB. Morphometric analysis of the lung in bronchopulmonary dysplasia. Am Rev Respir Dis : 391–400, 1991. [DOI] [PubMed] [Google Scholar]
- 13.Perry SF, Wilson RJ, Straus C, Harris MB, Remmers JE. Which came first, the lung or the breath? Comp Biochem Physiol A Mol Integr Physiol : 37–47, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Pieretti AC, Ahmed AM, Roberts JD, Kelleher CM. A novel in vitro model to study alveologenesis. Am J Respir Cell Mol Biol : 459–469, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rankin SA, Thi Tran H, Wlizla M, Mancini P, Shifley ET, Bloor SD, Han L, Vleminckx K, Wert SE, Zorn AM. A molecular atlas of Xenopus respiratory system development. Dev Dyn : 69–85, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandhaus RA, Turino G. Neutrophil elastase-mediated lung disease. COPD , Suppl 1: 60–63, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest : 1828–1834, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srisuma S, Bhattacharya S, Simon DM, Solleti SK, Tyagi S, Starcher B, Mariani TJ. Fibroblast growth factor receptors control epithelial-mesenchymal interactions necessary for alveolar elastogenesis. Am J Respir Crit Care Med : 838–850, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suki B, Sato S, Parameswaran H, Szabari MV, Takahashi A, Bartolak-Suki E. Emphysema and mechanical stress-induced lung remodeling. Physiology (Bethesda) : 404–413, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda SI, Ramanathan M, Estrera AS, Hsia CC. Postpneumonectomy alveolar growth does not normalize hemodynamic and mechanical function. J Appl Physiol : 491–497, 1999. [DOI] [PubMed] [Google Scholar]
- 21.Thibeault DW, Mabry SM, Ekekezie II, Truog WE. Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics : 1452–1459, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Wang W, Nguyen NM, Guo J, Woods JC. Longitudinal, noninvasive monitoring of compensatory lung growth in mice after pneumonectomy via (3)He and (1)H magnetic resonance imaging. Am J Respir Cell Mol Biol : 697–703, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West JB. Respiratory Physiology: The Essentials. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2012. [Google Scholar]
- 24.Ysasi AB, Belle JM, Gibney BC, Fedulov AV, Wagner W, AkiraTsuda, Konerding MA, Mentzer SJ. Effect of unilateral diaphragmatic paralysis on postpneumonectomy lung growth. Am J Physiol Lung Cell Mol Physiol : L439–L445, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng W, Wang Z, Collins JE, Andrews RM, Stemple D, Gong Z. Comparative transcriptome analyses indicate molecular homology of zebrafish swimbladder and mammalian lung. PLoS One : e24019, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]