

Abstract
The chemotherapeutic effect of doxorubicin (Dox) is limited by cumulative dose-dependent cardiotoxicity in cancer survivors. Dexrazoxane (DRZ) is approved to prevent Dox-induced cardiotoxicity. Humanin and its synthetic analog HNG have a cytoprotective effect on the heart. To investigate the cardioprotective efficacy of HNG alone or in combination with DRZ against Dox-induced cardiotoxicity, 80 adult male mice were randomly divided into 8 groups to receive the following treatments via intraperitoneal injection: saline daily, HNG (5 mg/kg) daily, DRZ (60 mg/kg) weekly, Dox (3 mg/kg) weekly, DRZ + HNG, Dox + HNG, Dox + DRZ, and Dox + HNG + DRZ. Echocardiograms were performed before and at 4, 8, and 9.5 wk after the beginning of treatment. All mice were euthanized at 10 wk. In the absence of Dox, HNG, DRZ, or DRZ + HNG had no adverse effect on the heart. Dox treatment caused decreases in ejection fraction and cardiac mass and increases in cardiomyocyte apoptosis and intracardiac fibrosis. HNG or DRZ alone blunted the Dox-induced decrease in left ventricle posterior wall thickness and modestly ameliorated the Dox-induced decrease in ejection fraction. HNG + DRZ significantly ameliorated Dox-induced decreases in ejection function, cardiac fibrosis, and cardiac mass. Using a targeted analysis for the mitochondrial gene array and protein expression in heart tissues, we demonstrated that HNG + DRZ reversed DOX-induced altered transcripts that were biomarkers of cardiac damage and uncoupling protein-2. We conclude that HNG enhances the cardiac protective effect of DRZ against Dox-induced cardiotoxicity. HNG + DRZ protects mitochondria from Dox-induced cardiac damage and blunts the onset of cardiac dysfunction. Thus, HNG may be an adjuvant to DRZ in preventing Dox-induced cardiotoxicity.
NEW & NOTEWORTHY Doxorubicin (Dox) is commonly used for treating a wide range of human cancers. However, cumulative dosage-dependent carditoxicity often limits its clinical applications. We demonstrated in this study that treating young adult male mice with synthetic humanin analog enhanced the cardiac protective effect of dexrazoxane against chemotherapeutic agent Dox-induced cardiac dysfunction. Thus, humanin analog can potentially serve as an adjuvant to dexrazoxane in more effectively preventing Dox-induced cardiac dysfunction and cardiomyopathy.
Keywords: cardiotoxicity, dexrazoxane, doxorubicin, humanin
INTRODUCTION
Doxorubicin (Dox), a member of the anthracycline family of anticancer agents, is commonly used for treating a wide range of human cancers, including advanced breast cancer and hematological malignancies. However, a cumulative dosage-dependent cardiotoxicity and late onset of congestive heart failure often limit its clinical applications (27, 29, 46). Dox treatment results in subclinical, progressive, irreversible cardiotoxicity causing significant morbidity and mortality in cancer survivors (22). Although Dox has been used clinically for over 40 yr, the underlying molecular mechanisms of its adverse effects on the induction of cardiomyopathy are only partially understood. Dox induces myocardial damage and dysfunction by stimulating reactive oxygen species (ROS) production by affecting redox homeostasis. In addition, free cellular iron-forming toxic Dox-iron complexes generate ROS, resulting in mitochondrial dysfunction (2, 41, 42). Dox binds both DNA and topoisomerase-2 (Top2) isoenzymes to form ternary Top2-Dox-DNA cleavage complexes, triggering DNA breaks. When Dox binds to Top2α, it inhibits DNA replication and results in cell cycle arrest and apoptosis of cancer cells. However, when Dox binds to Top2β in cardiomyocytes, it impairs Ca2+ handling and causes mitochondrial dysfunction and subsequent cardiomyocyte apoptosis (39, 49). These data suggest that mitochondria may be a primary target of Dox-induced cardiotoxicity (27).
Modified Dox analogs and liposomal Dox have been shown to lower cardiotoxicity rates. Other cardioprotective agents, including antioxidant N-acetylcystiene, β-blockers, angiotensin-converting enzyme inhibitors, and statins, have been tested in clinical trials with some efficacy for recovery of cardiac function but are not used prophylactically (7, 27). Dexrazoxane (DRZ) is a derivative of EDTA, functioning as a potent intracellular iron-chelating agent. The cardioprotective effect of DRZ is partially mediated through its iron-chelating activity against Dox-induced ROS generation in cardiomyocytes (11). DRZ also exerts its cardioprotective effect by depleting cardiac Top2β protein through proteasome-dependent degradation in cardiomyocytes (8, 26, 44). However, DRZ also depletes Top2α and may interfere with the cancer response to Dox (8). Clinical trials have shown that DRZ administered with Dox in women with advanced breast cancer and in children with acute leukemia reduced the rates of clinical and subclinical cardiotoxicity (1, 30). Although an earlier study showed a systemic treatment of hematological diseases with Dox, and high doses of DRZ might be associated with second malignancy, including acute myeloid leukemia/myelodysplasia (33), subsequent larger studies with a followup of 12 yr showed that DRZ did not compromise long-term survival and was not associated with death due to acute myeloid leukemia/myelodysplasia (6).
Our group has focused on a mitochondria-derived peptide, humanin, and its cytoprotective effect on chemotherapy-induced damage to normal tissues. Humanin is a 24-amino acid peptide encoded by an open reading frame from the mitochondrial 16S rRNA region (47), and it can be detected in the circulation in men (5, 14, 28). Humanin has cytoprotective, anti-inflammatory, antioxidative, and antiapoptotic properties (24, 20, 40). Extracellular humanin exerts its cytoprotective function through receptors composed of ciliary neurotrophic factor receptor-α (CNTFRα), IL-27 receptor (WSX-1), and glycoprotein (gp)130. Upon receptor binding, humanin activates a STAT3 or ERK1/2 intracellular signaling cascade to promote cell survival. Humanin also binds to IGF-binding protein-3 (IGFBP-3) and modulates IGF-I bioactivity to regulate cell growth, survival, and apoptosis. Intracellularly, humanin binds to Bax and other BH3 domain proteins, preventing their entry to mitochondria and inhibiting initiation of apoptosis induced by stress (18, 21, 45). Recent studies have shown that humanin protects the heart against cardiac ischemia/reperfusion-induced cardiomyopathy in rodents by decreasing mitochondrial ROS levels and reducing mitochondrial dysfunction (36, 37). We (19, 32) have previously demonstrated that humanin protects rodent male germ cells from apoptosis induced by intratesticular hormonal deprivation, increased temperature, and treatment with the chemotherapeutic agent cyclophosphamide. In addition, we demonstrated that administration of a potent humanin analog, S14G-humanin (HNG), not only protected germ cells and leucocytes against cyclophosphamide-induced damage but also enhanced cyclophosphamide-induced suppression of melanoma pulmonary metastases in male mice (25).
The objectives of this study were to investigate whether HNG alone or in combination with DRZ could 1) prevent Dox-induced cardiac dysfunction and 2) ameliorate Dox-induced mitochondrial damage in the heart.
MATERIALS AND METHODS
Animals and reagents.
Young adult (10 wk old, body weight: 24–28 g) male mice (C57BL/6J) purchased from the Jackson Laboratory (Bar Harbor, ME) were housed in a standard animal facility under controlled temperature (22°C) and photoperiod (12:12-h light-dark cycle) with free access to water and mouse chow. Animal handling and experimentation were in accordance with recommendations of the American Veterinary Medical Association and approved by the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center Animal Care and Use Review Committee (IACUC) and UCLA IACUC. Dox and DRZ were obtained from Tocris Bioscience (Bristol, UK) and dissolved in saline at a concentration of 1 mg/ml for Dox and 9 mg/ml for DRZ before use. Humanin analog HNG was synthesized by CPC Scientific (Sunnyvale, CA) and dissolved in saline at a concentration of 1 mg/ml just before use.
Study design.
A total of 80 adult male mice were randomly divided into the following 8 groups (10 mice/group) to receive the following treatments via intraperitoneal injection: daily saline injection as control (group 1), 5 mg/kg HNG daily (group 2), 3 mg/kg Dox weekly (group 3), 60 mg/kg DRZ weekly (group 4), DRZ + HNG (group 5), Dox + HNG (group 6), Dox + DRZ (group 7), and Dox + HNG + DRZ treatment (group 8). The selected doses of HNG (5 mg·kg−1·day−1), Dox (3 mg·kg−1·wk−1), and DRZ (60 mg·kg−1·wk−1) were based on previous mouse models to study HNG cytoprotection (25) and Dox cardiotoxicity (9, 16). Mice received eight doses of either Dox or DRZ or Dox + DRZ via weekly injection for 7 wk and a daily HNG injection for 10 wk. To monitor cardiac function, we performed an echocardiogram at baseline and repeated it at 4, 8, and 9.5 wk after the first injection of Dox/DRZ/HNG alone or in combinations. Dose adjustment of all drugs was based on weekly body weight of each mouse. At the end of 10 wk, mice were euthanized with an ip injection of pentobarbital sodium (200 mg/kg body weight). Blood samples were collected from the left ventricle (LV), and hearts were harvested, weighed, and divided into three portions by transverse sections through both the right ventricle (RV) and LV. One portion was immersed in 10% buffered formalin (Fisher Scientific), processed for histological examination, and assessment of cardiomyocyte apoptosis and fibrosis; another portion was snap frozen in liquid nitrogen for RNA isolation, quantitative RT-PCR array, and quantitative RT-PCR; and the remaining portion was frozen for protein isolation and analyses.
Echocardiography.
Echocardiography was performed as previously described (12, 43). Animals were under 1.5% isoflurane anesthesia. Echocardiography was performed using a Vevo2100 ultrasound system (VisualSonics, Toronto, ON, Canada). A parasternal long-axis B-mode image was obtained by positioning the probe parallel to the long axis of the LV with the ultrasound beam running perpendicular to the LV. The probe was rotated at 90° to obtain a parasternal short-axis view of the LV. The papillary muscle was used as a standard to ensure reproducible and similar images among animals. Using this short-axis view, an M-mode short video clip was stored to document LV dimensions for further analysis. Echocardiographic parameters for ejection fraction images were analyzed using the Vevo2100 cardiac analysis package.
Quantification of cardiomyocyte apoptosis by colocalization of tropomyosin and DNA fragmentation.
Formalin-fixed and paraffin-embedded transverse ventricular heart sections from each mouse were used to colocalize expression of tropomyosin, a myocardium marker, and TUNEL for the detection of apoptosis (13, 31). Briefly, after deparaffinization and rehydration, heart sections received antigen retrieval in citrate buffer (pH 6.0) with boiling for 30 min. Sections were then subjected to TUNEL procedures using an Apop Tag-peroxidase kit (Millipore). After TUNEL labeling, sections were treated with blocking serum (10% BSA) and subsequently incubated with a 1:200 dilution of a monoclonal anti-tropomyosin antibody (RRID:AB_261817, catalog no. T9283, Sigma-Aldrich) or 0.05% BSA-PBS (negative control) at 4°C overnight. Sections were then incubated with donkey anti-mouse Alexa fluor 594 (red)-labeled secondary antibody (Invitrogen, Walthan, MA) for 1 h and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Apoptosis was quantified in 6 mice/group and expressed as the apoptotic index (AI), which was the number of TUNEL-positive apoptotic cardiomyocytes per the number of cardiomyocyte nuclei in LV sections. Under a fluorescent microscope, 10 areas were randomly selected under the magnification of ×200, and a total of 3,000 cardiomyocyte nuclei were counted per mouse heart.
RNA extraction from cardiac tissue, reverse transcription, and real-time PCR.
Total RNA was extracted from LV tissue using TRIzol Reagent (Life Technologies). One microgram of RNA was reverse transcribed into first-strand cDNA using ProtoScript II Reverse Transcriptase (New England BioLabs) and Random Primer (Life Technologies). Real-time PCR was performed using the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA) using the iQ SYBR Green Supermix (Bio-Rad). Values were normalized to β-actin to calculate relative expression levels. The primers used for quantitative RT-PCR were as follows: 1) atrial natriuretic factor (ANF), mANF RT-forward (F) 5′-AGGCAGTCGATTCTGCTTGA-3′ and mANF RT-reverse (R) 5′-CGTGATAGATGAAGGCAGGAAG-3′ and 2) β-actin, mActB RT-F 5′-TGGCACCACACCTTCTACAA-3′ and mActB RT-R 5′-GTCTCCGGAGTCCATCACAA-3′.
Trichrome staining and quantification of fibrosis in hearts.
Formalin-fixed and paraffin-embedded transverse ventricular heart sections from 6 mice/group were stained with Masson’s trichrome procedures. Images of the trichrome-stained sections of LVs were made at ×200 magnification using a Nikon Eclipse TE 2000-U microscope. Images were analyzed for percent fibrosis per heart using the Nikon Image System Elements AR program. Fibrosis area was designated by blue staining, and the total tissue images were designated by both red and blue (17, 31, 48, 50). Fibrosis was quantified as the percentage of blue area over total area assessed.
Quantitative RT-PCR array and data analysis.
Nuclear gene-encoded mRNA for mitochondrial proteins of four mouse hearts from the control, Dox alone-, and Dox + HNG + DRZ-treated groups was assessed by mitochondrial gene-focused RT-PCR array (PAMM-087ZC RT2 ProfilerTM PCR array, Qiagen, Hilden, Germany). Total RNA was extracted from cardiac LV tissue using an RNeasy Fibrous Tissue Mini Kit (Qiagen). RNA concentration was measured by NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA). Isolated RNA from each heart was reverse transcribed into cDNA using the RT2 First Strand Kit (Qiagen). cDNA was then mixed with RT2 SYBR Green ROX qPCR Mastermix, placed into a mouse mitochondrial gene array, and subjected to run the real-time PCR cycling program (ABI one-step plus real-time PCR system) to detect the 84 nuclear gene-encoded mitochondrial gene expression. Gene expression data generated from arrays were analyzed by Qiagen software and compared among the control and treated groups.
Protein extraction and protein-simple assay.
LV samples from mice were sonicated in RIPA buffer (Sigma) supplemented with EDTA-free protease inhibitor cocktail tablets (Roche), and cell debris was removed by centrifugation. The protein concentration was analyzed by Bio-Rad Protein Assay and measured by a Beckman Du-640 spectrophotometer.
Protein measurement for uncoupling protein-2 (Ucp2) was performed using ventricle tissue lysates from control, HNG alone-, DRZ alone-, Dox alone-, Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mice. Protein expression was quantified by Wes Separation Module with 25 capillary cartridges for 12–230 kDa (Wes). Proteins (2 µg/sample) were separated through a size-resolving matrix in capillaries, immobilized to the inner capillary wall, incubated with anti-Ucp2 rabbit monoclonal antibody (RRID:AB_2721818, catalog no. 89326, Cell Signaling Technology) at a concentration of 1 μg/ml or antibody dilution buffer (negative control) and anti-rabbit secondary antibodies before detection using chemiluminescence. Signals reflected as the area under the curve of Ucp2 protein were generated automatically at the end of the run. Ucp2 protein levels were quantified as the ratio of the area under the curve from each sample that was normalized by the area under the curve of the corresponding total protein.
Statistical analysis.
We focused a priori on the comparison of effects of HNG + DRZ + Dox- vs. Dox- and HNG + Dox- or DRZ + Dox- vs. Dox-treated mice. Statistical analyses were carried out using the SigmaStat 12.0 Program (Systat Software). Results were tested for statistical significance using one-way ANOVA with Tukey’s post hoc test or Student’s t-test. For fibrosis quantification, the P value was calculated using Duncan’s multiple comparison tests. Differences were considered significant if P < 0.05.
RESULTS
Outcome and general health of treated mice.
Seventy-one of the 80 mice survived until the end of this study. Nine mice died before the termination of the experiment: one mouse from the control group died at week 8 of unknown cause, two mice from the Dox + DRZ group died at weeks 2 and 10, respectively, three mice from the Dox-treated group died at week 9, and three mice from the Dox + HNG group died at week 10. The cause of death in Dox-treated mice was most likely related to the general toxicity of Dox. At the end of 10 wk in the surviving mice, HNG, DRZ, or HNG + DRZ had no significant adverse effect on body weight or heart weight (Fig. 1). Dox treatment significantly decreased body weight compared with the control mice. Whereas HNG or DRZ in Dox-treated mice attenuated the Dox-induced body weight loss, combined treatment (Dox + HNG + DRZ) restored body weight to near control level (Fig. 1A). Dox alone significantly decreased heart weight by 14% compared with control mice; in contrast, there were no significant changes in heart weight in Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mice compared with control mice (Fig. 1B).
Fig. 1.

Body weight (A) and heart weight (B) from control, synthetic humanin analog (HNG) alone-, dexrazoxane (DRZ) alone-, DRZ + HNG-, doxorubicin (Dox) alone-, Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mice.
Dox-induced cardiac dysfunction.
HNG-, DRZ-, and DRZ + HNG-treated mice did not show any altered cardiac parameters measured compared with control mice across the treatment period. Dox treatment alone significantly decreased heart rate (Fig. 2A), LV posterior wall thickness (Fig. 2B), fractional shortening (Fig. 2C), and ejection fraction (Fig. 2D) at 9.5 wk after treatment. Dox + HNG treatment significantly attenuated the loss of LV posterior wall thickening but did not affect the other parameters (heart rate, fractional shortening, and ejection fraction) compared with Dox treatment alone. Dox + DRZ treatment significantly ameliorated the Dox-induced decreases in LV posterior wall thickening and ejection fraction. Most importantly, combined treatment of Dox + HNG + DRZ restored heart rate, LV posterior wall thickening, fractional shortening, and ejection fraction to control levels. While DRZ, HNG, or DRZ + HNG treatment did not significantly alter ANF transcripts from control, expression levels of ANF mRNA, a heart failure biomarker, were significantly increased in Dox alone-treated hearts; the addition of HNG, DRZ, or combined HNG + DRZ to Dox-treated mice returned expression of ANF to control levels (Fig. 3).
Fig. 2.

Representative M-mode echocardiography photo images of mouse hearts from different treatment groups. A−D: heart rate (A), left ventricle (LV) posterior wall thickening (B), fractional shortening (C), and ejection fraction (D) in control, synthetic humanin analog (HNG) alone-, dexrazoxane (DRZ) alone-, DRZ + HNG-, doxorubicin (Dox) alone, Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mice.
Fig. 3.
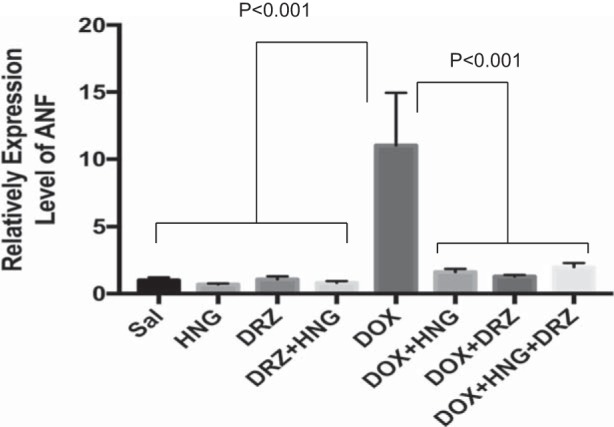
Expression level of atrial natriuretic factor (ANF) transcripts in control, synthetic humanin analog (HNG) alone-, dexrazoxane (DRZ) alone-, DRZ + HNG-, doxorubicin (Dox) alone, Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mice.
DOX-induced cardiomyocyte apoptosis.
Compared with control, Dox treatment alone significantly increased cardiomyocyte apoptosis. Whereas there was no markedly protective effect of HNG or DRZ treatment on Dox-induced apopotosis at 10 wk after treatment, combined treatment of DRZ + HNG significantly reduced Dox-induced cardiomyocyte apoptosis to control levels (Fig. 4).
Fig. 4.
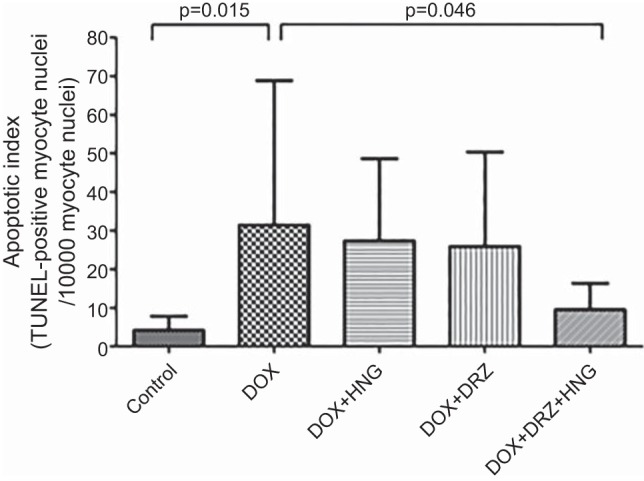
Apoptotic index (TUNEL-positive myocyte nuclei/1,000 myocyte nuclei) in control, synthetic humanin analog (HNG) alone-, dexrazoxane (DRZ) alone-, DRZ + HNG-, doxorubicin (Dox) alone, Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mice.
Dox-induced cardiac fibrosis.
Although there were no changes of fibrosis in hearts treated with HNG, DRZ, or HNG + DRZ, Dox alone treatment significantly induced cardiac fibrosis compared with controls. Although HNG or DRZ treatment did not reduce Dox-induced cardiac fibrosis in Dox-treated mice, the combined treatment of HNG and DRZ significantly decreased Dox-induced cardiac fibrosis to near control levels (Fig. 5).
Fig. 5.

Representative Masson trichrome staining for fibrosis in control, synthetic humanin analog (HNG) alone-, dexrazoxane (DRZ) alone-, DRZ + HNG-, doxorubicin (Dox) alone, Dox + HNG-, Dox + DRZ-, and Dox + HNG + DRZ-treated mouse hearts. Cardiac fibrosis is shown as blue. HNG + DRZ significantly attenuated Dox-induced cardiac fibrosis (A).
Cardiac mitochondria-related gene expression.
Since the echocardiogram demonstrated that Dox damaged cardiac function, and combined HNG+DRZ significantly ameliorated Dox-induced cardiac dysfunction, we used the RT-PCR array as a screening tool to explore the involvement of the mitochondria in the induction of Dox-induced cardiac dysfunction to seek mechanisms by which the combined HNG and DRZ treatment was effective in preventing Dox-induced cardiomyopathy. We compared the effect of Dox or Dox + HNG + DRZ on nuclear gene-encoded mitochondria gene expression to saline-treated control mice. Among the 84 genes examined, Dox induced upregulation of 78 genes and downregulation of 5 genes compared with control (threshold of 1.1-fold; Fig. 6). Using a threshold of 2-fold, we found Dox-induced upgulation of 12 genes, including Ucp2 and Ucp3, compared with control (table in Fig. 6). Compared with Dox alone treatment, combined treatment (Dox + HNG + DRZ) resulted in downregulation of 40 genes in the direction of the untreated controls, including Ucp2 and Ucp3 (Fig. 6). The data from quantitative RT-PCR showed that Dox treatment increased Ucp2 and Ucp3 mRNA expression, and HNG, DRZ, or combined treatment reversed Dox-induced Ucp2 and Ucp3 mRNA expression to control levels. We further assessed mitochondrial Ucp2 protein levels with a protein immunoassay. Inconsistent with mRNA expression, we found that Dox alone significantly decreased Ucp2 protein expression compared with control. Combined HNG + DRZ significantly restored Dox-induced Ucp2 expression to control levels (Fig. 7A). There were no significant differences in Ucp2 protein levels among control, HNG-alone, and DRZ-alone groups (Fig. 7B).
Fig. 6.

Heat map indicating coregulated genes across groups of control, doxorubicin (Dox) alone-, and Dox + synthetic humanin analog (HNG) + dexrazoxane (DRZ)-treated mice. Red or near-red color indicates upregulation; green or near-green color indicates downregulation. In the table, positive number indicates upregulation and negative number indicates downregulation.
Fig. 7.

A and B: uncoupling protein 2 (Ucp2) protein levels assessed by protein-simple assay in control, doxorubicin (Dox)-, Dox + synthetic humanin analog (HNG)-, Dox + dexrazoxane (DRZ)-, and Dox + HNG + DRZ-treated mice (A) and in control, HNG alone-, and DRZ alone-treated mice (B). HNG + DRZ significantly normalized Ucp2 protein suppressed by Dox to control levels. Values in the bar graph represent means ± SE of the area under the curve of Ucp2 signals normalized by that of the corresponding total proteins.
DISCUSSION
Dox causes a dose-dependent, cumulative, adverse effect on cardiac function in patients, limiting its effectiveness in cancer chemotherapy (22, 27, 29, 46, 29). Using a mouse model, we demonstrated that Dox-induced cardiac dysfunction (decreases in heart rate, LV posterior wall thickening, fractional shortening, and ejection fraction) did not occur until 8 wk after Dox treatment with an accumulative dose of 24 mg/kg body wt in mice. Dox treatment increased cardiomyocyte apoptosis and intracardiac fibrosis. These cumulative effects of Dox-induced cardiac dysfunction in mice mimicked the cardiac dysfunction in cancer patients treated with Dox. Using this Dox-induced cardiotoxicity mouse model, we investigated whether HNG, a synthetic humanin analog, alone or in combination with DRZ, a drug used clinically to prevent and treat Dox-induced cardiotoxicity, would prevent or ameliorate Dox-induced cardiac dysfunction and cardiomyopathy in mice. We found a small to moderate cytoprotective effect of HNG treatment alone on Dox-induced cardiac dysfunction. As expected, DRZ moderately improved heart function impaired by Dox treatment. Of note, we demonstrated in this study that combined HNG with DRZ treatment resulted in greater protection against Dox-induced cardiac dysfunction than either HNG or DRZ treatment alone. The protective effect of HNG + DRZ against Dox-induced cardiotoxicity was associated with maintenance of body weight to levels similar to those of control mice, and all 10 mice were healthy and alive at the end of study. Thus, HNG could potentially be developed as an addition to DRZ to prevent Dox-induced cardiac dysfunction and general toxicity. While previous studies have suggested that DRZ may be clinically useful for prevention of anthracycline-induced cardiotoxicity, the possibility of a reduced antineoplastic response (33) when DRZ is used has led the Food and Drug Administration in the United States to limit DRZ to a very narrow indication for its use and led to a generalized aversion to its use in the clinical oncology community. The data from this study provided evidence that it was possible that using HNG as an adjuvant agent might allow for lower doses of DRZ, which might or might not confer a reduced antineoplastic response. This notion was further supported by our previous data demonstrating that HNG alone had a moderate anticancer effect on metastatic pulmonary melanoma in mice (25).
To explore the underlying mechanisms of cardioprotection rendered by HNG + DRZ on Dox-induced cardiac dysfunction, we showed that the combined treatment of HNG+DRZ significantly decreased Dox-induced cardiomyocyte apoptosis and intracardiac fibrosis. Studies have shown that the cardiac protective effects of DRZ could be attributed to its iron-chelating, anti-ROS, and anti-Top2β actions (8, 26). Prior studies have also shown that HNG protected mitochondrial function by reducing stress-induced ROS production and prevented cardiomyocytes from apoptosis (3, 36−38). We proposed that the addition of HNG to DRZ might strengthen the anti-ROS function of DRZ and attenuate Dox-induced mitochondrial damage, resulting in greater cardiac protection. The results from our targeted analysis for mitochondrial related gene expression provided evidence showing that Dox treatment resulted in a global disturbance of mitochondrial gene expression, affecting 78 of 84 of measured genes compared with controls, and HNG + DRZ treatment ameliorated mitochondrial gene changes by reducing 40 of 84 genes that were elevated by Dox treatment. These results suggested that combined HNG + DRZ treatment blunted the Dox-induced mitochondrial disturbance in the heart as a possible mechanism of the observed cardiac protection.
The suggestion that combined HNG + DZR protected mitochondria from Dox-induced damage was further supported by the data that Dox-reduced Ucp2 protein abundance levels was reversed by HNG + DRZ administration. In contrast to the protein changes, our gene microarray data showed that Ucp2 mRNA was upregulated by Dox and reduced by HNG + DRZ cotreatment. We cannot explain the lack of concordance of mRNA expression and protein expression of Ucp2 in treated hearts. However, this discrepancy between mRNA and protein levels was in line with the general notion that mRNA levels were not sufficient to predict the corresponding protein levels (10, 23), and a posttranslational mechanism may have a significant contribution to the overall mitochondrial remodeling. It is important to note that HNG + DRZ reversed the Dox-induced decrease in Ucp2 protein or increased Ucp2 mRNA levels to control levels. Ucp2 protein is one of the members of mitochondrial transport proteins that regulate the mitochondrial membrane potential created by the proton gradient across the inner mitochondrial membrane. Increased Ucp2 induces proton conductance, and the enhanced proton conductance suppresses ROS production (4). Previous studies have shown that Dox treatment decreased cardiac Ucp2 protein expression that was associated with Dox-induced cardiac toxicity in rodents (15, 34, 35). Consistent with these reports, we demonstrated that Dox treatment decreased Ucp2 protein in the heart. HNG + DRZ treatment prevented the Dox-induced reduction of Ucp2, which might lead to suppressed ROS production and reduced DOX-induced cardiotoxicity. More studies will be needed to clarify the role of Ucp2 in Dox-induced ROS production in the heart and fully uncover the underlying mechanisms for the synergistic protection conferred by HNG and DRZ combined treatment.
We conclude that 1) HNG significantly enhances the cardioprotective effect of DRZ against Dox-induced cardiomyopathy and 2) this additive or synergistic cardioprotective effect of combined treatment by HNG and DRZ may be the results of preserved mitochondrial function in the heart. Thus, HNG can potentially serve as an adjuvant to DRZ in preventing Dox-induced cardiac dysfunction and cardiomyopathy.
GRANTS
This work was supported by the University of California-Los Angeles (UCLA) Clinical and Translational Science Institute (National Institutes of Health Grant UL1-TR-000124) to the Los Angeles Biomedical Research Institute and Harbor-UCLA Medical Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.L., R.S.S., Y.W., and C.W. conceived and designed research; Y.L., C.G., J.H., R.A., Y.J., M.R., S.R., V.A., J.Y., Y.Z., M.C., and Y.S. performed experiments; Y.L., C.G., J.H., R.A., M.R., and S.R. analyzed data; Y.L., R.S.S., and Y.W. interpreted results of experiments; Y.L. and C.G. prepared figures; Y.L. drafted manuscript; Y.L., C.G., R.S.S., Y.W., and C.W. edited and revised manuscript; Y.L., R.S.S., Y.W., and C.W. approved final version of manuscript.
REFERENCES
- 1.Asselin BL, Devidas M, Chen L, Franco VI, Pullen J, Borowitz MJ, Hutchison RE, Ravindranath Y, Armenian SH, Camitta BM, Lipshultz SE. Cardioprotection and safety of dexrazoxane in patients treated for newly diagnosed T-cell acute lymphoblastic leukemia or advanced-stage lymphoblastic non-hodgkin lymphoma: a report of the Children’s Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34: 854–862, 2016. doi: 10.1200/JCO.2015.60.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol 23: 15–25, 2007. doi: 10.1007/s10565-006-0140-y. [DOI] [PubMed] [Google Scholar]
- 3.Charununtakorn ST, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Potential roles of humanin on apoptosis in the heart. Cardiovasc Ther 34: 107–114, 2016. doi: 10.1111/1755-5922.12168. [DOI] [PubMed] [Google Scholar]
- 4.Cheng J, Nanayakkara G, Shao Y, Cueto R, Wang L, Yang WY, Tian Y, Wang H, Yang X. Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv Exp Med Biol 982: 359–370, 2017. doi: 10.1007/978-3-319-55330-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin YP, Keni J, Wan J, Mehta H, Anene F, Jia Y, Lue YH, Swerdloff R, Cobb LJ, Wang C, Cohen P. Pharmacokinetics and tissue distribution of humanin and its analogues in male rodents. Endocrinology 154: 3739–3744, 2013. doi: 10.1210/en.2012-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow EJ, Asselin BL, Schwartz CL, Doody DR, Leisenring WM, Aggarwal S, Baker KS, Bhatia S, Constine LS, Freyer DR, Lipshultz SE, Armenian SH. Late mortality after dexrazoxane treatment: a report from the Children’s Oncology Group. J Clin Oncol 33: 2639–2645, 2015. doi: 10.1200/JCO.2014.59.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conway A, McCarthy AL, Lawrence P, Clark RA. The prevention, detection and management of cancer treatment-induced cardiotoxicity: a meta-review. BMC Cancer 15: 366, 2015. doi: 10.1186/s12885-015-1407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Gödtel-Armbrust U, Wojnowski L. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer 14: 842, 2014. doi: 10.1186/1471-2407-14-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai VG, Herman EH, Moland CL, Branham WS, Lewis SM, Davis KJ, George NI, Lee T, Kerr S, Fuscoe JC. Development of doxorubicin-induced chronic cardiotoxicity in the B6C3F1 mouse model. Toxicol Appl Pharmacol 266: 109–121, 2013. doi: 10.1016/j.taap.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 10.Fortelny N, Overall CM, Pavlidis P, Freue GVC. Can we predict protein from mRNA levels? Nature 547: E19–E20, 2017. doi: 10.1038/nature22293. [DOI] [PubMed] [Google Scholar]
- 11.Gammella E, Maccarinelli F, Buratti P, Recalcati S, Cairo G. The role of iron in anthracycline cardiotoxicity. Front Pharmacol 5: 25, 2014. doi: 10.3389/fphar.2014.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao C, Ren S, Lee JH, Qiu J, Chapski DJ, Rau CD, Zhou Y, Abdellatif M, Nakano A, Vondriska TM, Xiao X, Fu XD, Chen JN, Wang Y. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Invest 126: 195–206, 2016. doi: 10.1172/JCI84015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao C, Howard-Quijano K, Rau C, Takamiya T, Song Y, Shivkumar K, Wang Y, Mahajan A. Inflammatory and apoptotic remodeling in autonomic nervous system following myocardial infarction. PLoS One 12: e0177750, 2017. doi: 10.1371/journal.pone.0177750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gidlund EK, von Walden F, Venojärvi M, Risérus U, Heinonen OJ, Norrbom J, Sundberg CJ. Humanin skeletal muscle protein levels increase after resistance training in men with impaired glucose metabolism. Physiol Rep 4: e13063, 2016. doi: 10.14814/phy2.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao E, Mukhopadhyay P, Cao Z, Erdélyi K, Holovac E, Liaudet L, Lee WS, Haskó G, Mechoulam R, Pacher P. Cannabidiol protects against doxorubicin-induced cardiomyopathy by modulating mitochondrial function and biogenesis. Mol Med 21: 38–45, 2015. doi: 10.2119/molmed.2014.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herman EH, Ferrans VJ. Preclinical animal models of cardiac protection from anthracycline-induced cardiotoxicity. Semin Oncol 25, Suppl 10: 15–21, 1998. [PubMed] [Google Scholar]
- 17.Hohensinner PJ, Takacs N, Kaun C, Thaler B, Krychtiuk KA, Pfaffenberger S, Aliabadi A, Zuckermann A, Huber K, Wojta J. Urokinase plasminogen activator protects cardiac myocytes from oxidative damage and apoptosis via hOGG1 induction. Apoptosis 22: 1048–1055, 2017. doi: 10.1007/s10495-017-1388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia Y, Lue YH, Swerdloff R, Lee KW, Cobb LJ, Cohen P, Wang C. The cytoprotective peptide humanin is induced and neutralizes Bax after pro-apoptotic stress in the rat testis. Andrology 1: 651–659, 2013. doi: 10.1111/j.2047-2927.2013.00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia Y, Ohanyan A, Lue YH, Swerdloff RS, Liu PY, Cohen P, Wang C. The effects of humanin and its analogues on male germ cell apoptosis induced by chemotherapeutic drugs. Apoptosis 20: 551–561, 2015. doi: 10.1007/s10495-015-1105-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karu I, Tahepold P, Ruusalepp A, Reimann E, Koks S, Starkopf J. Exposure to sixty minutes of hyperoxia upregulates myocardial humanins in patients with coronary artery disease−a pilot study. J Physiol Pharmacol 66: 899–906, 2015. [PubMed] [Google Scholar]
- 21.Lee C, Wan J, Miyazaki B, Fang Y, Guevara-Aguirre J, Yen K, Longo V, Bartke A, Cohen P. IGF-I regulates the age-dependent signaling peptide humanin. Aging Cell 13: 958–961, 2014. doi: 10.1111/acel.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipshultz SE, Franco VI, Miller TL, Colan SD, Sallan SE. Cardiovascular disease in adult survivors of childhood cancer. Annu Rev Med 66: 161–176, 2015. doi: 10.1146/annurev-med-070213-054849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell 165: 535–550, 2016. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Lue Y, Swerdloff R, Liu Q, Mehta H, Hikim AS, Lee KW, Jia Y, Hwang D, Cobb LJ, Cohen P, Wang C. Opposing roles of insulin-like growth factor binding protein 3 and humanin in the regulation of testicular germ cell apoptosis. Endocrinology 151: 350–357, 2010. doi: 10.1210/en.2009-0577. [DOI] [PubMed] [Google Scholar]
- 25.Lue Y, Swerdloff R, Wan J, Xiao J, French S, Atienza V, Canela V, Bruhn KW, Stone B, Jia Y, Cohen P, Wang C. The potent humanin analogue (HNG) protects germ cells and leucocytes while enhancing chemotherapy-induced suppression of cancer metastases in male mice. Endocrinology 156: 4511–4521, 2015. doi: 10.1210/en.2015-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67: 8839–8846, 2007. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 27.McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther 31: 63–75, 2017. doi: 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikolakopoulos P, Tzimagiorgis G, Goulis DG. Chatzopoulou F, Zepiridis L, Vavilis D. Serum humanin concentrations in women with pre-eclampsia compared to women with uncomplicated pregnancies. J Matern Fetal Neonatal Med 9: 1–7, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Sawyer DB. Anthracyclines and heart failure. N Engl J Med 368: 1154–1156, 2013. doi: 10.1056/NEJMcibr1214975. [DOI] [PubMed] [Google Scholar]
- 30.Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer 10: 337, 2010. doi: 10.1186/1471-2407-10-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun R, Wang J, Zheng Y, Li X, Xie T, Li R, Liu M, Cao Y, Lu L, Zhang Q, Zhang P. Traditional Chinese medicine baoxin decoction improves cardiac fibrosis of rats with dilated cardiomyopathy. Exp Ther Med 13: 1900–1906, 2017. doi: 10.3892/etm.2017.4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Surampudi P, Chang I, Lue Y, Doumit T, Jia Y, Atienza V, Liu PY, Swerdloff RS, Wang C. Humanin protects against chemotherapy-induced stage-specific male germ cell apoptosis in rats. Andrology 3: 582–589, 2015. doi: 10.1111/andr.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, Mendenhall NP, Sposto R, Chauvenet A, Schwartz CL. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J Clin Oncol 25: 493–500, 2007. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 34.Teshima Y, Akao M, Jones SP, Marbán E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res 93: 192–200, 2003. doi: 10.1161/01.RES.0000085581.60197.4D. [DOI] [PubMed] [Google Scholar]
- 35.Thompson KL, Rosenzweig BA, Zhang J, Knapton AD, Honchel R, Lipshultz SE, Retief J, Sistare FD, Herman EH. Early alterations in heart gene expression profiles associated with doxorubicin cardiotoxicity in rats. Cancer Chemother Pharmacol 66: 303−314, 2010. doi: 10.1007/s00280-009-1164-9. [DOI] [PubMed] [Google Scholar]
- 36.Thummasorn S, Apaijai N, Kerdphoo S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Humanin exerts cardioprotection against cardiac ischemia/reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc Ther 34: 404–414, 2016. doi: 10.1111/1755-5922.12210. [DOI] [PubMed] [Google Scholar]
- 37.Thummasorn S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. High-dose Humanin analogue applied during ischemia exerts cardioprotection against ischemia/reperfusion injury by reducing mitochondrial dysfunction. Cardiovasc Ther 35: e12289, 2017. doi: 10.1111/1755-5922.12289. [DOI] [PubMed] [Google Scholar]
- 38.Thummasorn S, Shinlapawittayatorn K, Khamseekaew J, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Humanin directly protects cardiac mitochondria against dysfunction initiated by oxidative stress by decreasing complex I activity. Mitochondrion 38: 31–40, 2018. doi: 10.1016/j.mito.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Vejpongsa P, Yeh ET. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol 64: 938–945, 2014. doi: 10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- 40.Voigt A, Jelinek HF. Humanin: a mitochondrial signaling peptide as a biomarker for impaired fasting glucose-related oxidative stress. Physiol Rep 4: e12796, 2016. doi: 10.14814/phy2.12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol 93: 105–115, 2003. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 42.Wallace KB. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc Toxicol 7: 101–107, 2007. doi: 10.1007/s12012-007-0008-2. [DOI] [PubMed] [Google Scholar]
- 43.Wang JJ-C, Rau C, Avetisyan R, Ren S, Romay MC, Stolin G, Gong KW, Wang Y, Lusis AJ. Genetic dissection of cardiac remodeling in an isoproterenol-induced heart failure mouse model. PLoS Genet 12: e1006038, 2016. doi: 10.1371/journal.pgen.1006038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu V. Dexrazoxane: a cardioprotectant for pediatric cancer patients receiving anthracyclines. J Pediatr Oncol Nurs 32: 178–184, 2015. doi: 10.1177/1043454214554008. [DOI] [PubMed] [Google Scholar]
- 45.Xiao J, Kim SJ, Cohen P, Yen K. Humanin: functional interfaces with IGF-I. Growth Horm IGF Res 29: 21–27, 2016. doi: 10.1016/j.ghir.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeh ET, Tong AT, Lenihan DJ, Yusuf SW, Swafford J, Champion C, Durand JB, Gibbs H, Zafarmand AA, Ewer MS. Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation 109: 3122–3131, 2004. doi: 10.1161/01.CIR.0000133187.74800.B9. [DOI] [PubMed] [Google Scholar]
- 47.Yen K, Lee C, Mehta H, Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol 50: R11–R19, 2013. doi: 10.1530/JME-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu W, Sun H, Zha W, Cui W, Xu L, Min Q, Wu J. Apigenin attenuates adriamycin-induced cardiomyocyte apoptosis via the PI3K/AKT/mTOR pathway. Evid Based Complement Alternat Med 2017: 2590676, 2017. doi: 10.1155/2017/2590676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18: 1639–1642, 2012. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Liang X, Liao S, Wang W, Wang J, Li X, Ding Y, Liang Y, Gao F, Yang M, Fu Q, Xu A, Chai Y-H, He J, Tse H-F, Lian Q. Potent paracrine effects of human induced pluripotent stem cell-derived mesenchymal stem cells attenuate doxorubicin-induced cardiomyopathy. 5: 11235, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]