

Abstract
Cardiac myosin-binding protein C (cMyBP-C) is involved in the regulation of cardiac myofilament contraction. Recent evidence showed that protein kinase D (PKD) is one of the kinases that phosphorylate cMyBP-C. However, the mechanism by which PKD-induced cMyBP-C phosphorylation affects cardiac contractile responses is not known. Using immunoprecipitation, we showed that, in contracting cardiomyocytes, PKD binds to cMyBP-C and phosphorylates it at Ser315. The effect of PKD-mediated phosphorylation of cMyBP-C on cardiac myofilament function was investigated in permeabilized ventricular myocytes, isolated from wild-type (WT) and from cMyBP-C knockout (KO) mice, incubated in the presence of full-length active PKD. In WT myocytes, PKD increased both myofilament Ca2+ sensitivity (pCa50) and maximal Ca2+-activated tension of contraction (Tmax). In cMyBP-C KO skinned myocytes, PKD increased pCa50 but did not alter Tmax. This suggests that cMyBP-C is not involved in PKD-mediated sensitization of myofilaments to Ca2+ but is essential for PKD-induced increase in Tmax. Furthermore, the phosphorylation of both PKD-Ser916 and cMyBP-C-Ser315 was contraction frequency-dependent, suggesting that PKD-mediated cMyBP-C phosphorylation is operational primarily during periods of increased contractile activity. Thus, during high contraction frequency, PKD facilitates contraction of cardiomyocytes by increasing Ca2+ sensitivity and by an increased Tmax through phosphorylation of cMyBP-C.
Keywords: cardiomyocyte contractility, calcium sensitivity, protein kinase A, phospho-cardiac myosin-binding protein C-serine-315
protein kinase d (PKD) belongs to a serine/threonine kinase family that is closely related to novel and conventional members of the protein kinase C (PKC) family, with which it shares the ability to be activated by diacylglycerol (DAG) (26). One of the mechanisms to activate PKD is via an A-kinase anchoring protein, AKAP-Lbc, which assembles a PKD activation complex and targets PKD to a specific subcellular location (3). AKAP-Lbc then recruits an upstream kinase, PKC, and coordinates protein kinase A (PKA) phosphorylation events that lead to the release of activated PKD at a specific location into the cell. Thus, AKAP-Lbc synchronizes PKA and PKC activities in a manner that leads to the activation of PKD (3).
Several proteins have been identified to be substrates of PKD, including cardiac troponin I (TnI), telethonin, and cardiac myosin-binding protein C (cMyBP-C) (9). cMyBP-C is a component of the cardiac sarcomere thick filaments that contributes to myosin assembly (1). cMyBP-C prevents weakly bound cross-bridges and increases force during the power stroke, thereby limiting ATP consumption per cross-bridge force production cycle (25). Furthermore, cMyBP-C has been shown to play a role in maintaining complete relaxation of cardiomyocytes (34). Several animal models have been developed to evaluate the role of cMyBP-C. Most of the introduced mutations in the cMyBP-C gene resulted in hypertrophic cardiomyopathy, indicative for a reduced contractile power (43). In rat, three phosphorylation sites have been identified in the MyBP-C motif located between C1 and C2, including Ser286, Ser295, and Ser315 (45, 46). In mouse myocardium, the cMyBP-C-Ser302 is structurally identical to the Ser315 phosphorylation site in rat. Upon phosphorylation of the MyBP-C motif, cMyBP-C interacts with the S2 region of myosin at its C1-C2-MyBP-C domains (1). cMyBP-C also binds to actin at the C1-MyBP-C motif, which could be important for the arrangement of myosin-actin filaments in the sarcomere (1). Phosphorylation of cMyBP-C by PKA appears to accelerate the rates of force development in systole and the rates of relaxation in diastole, thereby increasing cardiac contractility (8, 10). Recently, in connection with an observed binding of PKD to cMyBP-C in quiescent trabeculae, Bardswell and coworkers (1) demonstrated that the catalytic subunit of PKD interacts with cMyBP-C-Ser302 in skinned mouse myocardium. Independently, PKD has been shown to be activated during contraction of cardiomyocytes (12, 28). Whether PKD-mediated cMyBP-C phosphorylation is also increased upon contraction of cardiomyocytes remains to be established.
Phosphorylation of cMyBP-C is significantly reduced in patients with heart failure as it is in experimental models mimicking this pathology (14, 37). Therefore, it is relevant to study the physiological relevance of PKD-induced cMyBP-C phosphorylation during contraction, which is the natural state of living cardiomyocytes. In the present study, we analyzed the consequences of increased contraction frequency on PKD-mediated cMyBP-C phosphorylation and how this phosphorylation facilitates coping with increased contractile demands.
MATERIALS AND METHODS
Animals
Homozygous cardiac cMyBP-C-null (16) mice were generated in a Black Swiss background as previously described by Carrier et al. (4). Male Lewis rats (200–250 g) were obtained from Charles River. All animal studies were approved by the Committee on Animal Experimentation of Maastricht University and performed according to institutional guidelines and regulations.
Materials
Antibodies directed against PKD/PKCμ, phospho-PKD/PKC-μ (Ser916), and phospho-Troponin-I (Ser22/23) were obtained from Cell Signaling. The MYBPC3 antibody was obtained from Santa Cruz Biotechnology. The phospho-cardiac myosin light chain-2 (MLC2) (21) and the rat cMyBP-C-Ser315 antibody (36) were homemade. The full-length active PKD was synthesized in the laboratory of Prof. Johan Van Lint (42).
Oligomycin, staurosporine, calphostin-C, Gö-6983, protein kinase inhibitor (PKI), and isoproterenol were from Sigma (St. Louis, MO).
Isolation and Treatment of Adult Rat Cardiomyocytes
Cardiomyocytes were isolated from male Lewis rats (200–250 g) using a Langendorff perfusion system and a Krebs Henseleit bicarbonate medium equilibrated with a 95% O2-5% CO2 gas phase at 37°C as previously described (27). Cardiomyocytes were incubated for 6 min at 37°C with continuous shaking and electrically stimulated for 6 min using a commercially available Ionoptix stimulator (4 Hz, 40 V, pulse duration 5 ms).
Immunoprecipitation
Rat cardiomyocytes were pelleted and resuspended for 1 h in ice-cold lysis buffer: 50 mmol/l Tris·HCl (pH = 7.4), 150 mmol/l NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, Complete protease inhibitor (Roche), and PhosSTOP phosphatase inhibitor (Roche). Cells were pelleted by centrifugation for 10 min at 14,000 g, 4°C. Supernatant fractions (500 μl) were incubated with the PKD or MYBPC3 (K-16) antibody (5 μg) overnight at 4°C on a rotating wheel. Thereafter, 50 μl of Protein G Sepharose bead suspension (GE Healthcare) were added, and incubations were continued for 1 h at 4°C. The beads were collected by pulse centrifugation and washed. The final pellet was resuspended in 30 μl 1× sample buffer and heated to 95°C for 5 min. The beads were pelleted by centrifugation at 12,000 g for 20 s, and the supernatant was used for Western blot analysis.
Immunoblotting
Proteins were separated by using 4–12% SDS-PAGE gels and transferred to nitrocellulose membranes for Western Blotting. The membranes were probed with the relevant primary antibodies. The protein bands were visualized using enhanced chemiluminescence, and immunoblot intensities were analyzed by densitometry as described previously (28).
Permeabilized Myocyte Preparations
Ventricular mouse myocytes were dissociated mechanically as previously described (6). The heart was perfused retrogradely (1.6 ml/min) with a Ca2+-free Hanks-HEPES buffered solution for 5 min at room temperature then preskinned by perfusing with relaxing solution containing 1% Triton X-100 and protease inhibitors (complete cocktail tablets; Roche) (1 tablet was dissolved in 50 ml relaxing solution) for 10 min. Left ventricular strips (8 mm long, 2 mm width, 1 mm thick) were dissected from the same heart, frozen in liquid nitrogen for biochemistry, or further skinned for 10 min at 4°C in relaxing solution containing 1% Triton X-100 for mechanical experiments. The strips were then blended in fresh ice-cold relaxing solution at 6,098 g for 2–3 s (Polytron PT45–80 with a PTA 10TS shaft; KINEMATICA), resulting in a suspension of small clumps of myocyte-sized preparations and cell fragments. The homogenized tissue was filtered and centrifuged at 500 g for 1 min at 4°C. The pellet containing the myocytes was skinned in 0.3% Triton X-100 solution for 6 min to remove remaining membranes and then was extensively washed in relaxing solution. To prevent degradation, all solutions contained protease inhibitors (see above). Cells were kept on ice and used within the day. Some cells were used immediately after cell isolation for mechanical experiments; others were solubilized for immunoblotting. For PKD stimulation, cells were preincubated for 30 min at room temperature with 10 μg of full-length active PKD per milliliter of relaxing solution.
Solutions.
Ca2+-activating solutions were prepared by mixing relaxing (pCa 9.0) and maximal activating (pCa 4.5) stock solutions. The relaxing and activating solutions contained (in mmol/l) 12 phosphocreatine, 30 imidazole, 1 free Mg2+, 10 EGTA, 3.3 Na2ATP, and 0.3 dithiothreitol with pCa 9.0 (relaxing solution) and pCa 4.5 (maximal activating solution), pH 7.1.
Force measurements.
The procedure of cell attachment has been described previously (6). A skinned myocyte was attached to a piezo-resistive strain gauge (AE801 sensor; Memscap, Crolle, France; 500 Hz unloaded resonant frequency, compliance of the strain gauge 0.03 μm/μN) and to a stepper motor-driven micromanipulator (MP-285; Sutter Instrument), cardiomyocyte at 2.0 μm sarcomeric length (44). Contractions were induced by application of solutions containing various pCa (from 6.2 to 4.5).
Statistics
Data are presented as means ± SE. The paired Student's t-test and one-way ANOVA (for the mechanical experiments) was used. When significant interactions were found, a Bonferroni post hoc test was applied. A value of P < 0.05 indicates a statistical significance.
RESULTS
Contraction Increases PKD Binding and Phosphorylation on cMyBP-C
We previously demonstrated that contraction induced by electric field stimulation (EFS) activates PKD in isolated rat cardiomyocytes (28). Because cMyBP-C has been shown to be one of the targets of PKD (1), we first investigated whether phosphorylation of cMyBP-C is increased during contraction. We measured phosphorylation of cMyBP-C-Ser315 in electric field-stimulated rat cardiomyocytes, since this site has been shown to be preferentially phosphorylated by the catalytic subunit of PKD in quiescent mouse myocardium (1). Compared with quiescent cardiomyocytes, EFS increased the phosphorylation of cMyBP-C-Ser315 (Fig. 1A). During EFS of rat cardiomyocytes, PKD phopshorylates cMyBP-C-Ser315, and it does this by directly interacting with cMyBP-C, as shown by immunoprecipitation (Fig. 1B). Immunoprecipitation of PKD showed an increased binding to cMyBP-C-Ser315 with EFS, indicating that, during EFS, PKD phopsphorylates cMyBP-C-Ser315 by directly interacting with cMyBP-C (Fig. 1B). Reciprocally, after immunoprecipitation of cMyBP-C, PKD was co-immunoprecipitated with cMyBP-C, and the level of coimmunoprecipitated PKD was markedly higher in EFS-treated cardiomyocytes than in quiescent cardiomyocytes (Fig. 1B, second panel).
Fig. 1.
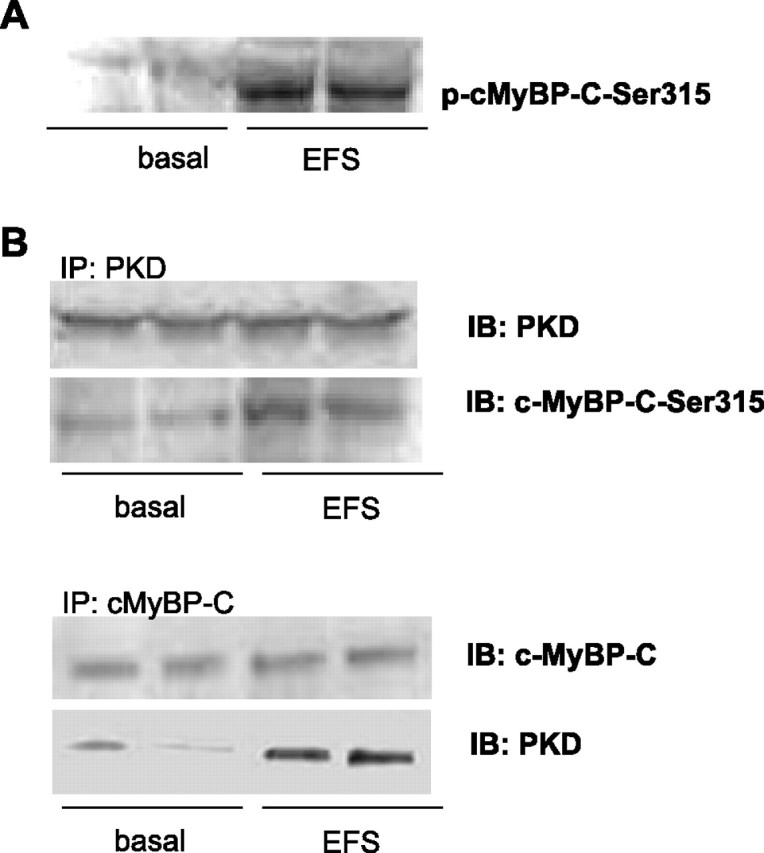
Interaction between protein kinase D (PKD) and cardiac myosin-binding protein C (cMyBP-C) upon increased contraction. Isolated rat cardiomyocytes were electrical field stimulated (EFS) at 4 Hz for 6 min and were compared with nonstimulated cardiomyocytes. A: cMyBP-C-Ser315 becomes phosphorylated (p) during EFS. B: upon PKD immunoprecipitation (IP), cMyBP-C-Ser315 and PKD were detected by immunoblotting (IB) (35). Upon cMyBP-C IP, cMyBP-C and PKD were detected by immunoblotting (35). Results shown are representative for three independent experiments.
We then applied a pharmacological approach to inhibit activation of PKD in EFS-treated cardiomyocytes by pretreating the cardiomyocytes with various PKC/PKD inhibitors (Fig. 2). We used staurosporine as a general PKC/PKD inhibitor, calphostin-C as an inhibitor of all diacylglycerol-activated PKC and PKD isoforms, and finally Gö-6983, which blocks conventional PKCs. The EFS-induced phosphorylation of cMyBP-C-Ser315 was prevented by staurosporine or calphostin-C, but not with Gö-6983. To further explore the PKC-dependent and PKC-independent activation of PKD following EFS, cardiomyocytes were treated with oligomycin, which has been shown to activate only PKD and not the main cardiac PKC isoforms (28). Similar to EFS, oligomycin treatment of cardiomyocytes increased the phosphorylation of cMyBP-C-Ser315, which was blocked by staurosporine or calphostin-C, but not with Gö-6983. Altogether the results suggest that, during contraction, cMyBP-C is a direct target of PKD.
Fig. 2.

Effects of protein kinase C/PKD inhibitors on cMyBP-C phosphorylation in cardiomyocytes. Isolated rat cardiomyocytes were preincubated for 15 min at 37°C with or without addition of 1 μmol/l staurosporine (Stau), 2 μmol/l calphostin-C (20), or 10 μmol/L Gö-6983. Subsequently, cardiomyocytes were electric field stimulated or treated with 5 μmol/l oligomycin. Samples were immunoblotted for Ser315 phosphorylation of cMyBP-C and caveolin 3 (Cav3) (loading control). The blots shown are representative for three independent experiments.
cMyBP-C-Ser315 Phosphorylation by PKD is Contraction Frequency Dependent
PKD-mediated cMyBP-C phosphorylation was determined at different EFS frequencies in isolated rat cardiomyocytes. Compared with quiescent rat cardiomyocytes, EFS at 1, 2, and 4 Hz increased PKD-Ser916 phosphorylation by 1.7-, 2.7-, and 3.2-fold and cMyBP-C-Ser315 phosphorylation by 3.1-, 4.4-, and 5.1-fold, respectively (Fig. 3). The parallel upregulation in contraction-induced PKD and cMyBP-C phosphorylation strongly supports the concept that PKD-dependent phosphorylation is instrumental for the cardiomyocyte to adjust to an increased contraction frequency.
Fig. 3.
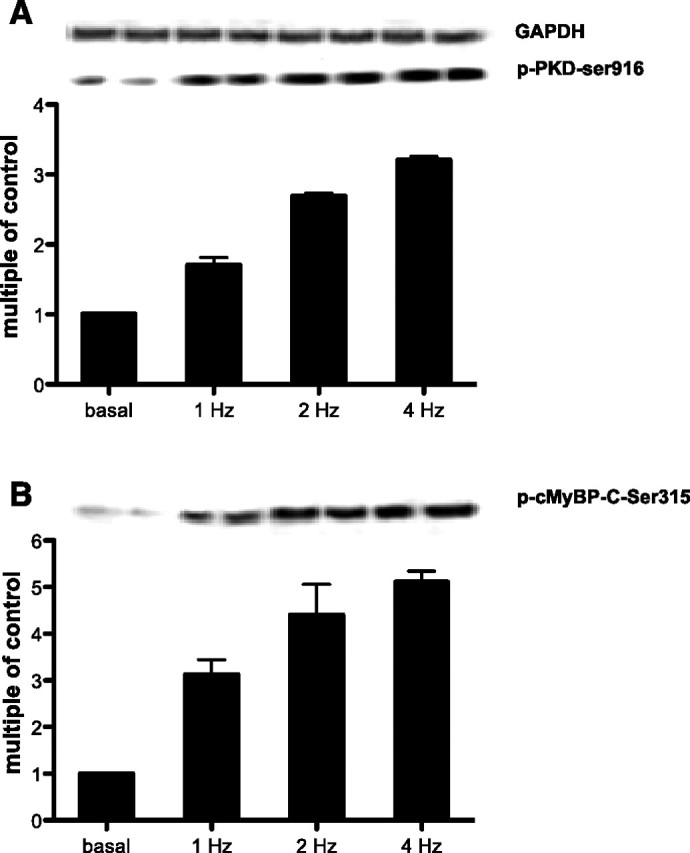
PKD-Ser916 (A) and cMyBP-C-Ser315 (B) phosphorylation increases upon increased contraction frequency. Isolated rat cardiomyocytes were electrically stimulated at 1, 2, and 4 Hz for 6 min and were compared with quiescent cells. Samples were probed against phospho-PKD-Ser916 and phospho-cMyBP-C-Ser315 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, loading control). Values are expressed as multiple of control. Results are representative of 3 independent experiments.
PKD-Dependent Phosphorylation of cMyBP-C Regulates Contractility in Cardiomyocytes
The effect of PKD-mediated phosphorylation of cMyBP-C on myofilament function was studied in permeabilized ventricular cardiomyocytes derived from cMyBP-C-knockout (KO) mice and wild-type (WT) littermates. Skinned cardiomyocytes were incubated with full-length active PKD to mimic as close as possible the in vivo conditions. The stimulation with PKD of the WT skinned myocytes induced an increase in cMyBP-C-Ser315 phosphorylation (4-fold), and to a lesser extent in TnI phosphorylation (2-fold) and MLC2 phosphorylation (2-fold) (Fig. 4A).
Fig. 4.

PKD increases myofilament Ca2+ sensitivity and maximal tension of contraction. A: Western blot analysis of levels of p-MyBP-Ser315, p-troponin I (TnI), and p-myosin light chain-2 (MLC2) in mouse myofilaments incubated for 30 min with full-length active PKD. The blots show that PKD is capable of phosphorylation of all three proteins. Caveolin-3 is used as a loading control. B: the relationship between Ca2+-activated tension and intracellular Ca2+ concentrations was measured in isolated, permeabilized cardiomyocytes at 2.0 μm sarcomere length. The relationship was fitted with a modified Hill equation, and the pCa at which half of the maximal tension is developed (pCa50) was determined as an index of myofilament Ca2+ sensitivity as described previously (5). Incubation of wild-type (WT) cardiomyocytes with full-length active PKD increased myofilament Ca2+ sensitivity, which is indicated by the shift toward the left of the curve and the increase of pCa50. Also, incubation of WT cardiomyocytes with PKD increased maximal tension of contraction (Tmax), which is indicated by the upward shift of the curve. In cMyBP-C knockout (KO) cardiomyocytes, incubation with full-length active PKD still increased pCa50, whereas PKD was not able anymore to affect Tmax. C: β-blocker treatment of WT mice did not interfere with PKD-induced cMyBP-C or MLC2 phosphorylation but prevented PKD-mediated TnI phosphorylation. D: in both WT and cMyBP-C KO cardiomyocytes from mice, pretreated with β-blockers, the PKD-mediated effects on pCa50 and Tmax were abolished. Results are expressed as means ± SE, n = 12 cells/group. *P < 0.05.
The physiological effect of cMyBP-C phosphorylation by PKD and the relationship between Ca2+-activated tension and internal Ca2+ concentrations (expressed as pCa = −log[Ca2+]) in permeabilized myocytes from WT and cMyBP-C KO mice were determined. Experiments were performed at 2.0 μm sarcomere length (SL), which is the length close to the conditions of the experiments in intact myocytes and also because a previous publication showed that cMyBP-C affects myofilament Ca2+ sensitivity at short SL and that stretch masks this effect at longer lengths (6). In WT myocytes, PKD increased the absolute maximal Ca2+-activated tension of contraction (Tmax) and submaximal active tension starting from pCa 5.875. These effects of PKD on Tmax were abolished in cMyBP-C KO myocytes, showing that cMyBP-C is involved in the PKD-mediated increase in the tension of the muscle fiber during contraction. This raised the question whether this increase in force was due to an increase in myofilament Ca2+ sensitivity. PKD induced a leftward shift of the relative tension-pCa relationship curve and therefore increased in pCa50 in both WT and KO myocytes (Fig. 4B), indicating that the PKD-mediated effect on pCa50 was independent of cMyBP-C (Fig. 4B). Other contractile parameters such as Hill coefficient and passive tension were not affected by PKD treatment (data not shown).
To test whether the PKD-mediated effects on myofilament function were dependent on prior PKA activation, cMyBP-C KO and WT mice were pretreated for 3 days before death with the nonselective β-blocker propranolol, which neutralizes PKA activity. In WT mice pretreated with β-blocker, full-length active PKD was able to phosphorylate cMyBP-C-Ser315 and MLC-2 but not TnI (Fig. 4C). In WT and KO mice pretreated with β-blocker, Ca2+ sensitivity was increased compared with nontreated mice (5.63 ± 0.02 to 5.78 ± 0.04 and 5.66 ± 0.02 to 5.83 ± 0.05 in WT and KO, respectively), which is an expected effect of the β-blocker treatment. Also, a reduction in phosphorylated TnI was observed. PKD did not alter either pCa50 or Tmax (Fig. 4D), indicating that the effect of PKD on pCa50 and Tmax is dependent on prior PKA activation. Because the β-blocker treatment neutralized the effect of PKD on Tmax, but not on cMyBP-C-Ser315 phosphoryation, we tested whether PKD was able to phosphorylate cMyBP-C-Ser295. Previously, it was shown that cMyBP-C-Ser282 in mouse, which is equivalent to cMyBP-C-Ser295 in rat (47), becomes phosphorylated upon PKA activation (15). However, PKD is not able to phosphorylate cMyBP-C-Ser295, neither in β-blocker-treated or in untreated mice (Fig. 5A). This is in agreement with an earlier publications (1, 47), which showed that, in mouse cardiomyocytes, PKD only phosphorylates Ser302 and not Ser282 or Ser273 on cMyBP-C (which are equivalent to cMyBP-C-Ser315, Ser295, and Ser273 in rat).
Fig. 5.
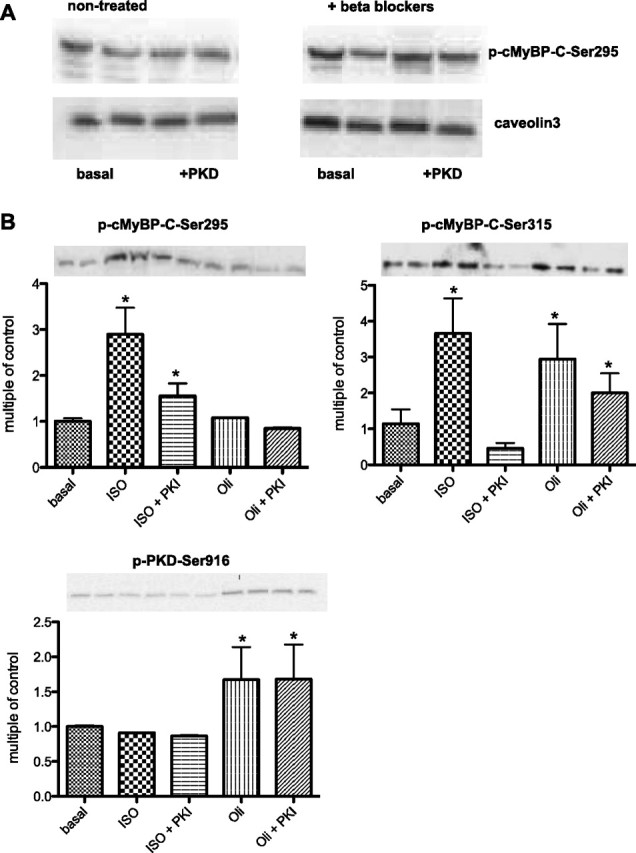
cMyBP-C-Ser315 phosphorylation is independent of acute PKA activation or inhibition. A: Western blot analysis of phosphorylation of MyBP-Ser295 in mouse myofilaments incubated for 30 min with full-length active PKD from untreated or β-blocker-pretreated mice. B: phosphorylation of cMyBP-C-Ser295, cMyBP-C-Ser315, and PKD-Ser916 was measured in rat cardiomyocytes after isoproterenol (ISO) or oligomycin stimulation in the absence or presence of 10 μmol/l protein kinase inhibitor (PKI). The data shown are representative for three independent experiments. *P < 0.05 vs. basal conditions.
For better understanding of the role of PKA in the PKD-mediated cMyBP-C phosphorylation, we finally tested whether acute PKA activation is necessary for PKD to be able to phosphorylate cMyBP-C. Isoproterenol (10 μM), a classic agonist, was used to activate PKA and increase the intracellular concentration of cAMP in cardiomyocytes (29). Here we show that isoproterenol was able to induce both cMyBP-C-Ser295 and cMyBP-C-Ser315 phosphorylation. However, oligomycin, which activates PKD signaling (14), was only able to induce cMyBP-C-Ser315 phosphorylation (Fig. 5B). Furthermore, specific blockade of PKA with the PKI did not prevent the oligomycin-induced cMyBP-C-Ser315 phosphorylation (Fig. 5B), whereas it largely blocked the isoproterenol-induced phosphorylation of both cMyBP-C-Ser315 and cMyBP-C-Ser295 (Fig. 5B). Thus, PKD activation (using oligomycin or a full-length PKD construct) results in phosphorylation of only cMyBP-C-Ser315 and not cMyBP-C-Ser295, whereas isoproterenol stimulation results in phosphorylation of both cMyBP-C-Ser315 and cMyBP-C-Ser295. Because isoproterenol is not able to phosphorylate PKD-Ser916 (Fig. 5B), and PKI was not able to inhibit oligomycin-stimulated cMyBP-C-Ser315 phosphorylation, we can conclude that PKA is not upstream of PKD but rather acts via a separate pathway. Together, these data suggest that prior PKA activation is necessary for PKD-mediated cMyBp-C-Ser315 phosphorylation to affect myofilament function.
DISCUSSION
Phosphorylation of cMyBP-C is markedly decreased in experimental models of cardiac pathology and in human heart failure, and as such it has been considered to be an important regulator of cardiac contractility (2, 14, 37). Understanding its regulation and the mechanisms by which cMyBP-C phosphorylation mediates cardiac contractile responses is critical in developing its potential as therapeutic target. We therefore investigated the PKD-mediated regulation of myofilament contraction through phosphorylation of cMyBP-C, and we show that cMyBP-C is involved in PKD-mediated increase in myofilament activation, but not in the PKD-mediated Ca2+ sensitization of the myofilaments.
During Contraction, PKD Phosphorylates cMyBP-C-Ser315 in Rat Cardiomyocytes
This study is the first to show that, during contraction, cMyBP-C is a direct physiological substrate of PKD. Previously, cMyBP-C has been described as a substrate of PKD in trabeculae under noncontracting conditions (1). Our data show that, during contraction, PKD is activated, which is characterized by its Ser916 phosphorylation. This activation leads to an increased binding of PKD to cMyBP-C. In response to increased cardiac contractile activity (evoked by EFS of the cells), the Ser315 site of cMyBP-C becomes phosphorylated by PKD. During increased contraction frequencies, phosphorylation of both PKD-Ser916 and cMyBP-C-Ser315 increases similarly. This concomitant phosphorylation in combination with direct physical interaction strongly suggests a functional relationship between the two proteins.
PKD-Directed cMyBP-C Phosphorylation Alters the Cardiac Contractility
Several studies have shown that exercise increases Ca2+ sensitivity of muscle tension and leads to a higher generated force (11, 32, 44). In exercising rats, Tmax of the ventricular papillary muscle at optimal length increased significantly (31). Also, in porcine cardiac myofilaments, phosphorylation of cMyBP-C was increased in response to endurance exercise training (22). Here we report that phosphorylation of cMyBP-C-Ser315, by application of an activated full-length PKD, results in an increase in submaximal and maximal Ca2+ activation. We showed that interaction of PKD with cMyBP-C increased during contraction of cardiomyocytes. Furthermore, during a rise in contraction frequency, phosphorylation of both PKD and cMyBP-C was increased. Also, the relevance of submaximal activation and Tmax increases with increasing contraction frequency. Thus, the PKD-mediated effect on cMyBP-C appears to become relevant during periods of high contraction. The involvement of PKD in the force-frequency relationship is supported by data in cardiac-specific PKD KO mice that displayed normal contractility but were unable to respond to stress (18).
In submaximal and maximal contraction, cMyBP-C seems to be directly involved as a mediator in the PKD-driven force development. On the other hand, the PKD-mediated change in Ca2+ sensitivity was independent of cMyBP-C, suggesting the involvement of other regulatory proteins. This is in agreement with a recent study in mouse trabeculae stimulated with the catalytic subunit of PKD that showed that cTnI phosphorylation at Ser22/23 is responsible for PKD-mediated regulation of myofilament Ca2+ sensitivity but not for the PKD-mediated acceleration of cross-bridge cycling kinetics (1). Recent studies using transgenic animals have shown that cMyBP-C plays an essential role in the PKA-mediated acceleration of cross-bridge cycling kinetics (39, 41) and is required for PKA-induced decrease of myofilament Ca2+ sensitivity (6, 7). The mechanism by which PKD activation leads to an increased pCa50 may imply phosphorylation of other contractile proteins, such as MLC2. Experiments in rabbit septal muscle preparations revealed a significant frequency-dependent increase in MLC2 phosphorylation, which is known to increase myofilament Ca2+ sensitivity (19, 40). Also, increased MLC2 phosphorylation and heart rate are attributed to different levels of treadmill exercise or β-adrenergic stimulation and inhibition (17). These data are in line with the concept that increased contraction frequency results in activation of PKD and subsequent Ca2+ sensitivity, independent of cMyBP-C. Nevertheless, Cuello and coworkers (9) showed a PKD-reduced myofilament Ca2+ sensitivity in mice pretreated with propranolol, which prevents PKA activation (9). This study also reported that PKD-mediated TnI phosphorylation decreased Ca2+ sensitivity. Their data are contradictory with our results. However, there are several methodological differences that could account for the discrepancies. For example, we used a full-length PKD, whereas the other study was performed with only the catalytic domain. It seems likely that full-length PKD and catalytic domain PKD have different binding characteristics and that these differences may affect the interaction between PKD and target proteins and/or the extent of phosphorylation. Also, the SL (2.0 vs. 2.2 μm) and the condition of PKD stimulation (separate vs. same batch of nonstimulated and PKD-stimulated cells) were different. Finally, the other investigators used only propranolol-pretreated animals.
In the present study, in propranolol-pretreated mice, PKD was still able to phosphorylate cMyBP-C-Ser315 but lost its effects on maximal activation and myofilament Ca2+ sensitivity in both WT and KO mice. Using a transgenic mouse model in which particular sites of TnI cannot be phosphorylated by PKA, Bardswell and coworkers (1) showed that PKD lost its ability to change myofilament Ca2+ sensitivity. This has also been described in human cardiomyocytes derived from donor and failing hearts in which the phoshorylation background of sarcomeric proteins affected PKA-dependent phosphorylation of TnI and myofilament Ca2+ sensitivity (23). Together with our results, this suggests that the effect of PKD on myofilament properties is dependent on prior PKA-dependent phosphorylation of regulatory contractile proteins. Upon activation, PKA is able to desensitize the muscle fiber for Ca2+-mediated force development and to increase cross-bridge cycle kinetics (1, 24, 30, 33). In our study, acute PKA activation does lead to direct phosphorylation of both cMyBP-C-Ser315 and cMyBP-Ser295, whereas PKD activation only leads to phosphorylation of cMyBP-Ser315 and not cMyBP-C-Ser295. Also, PKA inhibition by PKI did not affect phosphorylation of PKD-Ser916, which indicates that PKA does not function uptream of PKD. Most likely, a former PKA-mediated cMyBP-C phosphorylation of Ser295 may be necessary for the PKD-mediated effect on myofilament Tmax. Thus, PKD-mediated cMyBP-C phosphorylation alone appears to be insufficient to increase myofilament Tmax and may require preceding phosphorylation particular residues. Furthermore, application of the β-adrenergic blocker propranolol led to a marked increase in pCa50, which could not be further increased by PKD. Although propranolol pretreatment did not interfere with the PKD-mediated MLC2 phosphorylation, it did prevent PKD-induced TnI phosphorylation. Previously, it has been shown that isoproterenol increases phosphorylation of both TnI and MLC2. Also, it has been shown that MLC2 phosphorylation increases myofilament Ca2+ sensitivity (19, 33). Thus, PKD-stimulated increase in Ca2+ sensitivity is at least in part mediated by phosphorylation of TnI and MLC-2.
Because phosphorylation of both PKD-Ser916 and cMyBP-C-Ser315 increases during increased contraction frequencies, and the PKD-mediated effect on myofilament Tmax is inhibited by inhibiting the PKA pathway (using β-adrenergic blockers), our results suggest that combined PKA and PKD activation becomes necessary during increased contraction or exercise to coordinate the contraction mechanics. Interestingly, there is a basal β-adrenergic tone that is increased during various conditions of cardiac stress, such as exercise (38) and heart failure (5). Given that, during increased contraction, PKD is also involved in enhanced glucose uptake (12, 28), it can be suggested that PKD activation helps the cardiomyocytes to cope with an increased contraction frequency by 1) providing the cells with energy-rich substrate through an increase in glucose uptake and 2) phosphorylating cMyBP-C to increase Tmax. In this way, PKA is necessary for basal myofilament function, whereas PKD becomes important during increased contraction frequencies. Thus, PKD might provide a way for mutual adjustment of cardiac energetics and contractile mechanics during increased contraction frequencies (Fig. 6).
Fig. 6.
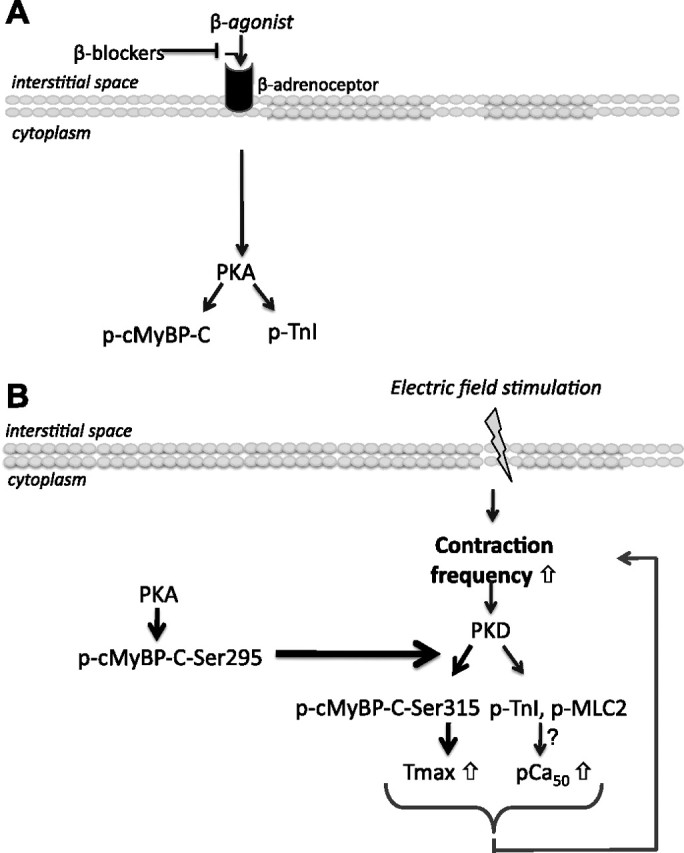
Schematic representation of the combined PKA and PKD action during increased contraction or exercise, needed to coordinate the contraction mechanics. A: a basal β-adrenergic tone leads to the activation of PKA and regulates contraction by phosphorylation of sarcomeric proteins, such as cMyBP-C and TnI. B: stimuli, like electric field stimulation, will increase the contraction frequency of the cardiomyocytes. This increase will lead to phosphorylation of PKD and cMyBP-C-Ser315. To cope with increased contractile demands, PKD facilitates contraction by increasing the Ca2+ sensitivity of contraction (pCa50) and by inducing cMyBP-C-Ser315 phosphorylation, which leads to an increase in Tmax. However, prior PKA activation of cMyBP-C-Ser295 is necessary for PKD-mediated cMyBP-C-Ser315 phosphorylation, so the combined action of both kinases is needed to come to a full deployment of contractile force.
In conclusion, an increase in contraction frequency induced by EFS leads to the phosphorylation of both PKD and cMyBP-C in isolated cardiomyocytes. To facilitate coping with increased contractile demands, PKD increases the myofilament Ca2+ sensitivity and induces cMyBP-C phosphorylation, which leads to an increase in maximal tension of contraction. In addition, PKD increase glucose uptake for energy production. Thus, PKD harmonizes contractile force with Ca2+ sensitivity and with energy supply and is therefore essential for the cardiomyocyte to properly respond to an increased contraction frequency, which happens during periods of increased contractile activity when both the contractile performance and energy demand of the cardiomyocytes are highest.
GRANTS
This study was funded by the transnational University Limburg and the European Community (Integrated Project LSHM-CT-2004-005272, Exgenesis).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: E.D., J.F.G., and J.J.L. conception and design of research; E.D., O.F.C., and I.L.-S. performed experiments; E.D., O.F.C., and I.L.-S. analyzed data; E.D., O.F.C., R.W.S., L.C., and J.J.L. interpreted results of experiments; E.D. prepared figures; E.D., O.F.C., L.C., G.J.v.E., and J.J.L. drafted manuscript; E.D., O.F.C., R.W.S., S.S., J.V.L., L.C., G.J.v.E., J.F.G., and J.J.L. edited and revised manuscript; E.D., O.F.C., R.W.S., I.L.-S., S.S., J.V.L., L.C., G.J.v.E., J.F.G., and J.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Thomas Eschenhagen (University Medical Center Hamburg-Eppendorf) for stimulating discussions and Elisabeth Krämer and Birgit Geertz (University Medical Center Hamburg-Eppendorf) for technical support.
REFERENCES
- 1. Bardswell SC , Cuello F , Rowland AJ , Sadayappan S , Robbins J , Gautel M , Walker JW , Kentish JC , Avkiran M. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem : 5674–5682, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barefield D , Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol : 866–875, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carnegie GK , Smith FD , McConnachie G , Langeberg LK , Scott JD. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol Cell : 889–899, 2004. [DOI] [PubMed] [Google Scholar]
- 4. Carrier L , Knoll R , Vignier N , Keller DI , Bausero P , Prudhon B , Isnard R , Ambroisine ML , Fiszman M , Ross J , Schwartz K , Chien KR. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res : 293–304, 2004. [DOI] [PubMed] [Google Scholar]
- 5. Cazorla O , Lucas A , Poirier F , Lacampagne A , Lezoualc'h F. The cAMP binding protein Epac regulates cardiac myofilament function. Proc Natl Acad Sci USA : 14144–14149, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cazorla O , Szilagyi S , Vignier N , Salazar G , Kramer E , Vassort G , Carrier L , Lacampagne A. Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res : 370–380, 2006. [DOI] [PubMed] [Google Scholar]
- 7. Chen PP , Patel JR , Rybakova IN , Walker JW , Moss RL. Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol : 615–627, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colson BA , Bekyarova T , Locher MR , Fitzsimons DP , Irving TC , Moss RL. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res : 244–251, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cuello F , Bardswell SC , Haworth RS , Yin X , Lutz S , Wieland T , Mayr M , Kentish JC , Avkiran M. Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ Res : 864–873, 2007. [DOI] [PubMed] [Google Scholar]
- 10. de Tombe PP. Myosin binding protein C in the heart. Circ Res : 1234–1236, 2006. [DOI] [PubMed] [Google Scholar]
- 11. Diffee GM , Chung E. Altered single cell force-velocity and power properties in exercise-trained rat myocardium. J Appl Physiol : 1941–1948, 2003. [DOI] [PubMed] [Google Scholar]
- 12. Dirkx E , Schwenk RW , Coumans WA , Hoebers N , Angin Y , Viollet B , Bonen A , van Eys GJ , Glatz JF , Luiken JJ. Protein kinase D1 is essential for contraction-induced glucose uptake but is not involved in fatty acid uptake into cardiomyocytes. J Biol Chem : 5871–5881, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El-Armouche A , Pohlmann L , Schlossarek S , Starbatty J , Yeh YH , Nattel S , Dobrev D , Eschenhagen T , Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol : 223–229, 2007. [DOI] [PubMed] [Google Scholar]
- 15. El-Armouche A , Pohlmann L , Schlossarek S , Starbatty J , Yeh YH , Nattel S , Dobrev D , Eschenhagen T , Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol : 223–229, 2007. [DOI] [PubMed] [Google Scholar]
- 16. Ellwanger K , Kienzle C , Lutz S , Jin ZG , Wiekowski MT , Pfizenmaier K , Hausser A. Protein kinase D controls voluntary-running-induced skeletal muscle remodelling. Biochem J : 327–324, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fentzke RC , Buck SH , Patel JR , Lin H , Wolska BM , Stojanovic MO , Martin AF , Solaro RJ , Moss RL , Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol : 143–157, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fielitz J , Kim MS , Shelton JM , Qi X , Hill JA , Richardson JA , Bassel-Duby R , Olson EN. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci USA : 3059–3063, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fitzsimons DP , Bodell PW , Baldwin KM. Phosphorylation of rodent cardiac myosin light chain 2: effects of exercise. J Appl Physiol : 2447–2453, 1989. [DOI] [PubMed] [Google Scholar]
- 20. Gao G , Vandenberghe LH , Alvira MR , Lu Y , Calcedo R , Zhou X , Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol : 6381–6388, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grimm M , Mahnecke N , Soja F , El-Armouche A , Haas P , Treede H , Reichenspurner H , Eschenhagen T. The MLCK-mediated alpha1-adrenergic inotropic effect in atrial myocardium is negatively modulated by PKCepsilon signaling. Br J Pharmacol : 991–1000, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hinken AC , Korte FS , McDonald KS. Porcine cardiac myocyte power output is increased after chronic exercise training. J Appl Physiol : 40–46, 2006. [DOI] [PubMed] [Google Scholar]
- 23. Kooij V , Saes M , Jaquet K , Zaremba R , Foster DB , Murphy AM , Dos Remedios C , van der Velden J , Stienen GJ. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol : 954–963, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Layland J , Solaro RJ , Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res : 12–21, 2005. [DOI] [PubMed] [Google Scholar]
- 25. Lecarpentier Y , Vignier N , Oliviero P , Guellich A , Carrier L , Coirault C. Cardiac Myosin-binding protein C modulates the tuning of the molecular motor in the heart. Biophys J : 720–728, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lint JV , Rykx A , Vantus T , Vandenheede JR. Getting to know protein kinase D. Int J Biochem Cell Biol : 577–581, 2002. [DOI] [PubMed] [Google Scholar]
- 27. Luiken JJ , van Nieuwenhoven FA , America G , van der Vusse GJ , Glatz JF. Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J Lipid Res : 745–758, 1997. [PubMed] [Google Scholar]
- 28. Luiken JJ , Vertommen D , Coort SL , Habets DD , El Hasnaoui M , Pelsers MM , Viollet B , Bonen A , Hue L , Rider MH , Glatz JF. Identification of protein kinase D as a novel contraction-activated kinase linked to GLUT4-mediated glucose uptake, independent of AMPK. Cell Signal : 543–556, 2008. [DOI] [PubMed] [Google Scholar]
- 29. Luiken JJ , Willems J , Coort SL , Coumans WA , Bonen A , Van Der Vusse GJ , Glatz JF. Effects of cAMP modulators on long-chain fatty-acid uptake and utilization by electrically stimulated rat cardiac myocytes. Biochem J : 881–887, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsuba D , Terui T , JOU , Tanaka H , Ojima T , Ohtsuki I , Ishiwata S , Kurihara S , Fukuda N. Protein kinase A-dependent modulation of Ca2+ sensitivity in cardiac and fast skeletal muscles after reconstitution with cardiac troponin. J Gen Physiol : 571–581, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mole PA. Increased contractile potential of papillary muscles from exercise-trained rat hearts. Am J Physiol Heart Circ Physiol : H421–H425, 1978. [DOI] [PubMed] [Google Scholar]
- 32. Natali AJ , Wilson LA , Peckham M , Turner DL , Harrison SM , White E. Different regional effects of voluntary exercise on the mechanical and electrical properties of rat ventricular myocytes. J Physiol : 863–875, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paroo Z , Noble EG. Isoproterenol potentiates exercise-induction of Hsp70 in cardiac and skeletal muscle. Cell Stress Chaperones : 199–204, 1999. [PMC free article] [PubMed] [Google Scholar]
- 34. Pohlmann L , Kroger I , Vignier N , Schlossarek S , Kramer E , Coirault C , Sultan KR , El-Armouche A , Winegrad S , Eschenhagen T , Carrier L. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res : 928–938, 2007. [DOI] [PubMed] [Google Scholar]
- 35. Ribe D , Sawbridge D , Thakur S , Hussey M , Ledent C , Kitchen I , Hourani S , Li JM. Adenosine A2A receptor signaling regulation of cardiac NADPH oxidase activity. Free Radic Biol Med : 1433–1442, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sadayappan S , Gulick J , Klevitsky R , Lorenz JN , Sargent M , Molkentin JD , Robbins J. Cardiac myosin binding protein-C phosphorylation in a β-myosin heavy chain background. Circulation : 1253–1262, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sadayappan S , Gulick J , Osinska H , Martin LA , Hahn HS , Dorn GW , Klevitsky R , Seidman CE , Seidman JG , Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res : 1156–1163, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Slatton ML , Eichhorn EJ. Beta-blocker therapy for heart failure. Curr Opin Cardiol : 263–268, 1996. [DOI] [PubMed] [Google Scholar]
- 39. Stelzer JE , Patel JR , Walker JW , Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res : 503–511, 2007. [DOI] [PubMed] [Google Scholar]
- 40. Sweeney HL , Bowman BF , Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol Cell Physiol : C1085–C1095, 1993. [DOI] [PubMed] [Google Scholar]
- 41. Tong CW , Stelzer JE , Greaser ML , Powers PA , Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res : 974–982, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Lint JV , Sinnett-Smith J , Rozengurt E. Expression and characterization of PKD, a phorbol ester and diacylglycerol-stimulated serine protein kinase. J Biol Chem : 1455–1461, 1995. [DOI] [PubMed] [Google Scholar]
- 43. Vignier N , Schlossarek S , Fraysse B , Mearini G , Kramer E , Pointu H , Mougenot N , Guiard J , Reimer R , Hohenberg H , Schwartz K , Vernet M , Eschenhagen T , Carrier L. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res : 239–248, 2009. [DOI] [PubMed] [Google Scholar]
- 44. Wisloff U , Loennechen JP , Currie S , Smith GL , Ellingsen O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res : 162–174, 2002. [DOI] [PubMed] [Google Scholar]
- 45. Wu SC , Solaro RJ. Protein kinase C zeta. A novel regulator of both phosphorylation and de-phosphorylation of cardiac sarcomeric proteins. J Biol Chem : 30691–30698, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yuan C , Guo Y , Ravi R , Przyklenk K , Shilkofski N , Diez R , Cole RN , Murphy AM. Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts–evidence for novel phosphorylation sites. Proteomics : 4176–4186, 2006. [DOI] [PubMed] [Google Scholar]
- 47. Yuan C , Sheng Q , Tang H , Li Y , Zeng R , Solaro RJ. Quantitative comparison of sarcomeric phosphoproteomes of neonatal and adult rat hearts. Am J Physiol Heart Circ Physiol : H647–H656, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]